
Abstract
There are 3 common physiological estrogens, of which estradiol (E2) is seen to decline rapidly over the menopausal transition. This decline in E2 has been associated with a number of changes in the brain, including cognitive changes, effects on sleep, and effects on mood. These effects have been demonstrated in both rodent and non-human preclinical models. Furthermore, E2 interactions have been indicated in a number of neuropsychiatric disorders, including Alzheimer’s disease, schizophrenia, and depression. In normal brain aging, there are a number of systems that undergo changes and a number of these show interactions with E2, particularly the cholinergic system, the dopaminergic system, and mitochondrial function. E2 treatment has been shown to ameliorate some of the behavioral and morphological changes seen in preclinical models of menopause; however, in clinical populations, the effects of E2 treatment on cognitive changes after menopause are mixed. The future use of sex hormone treatment will likely focus on personalized or precision medicine for the prevention or treatment of cognitive disturbances during aging, with a better understanding of who may benefit from such treatment.
Similar content being viewed by others

Introduction
This review will explore the effects of estrogens over the course of normal cognitive aging, and the potential implications for the development of cognitive disorders associated with aging. Here, we will discuss estrogens and their physiological signaling, before assessing the effects of reductions in estrogens in both clinical populations through menopause and in preclinical models. Next, this review will evaluate the interactions with systems seen to be dysregulated in aging. This is an important consideration as reductions in estrogens are most commonly seen during aging following menopause in females, or in males receiving androgen deprivation therapy for prostate cancer. Finally, the reported different effects of sex hormone treatment on cognition in aging will be considered. This is an area of significant controversy, with conflicting reports describing different effects and as such will be explored in detail.
There are three commonly considered estrogens, the most physiological relevant of which is estradiol (E2). The remaining two estrogens may be synthesized from estradiol, specifically estrone (E1) and estriol (E3). E2 has the highest affinity of the three for the intracellular estrogen receptors, ERα and ERβ [1]. In addition to these classical estrogen receptors, E2 also has a high affinity for the membrane-associated G protein coupled receptor, GPR30/GPER1 [2]. The synthesis of estrogens is quite unique, in that it involves the formation of an aromatic ring, a relative rarity in higher vertebrates, which commonly use amino acids with existing aromatic rings [3]. The ovary is the primary source of estrogens in females that are synthesized from acetate and cholesterol in a multi-step pathway utilizing a cytochrome P-450 aromatase as the final step. The aromatase is also expressed in non-ovarian sites, including the brain, testis, adrenals, fetal liver, skin, fat, breast, and bone marrow in which it acts on circulating precursor androgens [4]. In males, testosterone is aromatized to produce estrogen. This occurs in the testes at high levels in which estrogen action is important for fertility, as well as the non-ovarian sites described above [5]. In addition to these typical estrogens, there are a number of steroids that act through the estrogen receptors, including D5-androstenediol and 5a-androstanediol [6]. ERα and ERβ show differential expression throughout the body, with ERα being expressed in the epididymis and testis in males, the uterus in females, and pituitary, kidney and adrenals, as well as weak central nervous system expression in the thalamus and hypothalamus. In contrast, ERβ is expressed at highest levels in the ovary in females and prostate in males, with moderate expression in the testes in males, uterus in females, and bladder, and lung, and low levels of expression in the epididymis in males and pituitary, thymus and throughout the brain and spinal cord [1, 7]. Sex differences in ERα expression has been reported in the hypothalamus. E2 levels fluctuate throughout the female menstrual cycle, which consists of the follicular and luteal phases. The follicular phase starts on day 1 of menstrual bleeding, which is characterized by a consistent, low level of E2 that increases just prior to ovulation, around day 14. Following ovulation, the cycle enters the luteal phase, which displays increased E2 levels that return to low levels before the cycle re-enters the follicular phase around day 28 following regression of the corpus luteum [8]. GPR30/GPER1 is expressed broadly throughout the brain in rodents, with expression in the striatum, the hypothalamus, pituitary, hippocampus, substantia nigra, and areas of the brain stem [9].
As women age, they transition through natural menopause at an average age of 51 [10]. Natural menopause is defined to have occurred following 12 consecutive months of amenorrhrea with no obvious pathological or physiological cause, with perimenopause encompassing the time prior to menopause and 1 year postmenopause. The menopausal transition describes the time prior to menopause when there is increased menstrual cycle variability resulting in longer stretches of time in a low estrogen state [11]. Early or premature menopause may be induced surgically through bilateral oopherectomy, or following chemotherapy, or may be spontaneous due to premature ovarian failure (also called primary ovarian insufficiency) [12]. The process of menopausal transition takes 5–8 years on average and produces an array of variable clinical symptoms that have been investigated in observation studies such as the Study of Woman Across the Nation (SWAN) and the Penn Ovarian Aging Study [13, 14]. These studies have shown that 50–80% of women experience vasomotor symptoms (hot flushes, night sweats) at some point during the menopause transition [13, 14]. Increased sleep disturbances are reported during menopause [15], although some studies suggest these disruptions may be secondary to vasomotor symptoms. Somatic symptoms such as aches and pains have also been reported through the menopausal transition [14]. There have been an array of cognitive changes reported, including reduced processing speed and reduced verbal memory observed through the perimenopausal period [16]. Finally, depressive symptoms are also associated with menopausal stage [14]. In contrast, men do not experience a sudden decrease in E2 in the same way that women do. The decline in male testosterone and by extension bioavailable E2 is subtler and more gradual throughout normal aging [17], which correlates with decreasing E2 levels [18, 19].
Preclinical models have provided a vital platform for investigating the influence of E2 on an array of clinical symptoms, including cognition. However, rodents and non-human primates display a different lifetime pattern of estrogen change than what is seen in humans. Female rodents undergo estropause, during which cycles become irregular through midlife, before they become acyclic; however, the mechanisms differ from menopause. For example, menopause occurs due to a reduction in ovarian follicles throughout life, but in rodents, this occurs due to a reduction in the gonadotrophin surge which normally occurs prior to ovulation [20]. Ovariectomies of young and in some studies aged rodents have been used to study this estropause transition. For a translational model of menopause, 4-vinylcyclohexene diepoxide has been used, which produces a significant reduction in E2 levels rather than the complete loss seen with ovariectomies [21]. 4-Vinylcyclohexene diepoxide selectively destroys follicles over time, sparing granulosa cells, and oocytes [22], in a manner that is suggested to be comparible to the process seen in clinical patients during natural menopause [23]. Non-human primates proceed through a more typical menopausal transition [24], with a number of studies utilizing aged female non-human primates to model the menopause transition [25, 26]. In addition, ovariectomies are often performed to directly investigate the effects of E2 loss in non-human primates [27, 28]. It is important to note that although ovariectomy models the loss of E2, temporally this is markedly different from the changes that occur during natural menopause. In menopause, the less abrupt loss of E2 can produce cognitive deficits that continue to develop through later life. Furthermore, studies utilizing ovariectomy in young animals will produce a preclinical model that is inappropriate to interogate the interaction between decreased estrogen, the aging brain, and cognitive dysfunction. The aging brain and associated cognitive dysfunction is developing concurrently with the menopausal transition in clinical populations. These age-related changes will not be occurring in the preclinical model. Ovarectomizing older animals can partially assess this interaction. However, higher order cognitive functions that can be dysfunctional in aging are yet to be conclusively modeled in preclinical species.
Brain aging occurs alongside endocrine aging and the interactions of these processes have been implicated in cognitive decline and pathological conditions. Examination of the structural, cognitive, and behavioral effects of declining E2 levels and cognitive performance across aging preclinical species and clinical populations will be discussed. In the present review, three main predominant hypotheses of cognitive and brain aging will also be discussed in relation to changing estrogen levels, including the cholinergic hypothesis, the dopaminergic hypothesis, the mitochondrial aging hypothesis, and the effects of inflammation on cognitive aging. Here, we will explore how decreases in E2 levels impact these different systems and how therapeutic modulation of E2 levels may or may not augment normal and/or disease-related cognitive decline in preclinical species and both females and males.
Cognitive and Morphological Changes with Changing E2 Levels
This section will focus on changes in the brain that are associated a decline in E2.
-
A.
Cellular and morphological changes following E2 loss in the aging brains of rodents and non-human primates (NHPs)
E2 has both genomic and non-genomic effects, with the non-genomic effects through mitogen activated protein kinase (MAPK) activation being potentially relevant in synaptic plasticity in adults [29]. In vitro, E2 has been shown to have an effect on a timescale of minutes to an hour [30, 31], which is significantly faster than would be expected by classical transcriptional actions of steroids [32]. Through the use of genetic knock-out animals, it has been shown that ERα, ERβ, and GPR30/GPER1 mediate hippocampal synaptic plasticity and the cognitive effects of estrogens [33, 34]. Selective ERα and subsequently ERβ activation has displayed synaptic plasticity through interaction with metabotropic glutamate receptors [35]. Selective activation of ERα has shown increased dendritic spine density and cognitive performance [36], with selective activation of ERβ showing no change in dendritic spines and improvement in cognition at only certain doses, in specific tasks [36]. GPR30/GPER1 has been demonstrated to induce synaptic plasticity within the hippocampus following selective activation [37]. This suggests that ERα, ERβ, and GPR30/GPER1 are all positioned to modulate cognition. In addition to the classical estrogen receptors ERα, ERβ, and the GPCR GPR30/GPER1, there is evidence of two additional membrane localized estrogen receptors within the central nervous system with evidence of G protein coupling [38,39,40]. Modulation of E2 levels has been shown to be important in altering synaptic morphology across a number of preclinical species. In rodents, it has been shown that ovariectomy leads to a reduction in dendritic spines most notably on apical CA1 pyramidal cells that can be restored by E2 treatment [41]. In rhesus macaques, it has been shown that natural menopause promotes the selective loss of a specific synapse class, perforated synapses, in the dentate gyrus outer molecular layer. These synapses are purported to be important in maintaining synaptic long-term potentiation [26]. The same group later showed that E2 treatment increased synaptic density in the prefrontal cortex of aged, overectomised rhesus macaques [42] and that multisynaptic boutons decrease in the dorsolateral prefrontal cortex following ovariectomy in aged rhesus macaques that are rescued by E2 treatment [43].
-
B.
Structural and network changes secondary to E2 loss or replacement in clinical populations
In clinical populations, brain imaging techniques are useful in measuring the structural and network connectivity effects of E2, and changes that occur during menopause. For example, work from our laboratory utilizing magnetic resonance imaging (MRI) has shown that E2 treatment dose-dependently enhances hippocampal volume in postmenopausal women [44], which is supported by earlier studies suggesting decreases in hippocampal volumes over the menopausal transition when controlling for age [45]. By contrast, earlier work from the Woman’s Health Initiative Memory Study indicated that women treated with conjugated equine estrogens (CEE), with or without medroxyprogesterone acetate, experienced greater brain atrophy in frontal cortical and hippocampal regions than patients who did not receive treatment [46]. It should be noted that the greatest effect was seen in patients who experienced cognitive deficits prior to initiating hormone therapy. A further study demonstrated decreases in gray matter volume in the superior temporal gyrus, inferior frontal gyrus, olfactory cortex and supplementary motor area in postmenopausal women when correcting for age [47]. This provides conflicting evidence for the effects of E2 treatment on brain volume. The reasons for this discrepancy could include methodological differences in the studies, including differences in length of treatment and different treatment regimens such as E2 treatment alone, compared to CEE with or without medroxyprogesterone acetate. In particular, work assessing cognitive and imaging endpoints revealed that greater cognitive decline in postmenopausal women correlate with enhanced connectivity in the executive control network and the temporal lobe, but reduced connectivity in the frontal lobe [48]. Studies assessing the functional MRI changes with numerous different paradigms suggest that there is improved efficiency of brain function during a sustained attention task in postmenopausal females receiving E2 [49], and that during a working memory task postmenopausal women displayed increased frontal lobe activation as task difficulty increased following treatment with E2 [50].
-
C.
Effects on cognition of E2 loss or replacement
Given the array of cellular, morphological, and structural changes associated with declining E2 levels, reduced dendritic spines, decreased synaptic density, decreased numbers of specific synapses, changes in connectivity, and increases or reductions in gray matter volume in specific areas, it is unsurprising that there are associated cognitive changes. In preclinical models of menopause, ovariectomizing young rodents produced a reduction in performance in a spatial learning tasks, including the radial arm maze and the morris water maze [51,52,53]. In young and middle aged ovarectomised rodents, E2 supplementation has been shown to normalize these deficits [51, 54]. In aged overectomized rodents, E2 replacement had mixed results. One study described improvements in reference memory, but not in working memory [51], and other work suggested that acetylcholinesterase inhibitors restore the cognitive enhancing effects of E2 in aged overectomised rodents [53, 54]. In non-human primates, following menstrual cycle cessation and in ovariectomised aged females, there is a reduced performance on tasks such as the preclinical memory tasks the delayed response task and the delayed non-match to sample task [25,26,27]. In all these cases, treatment with E2 was shown to at least partially recover the abnormalities seen [27, 43]. Efforts to differentiate the effects of the different receptors have shown that all three receptors ERα [35], ERβ [35, 55] and GPR30/GPER1 are involved in modulating cognitive function. GPR30/GPER1 has shown involvement in cognitive function, particularly spatial and working memory tasks in rodents, with the selective agonist GPR30/GPER1 agonist G-1 improving performance in delayed match to position [56], novel object recognition [57], and Y-maze [58] in ovariectomized females.
The postmenopausal cognitive symptoms reported by clinical populations in the literature include self-reported forgetfulness and difficulty concentrating [59, 60]. Evidence from some these studies suggest that self-reported cognitive dysfunction correlates with declining cognitive performance [61]. However, confirming that subjective cognitive complaints correlate with objective impairment has proven complex. For example in previous studies, subjective memory complaints during the perimenopause did not correlate with decreased performance on verbal episodic memory tasks, but did correlate with working memory and vigilance tasks performance [62]. Furthermore, additional associations with depression, sleep disturbances, and somatic symptoms have been shown [62], with other studies suggesting a domain and transition stage specific association with cognitive performance. As such perimenopausal woman performs worse on verbal memory, with a reduction in processing speed during premenopause, early perimenopause, and postmenopause [63].
-
D.
Effects on sleep
Increased sleep disturbances are reported during menopause [15], with other studies suggesting this occurs predominantly in women who previously experienced poor sleep [64]. These sleep complaints are strongly correlated with vasomotor symptoms [64]. In a recent meta-analysis, E2 treatment with or without progesterone/progestin improved sleep quality in postmenopausal women who had vasomotor symptoms, but not in those that did not [65]. Hot flashes have been shown to precede awakenings in the first half of the night, whereas in the second half of the night, during which there is typically more rapid eye movement (REM) sleep, hot flashes follow awakening [66, 67]. During REM sleep, it has been shown that thermoregulatory responses are greatly decreased or abolished [68], which would explain why hot flashes no longer trigger awakenings in the second half of the night. REM sleep is controlled by cholinergic firing from both the basolateral forebrain and mid-pontine nuclei [69, 70], a system that E2 has been shown to modulate. However, a recent study displayed no difference between subjective sleep quality between pre- and postmenopausal women, suggesting that poor sleep may also be due to age-related changes rather than just menopause [71].
In preclinical species, there are few studies on the effects of E2 levels and sleep-wake architecture. It has been shown that ovariectomised female rats are more sensitive to the cognitive effects of REM sleep disruption [72]. However, the effects of E2 supplementation in ovariectomised animals on sleep has been complex. Previous studies showed that co-dosing E2 and progesterone increased wakefulness at baseline conditions, increased electroencephalogram delta power during recovery non-REM sleep following sleep deprivation, and decreased REM sleep in the 12-h following sleep deprivation [73, 74]. Whereas in rats that are ovariectomised but not sleep deprived, E2 treatment promotes wakefulness and reduces both REM and non-REM sleep during the awake phase [73,74,75].
-
E.
Effects on mood
Sex hormones may play an important role in mood regulation [76]. Women are at increased risk of depression from first menarche with the risk declining following menopause, though increased compared to males [77]. The mechanism through which E2 effects mood remains unclear; however, there are a number of hypothesis that may explain the increased incidence of depressive disorders during the reproductive years [78]. In perimenopausal women, it has been demonstrated that removal of E2 treatment results in increased depressive symptoms in a subset of individuals who had previously suffered from depressive symptoms [79]. E2 has been shown to interact with the brain serotonergic system, a system implicated in mood regulation and the treatment of depressive disorders, with E2 supplementation in oopherectomized women resulting in reduced binding potential of the serotonin transporter radioligand [11C]MADAM at the serotonin transporter in a PET study [80]. This may be due to increases of serotonin levels or direct E2 actions reducing expression of the serotonin transporter [80]. Moreover, studies reducing levels of brain serotonin via tryptophan depletion are more likely to produce depressive symptoms in women than in men [81], providing further evidence for the importance of the serotonin system in depression in women. Modulation of E2 levels has also been demonstrated to increase serotonin receptor responsivity to the agonist m-chlorophenylpiperazine through prolactin and cortisol response in postmenopausal woman [82], with later studies describing increases in 5-HT2A binding potential following E2 therapy [83], a receptor indicated in the prolactin and cortisol response [82]. In preclinical models, it has been demonstrated that treatment with E2 in ovariectomized rats results in an increase in serotonin 5-HT2A receptors in the dorsal raphe nucleus, anterior frontal, and cingulate regions [84] providing further evidence for the role of E2 in modulating the serotonergic system.
Alternatively, modulation of the stress response system by E2 may be important in the development of depressive symptoms. The stress response system modulates neuroendocrine, immune, and autonomic responses to stress through interactions with the hypothalamic pituitary axis. The resulting cortisol release may be altered by changing E2 levels [85]. Reduced cortisol responses are seen in premenopausal women compared to men [86], with alterations in the response seen during the menstrual cycle and pregnancy [87, 88]. This suggests that changes in E2 can alter the cortisol responses and may be responsible for the depressive symptoms seen through the menopause transition.
Estrogens and the Cholinergic System
The cholinergic hypothesis of cognitive aging was first proposed by Bartus in 1982 [89]; it states that the changes in the brain cholinergic system are responsible for the cognitive dysfunction seen in Alzheimer’s disease and normal aging. This hypothesis has been supported by an array of clinical and preclinical studies that show reduced nicotinic cholinergic binding sites in the cortex of patients with Alzheimer’s disease, and reduced cholinergic acetyltransferase activity (a marker of cholinergic neurons) [90]. These alterations in cholinergic neurons have been shown to correlate with reduced cognitive performance [91]. Moreover, reduction in cognitive performance and cholinergic signaling have been well established across aged preclinical species [92]. Further support of the cholinergic hypothesis of cognitive aging and Alzheimer’s disease stems from the treatments available for Alzheimer’s disease, specifically the FDA approved acetylcholinesterase inhibitors which act to enhance synaptic levels of acetylcholine and overall cholinergic signaling [93,94,95].
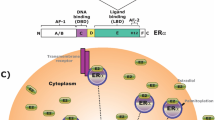
The cholinergic system has been hypothesized to interact with estrogen signaling, a schematic of this hypothesized interaction is shown in Fig. 1. From a cellular perspective, it has been shown that the G protein coupled estrogen receptor GPR30/GPER1 co-localizes with cholineacetyltransferase (ChAT) expressing neurons, particularly in the basal forebrain [96]. ChAT is a marker for cholinergic neurons, and as such this expression of GPR30/GPER1 on cholinergic neurons provides increasing evidence for an interaction of estrogens and the cholinergic system. Furthermore, ERα but not ERβ has been shown to co-localize with ChAT expressing neurons in the basal forebrain [97]. Gibbs and colleagues demonstrated that in aged rats, in which one would expect greater cholinergic decline that E2 treatment alone is unable to improve the deficits seen following ovariectomy. However, when E2 is combined with donepezil (an acetylcholinesterase inhibitor), the effects are similar to what is seen in a middle aged ovariectomized animal treated with E2 [54]. Similar results were reported in aged rats with another acetylcholinesterase inhibitor galantamine [53]. In the same study, the authors showed that E2 treatment increased cholinergic release in the hippocampus in response to food pellets [53]. Further evidence for the interaction of the E2 with the cholinergic system comes from a number of studies have explored the effect that E2 treatment has on modulating the effects of scopolamine, a nonselective muscarinic cholinergic antagonist. In ovarectomised rodents, E2 with or without progesterone following scopolamine treatment has been reported to improve spatial working memory [98,99,100], with evidence that this effect was observed on both acquisition and retention [101], but with no effect on reference memory [100].

Hypothesized effects of E2 signaling on cholinergic neurons
Finally, neurotrophins have been implicated in protection of cholinergic neurons, with brain-derived neurotrophic factor (BDNF) displaying protective effects on cholinergic neurons following axotomy [102, 103] and increased survival in vitro [104]. Nerve growth factor (NGF) displays protective effects following axotomy [105] and during aging in rats [106], with NGF believed to be the trophic factor for basal forebrain cholinergic neurons [107]. Estrogen receptors have been reported to co-localize with neurotrophin receptors [108, 109], notably the low affinity nerve growth factor receptors (p75NGFR) in cholinergic neurons [109], which binds NGF, BDNF, and neurotrophin 3 [109]. Following E2 treatment, it has been demonstrated that NGF and associated NGF receptor (p75NGFR) mRNA decrease in select brain areas, including the medial septum [110]. NGF also signals through trkA and it has been shown that trkA decreases in the basal forebrain (nucleus basalis of Maynert and medial septum) following ovariectomy in rodents and increases to normal levels following E2 treatment [111, 112]. This suggests that some of the cholinergic effects seen may be mediated through the protective effects of neurotrophin signaling (Fig. 1). It should be noted that in the nucleus basalis of Meynert increases in TrkA are seen with concurrent increases in ChAT following E2 treatment of ovariectomised rodents, however in the horizontal limb of the diagonal band of Broca, only ChAT increases are observed [113].
In humans, it has been shown that the length of hormone treatment positively correlates with vesicular acetylcholine transporter (VAChT) binding, particularly in frontal and temporal lobes, and anterior cingulate, and that age of menopause onset negatively correlates with VAChT binding [114]. E2/progesterone hormone therapy initiated early in the menopause transition was associated with greater cholinergic activity, as measured by acetylcholinesterase activity through PET techniques, in the hippocampus and posterior cingulate cortex; however, treatment with E2 alone did not produce these effects [115]. These data indicate that that treatment timing of E2 replacement is important due to a time-dependent decline in basal forebrain cholinergic neurons secondary to hormonal changes during/after the menopause transition. Clinical MRI imaging studies have shown that E2 treatment also modulates anti-muscarinic and anti-nicotinic induced brain activity [116]. In particular, treatment with tamoxifen, a selective estrogen receptor modulator, SERM, reduces anti-nicotinic and anti-muscarinic cognitive effects seen by performance improvements in reaction time and spatial memory tasks [117]. This combination of clinical data displaying increased cholinergic binding and activity following E2 treatment, alongside the experimental evidence whereby E2 treatment or SERMs attenuate the effects of anticholinergics provides a strong argument for the dependence of cholinergic system integrity on E2 levels.
Estrogens and the Dopaminergic System
An alternative literature proposes a dopaminergic hypothesis for cognitive aging [118, 119]. Reductions in dopamine binding [120,121,122], dopaminergic receptors [123], and dopamine transporter [119] are seen throughout aging. Preclinical studies in non-human primates and rodents have indicated that dopaminergic lesions may result in reduced cognitive performance [124, 125]. It has been observed that patients with diseases displaying a dysregulated dopaminergic system such as Huntington’s disease and Parkinson’s disease, present with cognitive abnormalities [126]. PET imaging studies have suggested that in individuals with the Huntington’s disease mutation that the amount of striatal dopamine receptor binding correlates with performance on cognitive tasks, with decreased binding corresponding to a poorer performance [127]. Further evidence for the importance of dopaminergic signaling in cognition is that dopaminergic antagonists decrease cognitive performance in humans [128].
The effects of E2 on dopaminergic signaling have been less well characterized than in the cholinergic system. In vitro, it has been shown that E2 protects against 6-hydroxydopamine toxicity in dopamine neurons [129]. In two different rodent models of menopause, ovariectomy, and 4-vinylcyclohexene diepoxide, it was demonstrated using a metabolomics approach that dopamine levels were found to be increased in the frontal cortex 6 weeks following ovariectomy with no change in norepinephrine, and increases in dopamine seen in the 4-vinylcyclohexene diepoxide model at 1-week post initiation. In contrast in the hippocampus and striatum, dopamine levels are unchanged in both models; however, norepinephrine was decreased at 1-week following initiation of both the models in the hippocampus, but normalized 6 weeks following initiation. In the striatum, norepinephrine is increased 1-week following ovariectomy, but normalizes by 6 weeks, and no change is seen in the 4-vinylcyclohexene diepoxide model [21]. In ovariectomised rats, E2 and progesterone treatment was implicated in increasing dopamine uptake into dopamine neurons [130]. In ovariectomized non-human primates, E2 supplementation has been demonstrated to preserve nigrostriatal dopaminergic tyrosine hydroxylase positive neurons, compared to vehicle ovariectomized controls that displayed around a 30% decrease in dopaminergic neurons [131]. In postmenopausal women, there is MRI evidence that dopaminergic agonists increase activity in the working memory network, suggesting that the dopaminergic system is still responsive to pharmacological manipulation after the menopausal transition [132].
It has been suggested that E2 may play a role in the modulating the development and onset of schizophrenia [133, 134], a disease in which dopaminergic dysfunction is implicated [135]. Women have a peak age of onset in early adulthood, with a second peak in midlife at 45–54 years [136]. Cohen and colleagues suggest that E2 may play a protective role in the development of schizophrenia with additional data revealing that earlier puberty results in later onset of clinical symptoms in women that display schizophrenic symptoms in early adulthood [137]. Additionally, symptoms in women tend to vary with the menstrual cycle and fewer symptoms are often seen during pregnancy, a time of high E2 [138]. In support of this hypothesis, a clinical study showed that a combination of E2 and haloperidol produced more positive outcomes in premenopausal females than haloperidol alone [139]. Trials utilizing the SERM raloxifene as a treatment for schizophrenia have demonstrated improved probabilistic learning and associated functional MRI activity in the hippocampus and parahippocampal regions [140]. Further cognitive improvements have been seen in attention and processing speed following treatment with ralixofene in schizophrenia [141]. It should be noted that there were no improvements in the psychotic symptoms [141].
It has been shown that men have a higher risk for Parkinson’s disease than women [142]. In addition, it has been suggested that the timing of the menopausal transition is a risk factor for developing Parkinson’s disease [143]. A retrospective study indicated that individuals who experienced early natural menopause, or had not used any estrogen treatment for at least 6 months after menopause, were at greater risk of Parkinson’s disease [143]. However, more recent meta-analyses of observational studies suggest that age of menopausal transition, the use of oral contraceptives, age of menarche, and reproductive life span did not convey additional risk for the development of Parkinson’s disease [144, 145]. In rodents, it has been suggested that E2, SERMs, and other sex hormones reduce dopaminergic neuron loss in the 1-methyl 4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) mouse model of Parkinson’s disease [146]. In the human literature, it has been suggested that E2 therapy is protective against the development of dementia in Parkinson’s patients [147].
Among the different hypotheses for the mechanism and receptor through which E2 exerts its neuroprotective effects on dopaminergic neurons, one suggestion is that this neuroprotection is mediated via actions on the local renin-angiotensin system [148]. The renin-angiotensin system is typically thought of as a systemic hormone signaling system involved in blood pressure control; however, it has been argued that a local system exists in many tissues [149]. Activation of this renin-angiotensin system contributes to oxidative stress [150]. A central renin-angiotensin system has been identified within the brain with high levels of localization in the dopaminergic system within the substantia nigra [151]. It can be shown that activation of this renin-angiotensin system results in toxicity to the dopaminergic neurons that can be reversed by inhibitors of the system [152, 153]. Recent studies suggest that E2 can exert neuroprotective effects on dopaminergic neurons following local renin-angiotensin activation after surgically induced menopause in rats [154]. E2 may mechanistically play an important role in protection of dopaminergic neurons and modulation of the neurotoxic effects of the renin-angiotensin system; however, there appears to be limited evidence that the hormonal changes associated with menopause produce any difference in the risk of developing Parkinson’s disease.
Estrogens and Mitochondrial Dysfunction
The mitochondrial aging hypothesis differs from the cholinergic and dopaminergic hypothesis in that it is a general hypothesis of aging suggesting that as mitochondria age, there is increased mitochondrial DNA damage, leading to increased reactive oxygen species damage and reduction in mitochondrial activity that manifests in signs of aging [155]. This hypothesis had been recently applied to cognitive aging and more specifically Alzheimer’s disease. It proposes that increases in reactive oxygen species promotes beta-sheet aggregation, apoptosis, or cell cycle arrest producing neurofibrillary tangles and the hallmarks of Alzheimer’s disease [156]. As Alzheimer’s disease progresses, it has been demonstrated that brain glucose uptake decreases leading to a switch from aerobic to anaerobic metabolism. This reduction in glucose uptake persists when correcting for loss of brain volume [157]. These changes in glucose utilization have been reported to precede clinical symptoms by a number of years in Alzheimer’s disease [158] and mitochondrial dysfunction precedes age-associated cognitive deficits in preclinical species [159]. The severity of brain hypometabolism has been correlated with dementia symptoms in later life and the pattern of hypometabolism has been linked with Braak staging of tau spread in AD [158].
Brinton and colleagues have performed clinical and preclinical studies assessing the effects of E2 on mitochondrial dysfunction and brain hypometabolism [160, 161]. In rodent studies, they have demonstrated a decrease in the expression of genes involved in normal glycolytic function in irregularly cycling animals. As the animals progress to an acyclic condition, the deficits in glycolytic genes remain; however, genes involved in mitochondrial function increase, potentially to compensate for the declining glucose availability [161]. In these acyclic animals, there is a concurrent increase in gene expression involved with fatty acid uptake, a transition from glucose to fatty acids/ketones as the energy source. Through the utilization of fluorodeoxygenase-positron emission tomography (FDG-PET) techniques, Yin and collegues were able to demonstrate reduced glucose uptake in the brain of irregularly cycling animals supporting the finding of a shift from glucose towards ketone metabolism [161]. This reduction in the ability to utilize glucose with declining E2 correlated with reduced synaptic plasticity, an indicator of cognitive performance [161], suggesting that this failure of glucose metabolism could influence cognitive deficits seen in aged animals. Further, it has been demonstrated that there substantial decreases in the amino acids tryptophan and tyrosine in the hippocampus [21]. These reductions may be due to the shift towards ketone metabolism as tyrosine and tryptophan can both be degraded to form ketones in hypoglycaemic conditions [21]. This proposed interaction of E2 and mitochondrial function is shown in Fig. 2.

Hypothesized effects of E2 loss on mitochondrial function and glucose utilization
Data from clinical studies support the hypothesis for the role of E2 in maintaining appropriate mitochondrial bioenergetics. One small longitudinal study (n = 9–11/treatment group) utilizing FDG-PET revealed that postmenopausal women that had not been taking E2 supplementation had reduced glucose uptake in prefrontal, parietal, cingulate, and temporal lobes 2 years after the initial baseline as compared to women that had been receiving E2 or conjugated equine estrogens with progesterone supplementation [162]. A recent study similarly demonstrated that glucose uptake in the temporal cortex, precuneus and frontal cortex declines during the menopause transition, reaching its lowest point following menopause. This was shown to correlate with platelet mitochondrial cyclochrome oxidase activity, an important step in ATP synthesis [160]. These findings suggest that as glucose uptake decreases, ATP production will also decrease consistent with reductions in the substrates for aerobic cellular respiration.
Inflammation and Cognitive Aging
Similarly to the mitochondrial hypothesis of aging, low-grade inflammation has been identified as a possible cause of systemic aging, and more recently specifically for cognitive decline and neurodegenerative diseases [163]. Increased levels of inflammatory markers such as IL-6 are linked to reduced cognitive performance in clinical populations [164]. In preclinical species, administering inflammatory cytokines peripherally produce impaired cognitive performance which is normalized if a neutralizing antibody is administered prior to the inflammatory cytokine [165].
In a model of inflammation in rats it has been demonstrated ERβ down regulates the P2X3 receptor in the periphery, producing anti-inflammatory effects [166]. Following ovariectomy in rodents a significant increase in blood brain barrier permeability is observed, which is normalized by a combined E2/estriol treatment [167]. In a peripheral inflammatory model in female rodents, ovarectomized subjects were seen to have increased in concentrations of cytokines within the brain, which was normalized by E2 treatment [168]. Women following menopause display an increase in inflammatory markers such as IL-1, IL-6, and TNF-alpha [169]. These increases in inflammatory markers appear to be normalized following treatment with sex hormones [170]. Women that undergo a premenopausal oophorectomy have been shown to display increased levels of the inflammatory marker C-reactive protein when compared to women that undergo hysterectomy with ovaries intact [171].
Inflammation is shown to produce a reduction in cognitive performance in clinical populations and preclinical species, and that E2 is an important anti-inflammatory factor. Following menopause the levels of inflammatory cytokines increase suggesting, and the permeability of the blood brain barrier increases suggesting this increased generalized inflammatory state could contribute to some of the cognitive abnormalities seen. Furthermore, chronic inflammation with glial priming, release of proinflammatory factors, and neuronal damage may play a key role in the progression of Alzheimer’s disease [172, 173] for which there is increased female risk.
Estrogen Support for the Aging Brain
The use of sex hormones to treat the symptoms associated with menopause has been an area of controversy, with some research suggesting E2 or conjugated equine estrogen (CEE) supplementation has limited beneficial effects and an array of negative risks. Others suggest that E2 treatment alone or in combination with progesterone or a progestin provides a significant benefit when treating a number of the symptoms associated with menopause.
Some studies have suggested a range of negative effects with sex hormone treatments. A number of studies from data collected by the Woman’s Health Initiative suggested that long-term treatment with CEE or CEE plus the progestin medroxyprogesterone acetate (MPA) increased the risk of developing mild cognitive impairment or dementia [174, 175]. However, it should be noted that these studies were limited by the time of estrogen treatment initiation (later than typical for menopausal symptom relief), the neurocognitive domains being tested, the type of estrogen treatment and the effects of progestins on cognition [176]. In contrast, there are a number of studies indicating positive effects of sex hormone treatments on cognitive function. For example, E2 treatment with or without progesterone has shown improved verbal [177] and visual memory [178].
Preclinical data from aged ovariectomised non-human primates demonstrated that E2 alone (continuously administered) or E2 (continuously or cyclically administered) in conjunction with progesterone (continuously or cyclically administered) failed to produce positive cognitive effects, but was associated with negative effects in a delayed response or delayed non-match to sample working memory task [28]. These data contrasted with earlier data published by the same group suggesting cyclic E2 treatment had a positive effect on the delayed response task in these non-human primate models [179]. Collectively, these studies suggests that the precise regimen of E2 may be important in modulating cognitive performance in this preclinical model. For example, a study in non-human primates of E2 or E2 plus cyclic progesterone dosed over a prolonged period (12 months) showed that continuous E2 administration was effective in improving performance in a delayed response task [180].
Critical Window Hypothesis
In light of difficulties in reconciling apparently contradictory data, the critical period or window hypothesis has been developed, suggesting that there is a specific time when treatment with sex hormones are most effective in treating the cognitive symptoms associated with menopause or in producing long-term cognitive benefit [176]. This critical period is thought to be around the time of the menopausal transition, when E2 is initially declining or shortly thereafter. It is during this transitional phase that initial changes in estrogen receptors are also seen. In rats, ERβ is seen to decline during the transition before rebounding whereas ERα is unchanged [161]. In other rodent studies, it has been shown that delaying E2 supplementation until 10 months following overiectomy (so having an extended period of time with no estrogen) leads to a reduction in the positive cognitive effects of E2 treatment [181]. Interestingly, treating with the acetylcholinesterase inhibitor donepezil restored the ability of E2 to enhance cognition when there was a long delay in treatment [54]. It has thus been suggested that this critical period exist at least in part due to the declining function of basal forebrain cholinergic neurons through the menopausal transition [182]. Consistent with these findings, treating with E2 close to the time of ovariectomy enhances cognitive performance in a range of cognitive domains in rodents [54, 181] and non-human primates [27, 43, 179]. A hypothesized progression is of cognitive performance dependent on the time of sex hormone treatment is shown in Fig. 3.

Hypothesized effect on cognitive performance of initiating sex hormone therapy at different menopausal stages
Beyond hormone therapy, there are novel treatments that could be considered when discussing the possible treatments for cognitive symptoms associated with the menopausal transition. SERMs, such as tamoxifen and raloxifene, have shown promise, interacting with ERα, ERβ, and GPR30 [183] and expressing neuroprotective effects [184]. For example, SERMs have been demonstrated to reduce activation of microglial, in turn reducing proinflammatory cytokines and chemokines following lipopolysaccharide administration in primary tissue culture [185]. Further, in postmenopausal women, SERMs have demonstrated efficacy in reducing anticholinergic effects on cognitive performance in some cognitive domains [117].
Relationship to Alzheimer’s Disease Risk
Alzheimer’s disease diagnosis is based on clinical history and signs with the presence of β-amyloid plaque and tau deposition being confirmed via spinal fluid measures, positron emission tomography imaging of β-amyloid plaque or tau deposits, and post-mortem. With advances in imaging and biomarker techniques, it is becoming increasingly understood that many of the pathological changes occur many years before the clinical symptoms prevent [186]. A recent meta-analysis revealed that female sex increases the risk of developing Alzheimer’s disease compared to men, regardless of genotype; this is particularly apparent for late onset Alzheimer’s disease [187]. In postmenopausal women, reduced glucose metabolism as measured by FDG-PET is seen in Alzheimer’s disease vulnerable areas, such as the posterior cingulate, frontal, parietal medial, and lateral cortices [160]. In women who had undergone surgical menopause, an earlier age of procedure was associated with increased risk of cognitive decline and Alzheimer’s associated pathology [188]. Certain observational studies have demonstrated a reduction in the risk of Alzheimer’s disease with sex hormone treatment after menopause [189, 190]. A recent pilot study indicated that transdermal E2 treatment reduced the β-amyloid deposition in recently menopausal women [191], suggesting that E2 may play a role modulating β-amyloid plaque formation.
Preclinical studies have demonstrated that reducing E2 levels results in decreased expression of genes associated with amyloid processing [161]. Merlo and colleagues [192] thoroughly review the mechanisms through which estrogens may modulate Alzheimer’s disease development. In brief, a number of in vitro studies have demonstrated that E2 stimulation results in amyloid being preferentially processed to the soluble form [193,194,195]. E2 also regulates β-amyloid degrading enzymes, for example, metalloproteases [196] and neprylisin [197, 198], which may reduce β-amyloid plaque formation in vivo. Further in vivo studies showed that E2 treatment can reduce Tau phosphorylation [199] and pharmacologically attenuate tau hyperphosphorylation [200]. This reduction in phosphorylation may also play a role in reducing Alzheimer’s associated pathology.
It is important to recognize that two of the systems we have discussed in this review are dysregulated in Alzheimer’s disease. The basal forebrain cholinergic system declines as Alzheimer’s disease progresses [89], and E2 therapy appears to support the cholinergic system [53, 54]. Likewise, the mitochondrial bioenergetic system is altered in Alzheimer’s disease, resulting in increased ketone metabolism and production of reactive oxygen species, a process which is also seen following menopause preclinically and clinically [160, 161].
Estrogens in the Male Brain
As previously discussed, men tend to experience a more gradual decline in sex hormones as they age when compared to women. However, in prostate cancer patients receiving androgen deprivation therapy, the decline in sex hormones is more rapid, providing a model for investigating the effects of testosterone and E2 withdrawal in men. In rodents, it has been demonstrated that following androgen deprivation therapy there is a reduction in estrogen receptors in a number of brain areas, including the hippocampus and prefrontal cortex, areas which are important for cognition [201]. Prospective randomized trials of prostate cancer patients who experienced reduced E2 levels due to treatment failed to show any effect of E2 loss or E2 supplementation [202, 203]. Results from a smaller study utilizing a semi-structured interview rather than performance assessments suggested that androgen deprivation therapy for prostate cancer produced worsening concentration, information processing, verbal fluency, visuospatial function, memory, and executive function [204]. There are also studies in men correlating declining E2 levels in aging with decreasing cognitive function, including decreased verbal fluency and information processing [205, 206]. One of these studies found that the greatest effect was seen in men younger than 72 years old, suggesting there may be a temporal window during the aging process when E2 is important in modulating cognitive function in men [205]. However, another study indicated that when correcting for age and education, cognitive examination scores declined (indicating worse cognition) with increasing E2 levels, whereas decreased testosterone levels correlated with reduced memory and concentration [207].
To date, the effects of E2 in the male brain remain less well characterized than their effects on the female brain. The discrepancies seen between the studies above whereby some studies displayed a positive correlation between E2 levels and cognitive performance, with the other revealing a negative correlation may be due a number of reasons. A temporal window when the effects of E2 are most apparent, the difficulty in separating the effect of androgens from E2 effects, the lack of an abrupt change in E2 levels as seen in women, and small sample sizes in a number of the studies.
Discussion
This review has explored the effects of changing E2 level on cognitive functioning with advancing age. We have identified that E2 may interact with three systems within the brain important to cognitive aging: the basal forebrain cholinergic system, the dopaminergic system, and the mitochondrial bioenergetic system. Declining E2 levels have been linked with decreased cognitive performance in clinical and preclinical studies, with E2 treatment attenuating these symptoms. In both clinical and preclinical studies, the timing of the treatment may be important, and this critical window may be linked to the declining cholinergic system. Some later life disorders appear to show a link to reduced E2 levels and the timing of menopause, particularly Alzheimer’s disease and depression. Alzheimer’s disease shows a higher incidence in women, particularly in women who have experienced an earlier menopause. In addition, schizophrenia displays a second peak of onset in midlife in women, around the time of menopause, and an early age of first menarche appears protective against developing schizophrenia.
Given the cholinergic, dopaminergic, and mitochondrial systems are dysregulated in a number of neurodegenerative diseases associated with aging (e.g., Alzheimer’s disease) utilization of E2 therapy has been explored in with a number of clinical trials without success to date [174, 208, 209]. However, it may be that E2 treatment needs to be specifically tailored based on the patients risk profile. The preclinical studies assessing time of E2 delivery with reference to time of reduced E2, and method of E2 delivery, cyclic versus non-cyclic, with or without progesterone provide possible explanations of the disparate results that are seen in the clinical literature. It should also be noted that the nature of the menopause (natural or induced) may be important in dictating the efficacy of potential treatment. Further, not only are the majority of the positive effects of treatment on cognitive endpoints seen when the treatment is given around the time of the menopause transition, but this is also the time when the greatest structural and estrogen receptor changes are seen.
One concept that will become increasingly important over the coming years is that of personalized or precision medicine. Specifically, identification of which individuals may benefit from E2 therapy and which individuals will not. To enable this, genomics approaches assessing the effects of genetic variables and different phenotypic presentations on response to different treatments should be utilized. This strategy will enable the maximal benefit to be obtained without treating individuals that do not require treatment and/or leading to increased risk without corresponding benefit. The presence of subjective cognitive decline (SCD), a known risk factor for the development of cognitive decline and dementia, that first occurs during or after menopause may indicate a higher risk population for which specific therapies may be helpful [188]. Additionally, it has been suggested that a neuroprotective effect of E2 may be dependent on apolipoprotein E genotype, although the specifics of this interaction are still not fully understood [210]. This does however provide evidence that based on age, phenotype, and genotype, it may be possible to identify patients that are at a higher risk of developing Alzheimer’s disease and may potentially benefit from E2 therapy.
With regard to depressive disorders, it is known that previous depressive episodes prior to the menopausal transition increases the risk of further depressive episodes following menopause in the absence of E2 therapy [79]. Given the highly polymorphic nature of presence or absence of clinical signs following menopause, it is possible that specific phenotypes or genotypes predispose women to developing mood dysfunction following the menopause transition. Future studies that investigate whether specific phenotypes or genotypes predispose to cognitive or mood dysfunction following menopause, or whether specific genotypes predispose to responsiveness to specific therapies, would allow for greatly improved targeting of therapies by identifying only those individuals that will likely be responsive to treatment.
Since the publication of the Women’s Health Initiative studies beginning in the early 2000s, there has been a decline in the prescription of sex hormone treatment for postmenopausal symptomatology and presumed disease prevention. This decline was in part appropriate but the subtleties of the WHI studies and their findings have not been as widely appreciated by clinicians and thus the risks of sex hormone prescription may be perceived as larger than the actual data would support. Future research will help clarify which hormone preparations may be potentially useful and at what stage of life. Rather than ubiquitous prescription for all postmenopausal women, selective use of sex hormone treatment will be aided by better differentiation of which women have higher or lower risk for late life cognitive or emotional impairment. In addition, novel hormone preparations or SERMs may be developed that allow for brain-specific effects without risks to peripheral tissues.
With the interaction of E2 and cholinergic, dopaminergic, and mitochondrial bioenergic systems, it should be considered that targeting of these systems may have E2 sparing effects, or potentiate E2 beneficial effects. Acetylcholinesterase inhibitors are the leading treatment for Alzheimer’s disease, which function through boosting cholinergic synaptic signaling. Gibbs and colleagues [53, 54] demonstrated acetylcholinesterase inhibitors can potentiate the effects of E2 in aged rats following prolonged estrogen deprivation. In clinical populations, it has been demonstrated that females who received the acetylcholinesterase inhibitors tacrine in conjunction with estradiol displayed a greater response on cognitive assessment that females who were not receiving concurrent estradiol therapy [211]. Thus combined hormonal and neurotransmitter-based approaches may increase the beneficial effects of both individual therapeutic strategies. Direct targeting of the specific cholinergic receptors is also becoming an area of interest for boosting the failing cholinergic system in Alzheimer’s disease [212,213,214,215,216]. For example, selective M1 acetylcholine receptor positive allosteric modulators (PAMs) has been of particular interest in Alzheimer’s disease, with efficacy seen across cognitive and biomarker measures in preclinical species [214,215,216,217,218,219]. Identification of at-risk individuals, for example individuals who are displaying SCD, asymptomatic, or early symptomatic increased β-amyloid plaque deposition could provide a population for combined treatment approaches. In future studies, it will be critical to understand the potential for M1 PAMs to act synergistically with E2 replacement (or similar hormonal approaches) to restore the cognitive and affective deficits associated with aging and pathologic decline such as Alzheimer’s disease in preclinical species and clinical populations.
References
Kuiper GGJM, Carlsson B, Grandien K, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors and α and β. Endocrinology. 1997;138:863–870.
Thomas P, Pang Y, Filardo EJ, et al. Identity of an Estrogen Membrane Receptor Coupled to a G Protein in Human Breast Cancer Cells. Endocrinology. 2005;146:624–632.
Ryan KJ. Biological aromatization of steroids. J. Biol. Chem. 1959;234:268–272.
Ryan KJ. Biochemistry of aromatase: significance to female reproductive physiology. Cancer Res. 1982;42:3342s–3344s.
Cooke PS, Nanjappa MK, Ko C, et al. Estrogens in Male Physiology. Physiol. Rev. 2017;97:995–1043.
Baker ME. What are the physiological estrogens? Steroids. 2013;337–340.
Simerly RB, Swanson LW, Chang C, et al. Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: An in situ hybridization study. J. Comp. Neurol. 1990;294:76–95.
Jabbour HN, Kelly RW, Fraser HM, et al. Endocrine Regulation of Menstruation. Endocr. Rev. 2006;27:17–46.
Brailoiu E, Dun SL, Brailoiu GC, et al. Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 2007;193:311–321.
McKinlay SM, Brambilla DJ, Posner JG. The normal menopause transition. Maturitas. 1992;14:103–115.
World Health Organization. Research on the Menopause in the 1990s: Report of a WHO Scientific Group. WHO Tech. Rep. Ser. No. 866. 1996;12–14.
Shuster LT, Rhodes DJ, Gostout BS, et al. Premature menopause or early menopause: Long-term health consequences. Maturitas. 2010;65:161–166.
Gold EB, Colvin A, Avis N, et al. Longitudinal analysis of the association between vasomotor symptoms and race/ethnicity across the menopausal transition: study of women’s health across the nation. Am. J. Public Health. 2006;96:1226–1235.
Freeman EW, Sammel MD, Lin H, et al. Symptoms Associated With Menopausal Transition and Reproductive Hormones in Midlife Women. Obstet. Gynecol. 2007;110:230–240.
Kravitz HM, Janssen I, Bromberger JT, et al. Sleep Trajectories Before and After the Final Menstrual Period in The Study of Women’s Health Across the Nation (SWAN). Curr. sleep Med. reports. 2017;3:235–250.
Lamar M, Resnick SM, Zonderman AB, et al. Longitudinal changes in verbal memory in older adults: distinguishing the effects of age from repeat testing. Neurology. 2003;60:82–86.
Morley JE, Kaiser FE, Perry HM, et al. Longitudinal changes in testosterone, luteinizing hormone, and follicle-stimulating hormone in healthy older men. Metabolism. 1997;46:410–413.
Ferrini RL, Barrett-Connor E. Sex Hormones and Age: A Cross-sectional Study of Testosterone and Estradiol and Their Bioavailable Fractions in Community-dwelling Men. Am. J. Epidemiol. 1998;147:750–754.
van den Beld AW, de Jong FH, Grobbee DE, et al. Measures of Bioavailable Serum Testosterone and Estradiol and Their Relationships with Muscle Strength, Bone Density, and Body Composition in Elderly Men. J. Clin. Endocrinol. Metab. 2000;85:3276–3282.
Finch CE. The menopause and aging, a comparative perspective. J. Steroid Biochem. Mol. Biol. 2014;142:132–141.
Long T, Yao JK, Li J, et al. Comparison of transitional vs surgical menopause on monoamine and amino acid levels in the rat brain. Mol. Cell. Endocrinol. 2018;476:139–147.
Springer LN, McAsey ME, Flaws JA, et al. Involvement of Apoptosis in 4-Vinylcyclohexene Diepoxide- Induced Ovotoxicity in Rats. Toxicol. Appl. Pharmacol. 1996;139:394–401.
Mayer LP, Pearsall NA, Christian PJ, et al. Long-term effects of ovarian follicular depletion in rats by 4-vinylcyclohexene diepoxide. Reprod. Toxicol. 2002;16:775–781.
Gilardi K V, Shideler SE, Valverde CR, et al. Characterization of the onset of menopause in the rhesus macaque. Biol. Reprod. 1997;57:335–340.
Roberts JA, Gilardi KVK, Lasley B, et al. Reproductive senescence predicts cognitive decline in aged female monkeys. Neuroreport. 1997;8:2047–2051.
Hara Y, Park CS, Janssen WGM, et al. Synaptic correlates of memory and menopause in the hippocampal dentate gyrus in rhesus monkeys. Neurobiol. Aging. 2012;33:421.e17–421.e28.
Rapp PR, Morrison JH, Roberts JA. Cyclic estrogen replacement improves cognitive function in aged ovariectomized rhesus monkeys. J. Neurosci. 2003;23:5708–5714.
Baxter MG, Roberts MT, Gee NA, et al. Multiple clinically-relevant hormone therapy regimens fail to improve cognitive function in aged ovariectomized rhesus monkeys. Neurobiol Aging. 2013;34:1882–1890.
Hasegawa Y, Hojo Y, Kojima H, et al. Estradiol rapidly modulates synaptic plasticity of hippocampal neurons: Involvement of kinase networks. Brain Res. 2015;1621:147–161.
Pietras RJ, Szego CM. Endometrial cell calcium and oestrogen action. Nature. 1975;253:357–359.
Perret S, Dockery P, Harvey B. 17β-Oestradiol stimulates capacitative Ca2+ entry in human endometrial cells. Mol. Cell. Endocrinol. 2001;176:77–84.
Lösel R, Wehling M. Nongenomic actions of steroid hormones. Nat. Rev. Mol. Cell Biol. 2003;4:46–55.
Liu F, Day M, Muñiz LC, et al. Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008;11:334–343.
Spencer-Segal JL, Tsuda MC, Mattei L, et al. Estradiol acts via estrogen receptors alpha and beta on pathways important for synaptic plasticity in the mouse hippocampal formation. Neuroscience. 2012;202:131–146.
Boulware MI, Heisler JD, Frick KM. The Memory-Enhancing Effects of Hippocampal Estrogen Receptor Activation Involve Metabotropic Glutamate Receptor Signaling. J. Neurosci. 2013;33:15184–15194.
Phan A, Lancaster KE, Armstrong JN, et al. Rapid Effects of Estrogen Receptor α and β Selective Agonists on Learning and Dendritic Spines in Female Mice. Endocrinology. 2011;152:1492–1502.
Briz V, Liu Y, Zhu G, et al. A novel form of synaptic plasticity in field CA3 of hippocampus requires GPER1 activation and BDNF release. J. Cell Biol. 2015;210:1225–1237.
Revankar CM, Cimino DF, Sklar LA, et al. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science. 2005;307:1625–1630.
Toran-Allerand CD, Guan X, MacLusky NJ, et al. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 2002;22:8391–8401.
Qiu J, Bosch MA, Tobias SC, et al. Rapid signaling of estrogen in hypothalamic neurons involves a novel G-protein-coupled estrogen receptor that activates protein kinase C. J. Neurosci. 2003;23:9529–9540.
Gould E, Woolley CS, Frankfurt M, et al. Gonadal steroids regulate dendritic spine density in hippocampal pyramidal cells in adulthood. J. Neurosci. 1990;10:1286–1291.
Hao J, Rapp PR, Leffler AE, et al. Estrogen Alters Spine Number and Morphology in Prefrontal Cortex of Aged Female Rhesus Monkeys. J. Neurosci. 2006;26:2571–2578.
Hara Y, Yuk F, Puri R, et al. Estrogen Restores Multisynaptic Boutons in the Dorsolateral Prefrontal Cortex while Promoting Working Memory in Aged Rhesus Monkeys. J. Neurosci. 2016;36:901–910.
Albert K, Hiscox J, Boyd B, et al. Estrogen enhances hippocampal gray-matter volume in young and older postmenopausal women: a prospective dose-response study. Neurobiol. Aging. 2017;56:1–6.
Goto M, Abe O, Miyati T, et al. 3 Tesla MRI detects accelerated hippocampal volume reduction in postmenopausal women. J. Magn. Reson. Imaging. 2011;33:48–53.
Resnick SM, Espeland MA, Jaramillo SA, et al. Postmenopausal hormone therapy and regional brain volumes: the WHIMS-MRI Study. Neurology. 2009;72:135–142.
Kim G-W, Park K, Jeong G-W. Effects of Sex Hormones and Age on Brain Volume in Post-Menopausal Women. J. Sex. Med. 2018;15:662–670.
Vega JN, Zurkovsky L, Albert K, et al. Altered Brain Connectivity in Early Postmenopausal Women with Subjective Cognitive Impairment. Front. Neurosci. 2016;10:433.
Stevens MC, Clark VP, Prestwood KM. Low-dose estradiol alters brain activity. Psychiatry Res. Neuroimaging. 2005;139:199–217.
Dumas JA, Kutz AM, Naylor MR, et al. Increased memory load-related frontal activation after estradiol treatment in postmenopausal women. Horm. Behav. 2010;58:929–935.
Heikkinen T, Puoliväli J, Tanila H. Effects of long-term ovariectomy and estrogen treatment on maze learning in aged mice. Exp. Gerontol. 2004;39:1277–1283.
da Rocha JT, Sampaio TB, Santos Neto JS, et al. Cognitive effects of diphenyl diselenide and estradiol treatments in ovariectomized mice. Neurobiol. Learn. Mem. 2013;99:17–24.
Gibbs RB, Chipman AM, Hammond R, et al. Galanthamine plus estradiol treatment enhances cognitive performance in aged ovariectomized rats. Horm. Behav. 2011;60:607–616.
Gibbs RB, Mauk R, Nelson D, et al. Donepezil treatment restores the ability of estradiol to enhance cognitive performance in aged rats: evidence for the cholinergic basis of the critical period hypothesis. Horm. Behav. 2009;56:73–83.
Jacome LF, Gautreaux C, Inagaki T, et al. Estradiol and ERβ agonists enhance recognition memory, and DPN, an ERβ agonist, alters brain monoamines. Neurobiol. Learn. Mem. 2010;94:488–498.
Hammond R, Mauk R, Ninaci D, et al. Chronic treatment with estrogen receptor agonists restores acquisition of a spatial learning task in young ovariectomized rats. Horm. Behav. 2009;56:309–314.
Kim J, Szinte JS, Boulware MI, et al. 17β-Estradiol and Agonism of G-protein-Coupled Estrogen Receptor Enhance Hippocampal Memory via Different Cell-Signaling Mechanisms. J. Neurosci. 2016;36:3309–3321.
Hawley WR, Grissom EM, Moody NM, et al. Activation of G-protein-coupled receptor 30 is sufficient to enhance spatial recognition memory in ovariectomized rats. Behav. Brain Res. 2014;262:68–73.
Gold EB, Sternfeld B, Kelsey JL, et al. Relation of Demographic and Lifestyle Factors to Symptoms in a Multi-Racial/Ethnic Population of Women 40-55 Years of Age. Am. J. Epidemiol. 2000;152:463–473.
Woods NF, Mitchell ES, Adams C. Memory functioning among midlife women: Observations from the Seattle Midlife Women’s Health Study. Menopause. 2000. p. 257–265.
Drogos LL, Rubin LH, Geller SE, et al. Objective cognitive performance is related to subjective memory complaints in midlife women with moderate to severe vasomotor symptoms. Menopause. 2013;20:1236–1242.
Weber MT, Mapstone M, Staskiewicz J, et al. Reconciling subjective memory complaints with objective memory performance in the menopausal transition. Menopause. 2012;19:735–741.
Greendale GA, Huang M-H, Wight RG, et al. Effects of the menopause transition and hormone use on cognitive performance in midlife women. Neurology. 2009;72:1850–1857.
Freeman EW, Sammel MD, Gross SA, et al. Poor sleep in relation to natural menopause: a population-based 14-year follow-up of midlife women. Menopause. 2015;22:719–726.
Cintron D, Lipford M, Larrea-Mantilla L, et al. Efficacy of menopausal hormone therapy on sleep quality: systematic review and meta-analysis. Endocrine. 2017;55:702–711.
Freedman RR, Roehrs TA. Effects of REM sleep and ambient temperature on hot flash-induced sleep disturbance. Menopause J. North Am. Menopause Soc. 2006;13:576–583.
Freedman RR, Roehrs TA. Sleep disturbance in menopause. Menopause J. North Am. Menopause Soc. 2007;14:826–829.
Parmeggiani PL, Zamboni G, Cianci T, et al. Absence of thermoregulatory vasomotor responses during fast wave sleep in cats. Electroencephalogr. Clin. Neurophysiol. 1977;42:372–380.
Van Dort CJ, Zachs DP, Kenny JD, et al. Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc. Natl. Acad. Sci. U. S. A. 2015;112:584–589.
Han Y, Shi Y, Xi W, et al. Selective Activation of Cholinergic Basal Forebrain Neurons Induces Immediate Sleep-wake Transitions. Curr. Biol. 2014;24:693–698.
Tao MF, Sun DM, Shao HF, et al. Poor sleep in middle-aged women is not associated with menopause per se. Brazilian J. Med. Biol. Res. 2016;49:e4718.
Hajali V, Sheibani V, Esmaeili-Mahani S, et al. Female rats are more susceptible to the deleterious effects of paradoxical sleep deprivation on cognitive performance. Behav. Brain Res. 2012;228:311–318.
Deurveilher S, Seary ME, Semba K. Ovarian hormones promote recovery from sleep deprivation by increasing sleep intensity in middle-aged ovariectomized rats. Horm. Behav. 2013;63:566–576.
Schwartz MD, Mong JA. Estradiol suppresses recovery of REM sleep following sleep deprivation in ovariectomized female rats. Physiol. Behav. 2011;104:962–971.
Paul KN, Laposky AD, Turek FW. Reproductive hormone replacement alters sleep in mice. Neurosci. Lett. 2009;463:239–243.
Albert K, Newhouse P. Estrogen, stress, and depression: Cognitive and biological interactions. Annu. Rev. Psychol.
Kessler RC, Berglund P, Demler O, et al. Lifetime Prevalence and Age-of-Onset Distributions of DSM-IV Disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry. 2005;62:593.
Newhouse P, Albert K. Estrogen, Stress, and Depression. JAMA Psychiatry. 2015;72:727.
Schmidt PJ, Ben Dor R, Martinez PE, et al. Effects of Estradiol Withdrawal on Mood in Women With Past Perimenopausal Depression. JAMA Psychiatry. 2015;72:714.
Jovanovic H, Kocoska-Maras L, Rådestad AF, et al. Effects of estrogen and testosterone treatment on serotonin transporter binding in the brain of surgically postmenopausal women – a PET study. Neuroimage. 2015;106:47–54.
Booij L, Does W Van der, Benkelfat C, et al. Predictors of Mood Response to Acute Tryptophan Depletion A Reanalysis. Neuropsychopharmacology. 2002;27:852–861.
Halbreich U, Rojansky N, Palter S, et al. Estrogen augments serotonergic activity in postmenopausal women. Biol. Psychiatry. 1995;37:434–441.
Moses-Kolko EL, Berga SL, Greer PJ, et al. Widespread increases of cortical serotonin type 2A receptor availability after hormone therapy in euthymic postmenopausal women. Fertil. Steril. 2003;80:554–559.
Fink G, Sumner BE, Rosie R, et al. Estrogen control of central neurotransmission: effect on mood, mental state, and memory. Cell. Mol. Neurobiol. 1996;16:325–344.
Stokes PE. The potential role of excessive cortisol induced by HPA hyperfunction in the pathogenesis of depression. Eur. Neuropsychopharmacol. 1995;5:77–82.
Kirschbaum C, Wust S, Hellhammer D. Consistent sex differences in cortisol responses to psychological stress. Psychosom. Med. 1992;54:648–657.
Altemus M. Sex differences in depression and anxiety disorders: potential biological determinants. Horm. Behav. 2006;50:534–538.
Kirschbaum C, Kudielka BM, Gaab J, et al. Impact of gender, menstrual cycle phase, and oral contraceptives on the activity of the hypothalamus-pituitary-adrenal axis. Psychosom. Med. 61:154–162.
Bartus RT, Dean III RL, Beer B, et al. The Cholinergic Hypothesis of Geriatric Memory Dysfunction. Science (80-. ). 1982;217:408–417.
Nordberg A, Winblad B. Reduced number of [3H]nicotine and [3H]acetylcholine binding sites in the frontal cortex of Alzheimer brains. Neurosci. Lett. 1986;72:115–120.
Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006;113:1625–1644.
Colombo PJ, Gallagher M. Individual Differences in Spatial Memory and Striatal ChAT Activity among Young and Aged Rats. Neurobiol. Learn. Mem. 1998;70:314–327.
Rogers SL, Farlow MR, Doody RS, et al. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Donepezil Study Group. Neurology. 1998;50:136–145.
Raskind MA, Peskind ER, Wessel T, et al. Galantamine in AD: A 6-month randomized, placebo-controlled trial with a 6-month extension. The Galantamine USA-1 Study Group. Neurology. 2000;54:2261–2268.
Rösler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomised controlled trial. BMJ. 1999;318:633–638.
Hammond R, Gibbs RB. GPR30 is positioned to mediate estrogen effects on basal forebrain cholinergic neurons and cognitive performance. Brain Res. 2011;1379:53–60.
Shughrue PJ, Scrimo PJ, Merchenthaler I. Estrogen binding and estrogen receptor characterization (ERalpha and ERbeta) in the cholinergic neurons of the rat basal forebrain. Neuroscience. 2000;96:41–49.
Dohanich GP, Fader AJ, Javorsky DJ. Estrogen and estrogen-progesterone treatments counteract the effect of scopolamine on reinforced T-maze alternation in female rats. Behav. Neurosci. 1994;108:988–992.
Tanabe F, Miyasaka N, Kubota T, et al. Estrogen and progesterone improve scopolamine-induced impairment of spatial memory. J. Med. Dent. Sci. 2004;51:89–98.
Fader AJ, Johnson PE., Dohanich GP. Estrogen Improves Working But Not Reference Memory and Prevents Amnestic Effects of Scopolamine on a Radial-Arm Maze. Pharmacol. Biochem. Behav. 1999;62:711–717.
Gibbs RB, Burke AM, Johnson DA. Estrogen Replacement Attenuates Effects of Scopolamine and Lorazepam on Memory Acquisition and Retention. Horm. Behav. 1998;34:112–125.
Knüsel B, Beck KD, Winslow JW, et al. Brain-derived neurotrophic factor administration protects basal forebrain cholinergic but not nigral dopaminergic neurons from degenerative changes after axotomy in the adult rat brain. J. Neurosci. 1992;12:4391–4402.
Morse JK, Wiegand SJ, Anderson K, et al. Brain-derived neurotrophic factor (BDNF) prevents the degeneration of medial septal cholinergic neurons following fimbria transection. J. Neurosci. 1993;13:4146–4156.
Alderson RF, Alterman AL, Barde Y-A, et al. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron. 1990;5:297–306.
Hefti F. Nerve growth factor promotes survival of septal cholinergic neurons after fimbrial transections. J. Neurosci. 1986;6:2155–2162.
Fischer W, Wictorin K, Björklund A, et al. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329:65–68.
Gibbs RB. Estrogen and Nerve Growth Factor-related Systems in Brain. Ann. N. Y. Acad. Sci. 1994;743:165–196.
Singh M, Sétáló G, Guan X, et al. Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J. Neurosci. 1999;19:1179–1188.
Toran-Allerand CD, Miranda RC, Bentham WD, et al. Estrogen receptors colocalize with low-affinity nerve growth factor receptors in cholinergic neurons of the basal forebrain. Proc. Natl. Acad. Sci. U. S. A. 1992;89:4668–4672.
Gibbs RB, Wu D, Hersh LB, et al. Effects of Estrogen Replacement on the Relative Levels of Choline Acetyltransferase, trkA, and Nerve Growth Factor Messenger RNAs in the Basal Forebrain and Hippocampal Formation of Adult Rats. Exp. Neurol. 1994;129:70–80.
McMillan PJ, Singer CA, Dorsa DM. The effects of ovariectomy and estrogen replacement on trkA and choline acetyltransferase mRNA expression in the basal forebrain of the adult female Sprague-Dawley rat. J. Neurosci. 1996;16:1860–1865.
Gibbs RB. Levels of trkA and BDNF mRNA, but not NGF mRNA, fluctuate across the estrous cycle and increase in response to acute hormone replacement. Brain Res. 1998;787:259–268.
Singer CA, McMillan PJ, Dobie DJ, et al. Effects of estrogen replacement on choline acetyltransferase and trkA mRNA expression in the basal forebrain of aged rats. Brain Res. 1998;789:343–346.
Smith YR, Minoshima S, Kuhl DE, et al. Effects of Long-Term Hormone Therapy on Cholinergic Synaptic Concentrations in Healthy Postmenopausal Women. J. Clin. Endocrinol. Metab. 2001;86:679–684.
Smith YR, Bowen L, Love TM, et al. Early Initiation of Hormone Therapy in Menopausal Women Is Associated with Increased Hippocampal and Posterior Cingulate Cholinergic Activity. J. Clin. Endocrinol. Metab. 2011;96:E1761–E1770.
Dumas JA, Kutz AM, Naylor MR, et al. Estradiol treatment altered anticholinergic-related brain activation during working memory in postmenopausal women. Neuroimage. 2012;60:1394–1403.
Newhouse P, Albert K, Astur R, et al. Tamoxifen Improves Cholinergically Modulated Cognitive Performance in Postmenopausal Women. Neuropsychopharmacology. 2013;38:2632–2643.
Bäckman L, Ginovart N, Dixon RA, et al. Age-Related Cognitive Deficits Mediated by Changes in the Striatal Dopamine System. Am. J. Psychiatry. 2000;157:635–637.
Erixon-Lindroth N, Farde L, Robins Wahlin T-B, et al. The role of the striatal dopamine transporter in cognitive aging. Psychiatry Res. Neuroimaging. 2005;138:1–12.
Rinne JO, Lönnberg P, Marjamäki P. Age-dependent decline in human brain dopamine D1 and D2 receptors. Brain Res. 1990;508:349–352.
Wang Y, Chan GLY, Holden JE, et al. Age-dependent decline of dopamine D1 receptors in human brain: A PET study. Synapse. 1998;30:56–61.
Kish SJ, Shannak K, Rajput A, et al. Aging Produces a Specific Pattern of Striatal Dopamine Loss: Implications for the Etiology of Idiopathic Parkinson’s Disease. J. Neurochem. 1992;58:642–648.
Seeman P, Bzowej NH, Guan H-C, et al. Human brain dopamine receptors in children and aging adults. Synapse. 1987;1:399–404.
Simon H, Taghzouti K, Le Moal M. Deficits in spatial-memory tasks following lesions of septal dopaminergic terminals in the rat. Behav. Brain Res. 1986;19:7–16.
Baunez C, Robbins TW. Effects of dopamine depletion of the dorsal striatum and further interaction with subthalamic nucleus lesions in an attentional task in the rat. Neuroscience. 1999;92:1343–1356.
Brown RG, Marsden CD. ‘Subcorttcal dementia’: The neuropsychological evidence. Neuroscience. 1988;25:363–387.
Lawrence A, Weeks RA, Brooks DJ, et al. The relationship between striatal dopamine receptor binding and cognitive performance in Huntington’s disease. Brain. 1998;121:1343–1355.
Luciana M, Collins PF. Dopaminergic Modulation of Working Memory for Spatial but Not Object Cues in Normal Humans. J. Cogn. Neurosci. 1997;9:330–347.
Callier S, Le Saux M, Lhiaubet A-M, et al. Evaluation of the protective effect of oestradiol against toxicity induced by 6-hydroxydopamine and 1-methyl-4-phenylpyridinium ion (MPP+) towards dopaminergic mesencephalic neurones in primary culture. J. Neurochem. 2002;80:307–316.
Morissette M, Paolo T Di. Effect of Chronic Estradiol and Progesterone Treatments of Ovariectomized Rats on Brain Dopamine Uptake Sites. J. Neurochem. 1993;60:1876–1883.
Leranth C, Roth RH, Elsworth JD, et al. Estrogen is essential for maintaining nigrostriatal dopamine neurons in primates: implications for Parkinson’s disease and memory. J. Neurosci. 2000;20:8604–8609.
Dumas JA, Filippi CG, Newhouse PA, et al. Dopaminergic contributions to working memory-related brain activation in postmenopausal women. Menopause. 2017;24:163–170.
Kambeitz J, Abi-Dargham A, Kapur S, et al. Alterations in cortical and extrastriatal subcortical dopamine function in schizophrenia: systematic review and meta-analysis of imaging studies. Br. J. Psychiatry. 2014;204:420–429.
Howes OD, McCutcheon R, Owen MJ, et al. The Role of Genes, Stress, and Dopamine in the Development of Schizophrenia. Biol. Psychiatry. 2017;81:9–20.
Howes OD, Montgomery AJ, Asselin M-C, et al. Elevated Striatal Dopamine Function Linked to Prodromal Signs of Schizophrenia. Arch. Gen. Psychiatry. 2009;66:13.
Häfner H, Riecher-Rössler A, An Der Heiden W, et al. Generating and testing a causal explanation of the gender difference in age at first onset of schizophrenia. Psychol. Med. 1993;23:925–940.
Cohen RZ, Seeman M V, Gotowiec A, et al. Earlier puberty as a predictor of later onset of schizophrenia in women. Am. J. Psychiatry. 1999;156:1059–1064.
Grigoriadis S, Seeman M V. The Role of Estrogen in Schizophrenia: Implications for Schizophrenia Practice Guidelines for Women. Can. J. Psychiatry. 2002;47:437–442.
Akhondzadeh S, Nejatisafa AA, Amini H, et al. Adjunctive estrogen treatment in women with chronic schizophrenia: a double-blind, randomized, and placebo-controlled trial. Prog. Neuro-Psychopharmacology Biol. Psychiatry. 2003;27:1007–1012.
Kindler J, Weickert CS, Skilleter AJ, et al. Selective Estrogen Receptor Modulation Increases Hippocampal Activity during Probabilistic Association Learning in Schizophrenia. Neuropsychopharmacology. 2015;40:2388–2397.
Weickert TW, Weinberg D, Lenroot R, et al. Adjunctive raloxifene treatment improves attention and memory in men and women with schizophrenia. Mol. Psychiatry. 2015;20:685–694.
Van Den Eeden SK, Tanner CM, Bernstein AL, et al. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003;157:1015–1022.
Benedetti MD, Maraganore DM, Bower JH, et al. Hysterectomy, menopause, and estrogen use preceding Parkinson’s disease: An exploratory case-control study. Mov. Disord. 2001;16:830–837.
Lv M, Zhang Y, Chen G, et al. Reproductive factors and risk of Parkinson’s disease in women: A meta-analysis of observational studies. Behav. Brain Res. 2017;335:103–110.
Liu R, Baird D, Park Y, et al. Female reproductive factors, menopausal hormone use, and Parkinson’s disease. Mov. Disord. 2014;29:889–896.
Litim N, Morissette M, Di Paolo T. Neuroactive gonadal drugs for neuroprotection in male and female models of Parkinson’s disease. Neurosci. Biobehav. Rev. 2016;67:79–88.
Marder K, Tang MX, Alfaro B, et al. Postmenopausal estrogen use and Parkinson’s disease with and without dementia. Neurology. 1998;50:1141–1143.
Labandeira-Garcia JL, Rodriguez-Perez AI, Valenzuela R, et al. Menopause and Parkinson’s disease. Interaction between estrogens and brain renin-angiotensin system in dopaminergic degeneration. Front. Neuroendocrinol. 2016;43:44–59.
Re RN. Tissue renin angiotensin systems. Med. Clin. North Am. 2004;88:19–38.
Garrido AM, Griendling KK. NADPH oxidases and angiotensin II receptor signaling. Mol. Cell. Endocrinol. 2009;302:148–158.
Garrido-Gil P, Valenzuela R, Villar-Cheda B, et al. Expression of angiotensinogen and receptors for angiotensin and prorenin in the monkey and human substantia nigra: an intracellular renin-angiotensin system in the nigra. Brain Struct. Funct. 2013;218:373–388.
Muñoz A, Rey P, Guerra MJ, et al. Reduction of dopaminergic degeneration and oxidative stress by inhibition of angiotensin converting enzyme in a MPTP model of parkinsonism. Neuropharmacology. 2006;51:112–120.
Joglar B, Rodriguez-Pallares J, Rodriguez-Perez AI, et al. The inflammatory response in the MPTP model of Parkinson’s disease is mediated by brain angiotensin: relevance to progression of the disease. J. Neurochem. 2009;109:656–669.
Rodriguez-Perez AI, Borrajo A, Valenzuela R, et al. Critical period for dopaminergic neuroprotection by hormonal replacement in menopausal rats. Neurobiol. Aging. 2015;36:1194–1208.
Harman D. Free radical theory of aging: Consequences of mitochondrial aging. Age (Omaha). 1983;6:86–94.
Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses. 2004;63:8–20.
Croteau E, Castellano CA, Fortier M, et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018;107:18–26.
Mosconi L, Mistur R, Switalski R, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:811–822.
Yao J, Irwin RW, Zhao L, et al. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14670–14675.
Mosconi L, Berti V, Quinn C, et al. Perimenopause and emergence of an Alzheimer’s bioenergetic phenotype in brain and periphery. PLoS One. 2017;12:e0185926.
Yin F, Yao J, Sancheti H, et al. The perimenopausal aging transition in the female rat brain: decline in bioenergetic systems and synaptic plasticity. Neurobiol. Aging. 2015;36:2282–2295.
Rasgon NL, Silverman D, Siddarth P, et al. Estrogen use and brain metabolic change in postmenopausal women. Neurobiol. Aging. 2005;26:229–235.
Schmidt R, Schmidt H, Curb JD, et al. Early inflammation and dementia: A 25-year follow-up of the Honolulu-Asia aging study. Ann. Neurol. 2002;52:168–174.
Teunissen CE, van Boxtel MPJ, Bosma H, et al. Inflammation markers in relation to cognition in a healthy aging population. J. Neuroimmunol. 2003;134:142–150.
Banks WA, Farr SA, La Scola ME, et al. Intravenous human interleukin-1alpha impairs memory processing in mice: dependence on blood-brain barrier transport into posterior division of the septum. J. Pharmacol. Exp. Ther. 2001;299:536–541.
Jiang Q, Li W, Sun J, et al. Inhibitory effect of estrogen receptor beta on P2X3 receptors during inflammation in rats. Purinergic Signal. 2017;13:105–117.
Cipolla MJ, Godfrey JA, Wiegman MJ. The effect of ovariectomy and estrogen on penetrating brain arterioles and blood-brain barrier permeability. Microcirculation. 2009;16:685–693.
Brown CM, Mulcahey TA, Filipek NC, et al. Production of proinflammatory cytokines and chemokines during neuroinflammation: novel roles for estrogen receptors alpha and beta. Endocrinology. 2010;151:4916–4925.
Pfeilschifter J, Köditz R, Pfohl M, et al. Changes in Proinflammatory Cytokine Activity after Menopause. Endocr. Rev. 2002;23:90–119.
Störk S, von Schacky C, Angerer P. The effect of 17beta-estradiol on endothelial and inflammatory markers in postmenopausal women: a randomized, controlled trial. Atherosclerosis. 2002;165:301–307.
Kalyan S, Hitchcock CL, Pudek M, et al. Acute Effects of Premenopausal Hysterectomy with Bilateral Oophorectomy on Serum Lipids, Hormonal Values, Inflammatory Markers, and Metabolism. J. Gynecol. Surg. 2011;27:9–15.
Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016;12:719–732.
Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934.
Shumaker SA, Legault C, Kuller L, et al. Conjugated Equine Estrogens and Incidence of Probable Dementia and Mild Cognitive Impairment in Postmenopausal Women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2947.
Espeland MA, Rapp SR, Shumaker SA, et al. Conjugated Equine Estrogens and Global Cognitive Function in Postmenopausal Women: Women’s Health Initiative Memory Study. JAMA. 2004;291:2959.
Sherwin BB. Estrogen therapy: is time of initiation critical for neuroprotection? Nat. Rev. Endocrinol. 2009;5:620–627.
Maki PM, Zonderman AB, Resnick SM. Enhanced Verbal Memory in Nondemented Elderly Women Receiving Hormone-Replacement Therapy. Am. J. Psychiatry. 2001;158:227–233.
Resnick SM, Metter EJ, Zonderman AB. Estrogen replacement therapy and longitudinal decline in visual memory. A possible protective effect? Neurology. 1997;49:1491–1497.
Hao J, Rapp PR, Janssen WGM, et al. Interactive effects of age and estrogen on cognition and pyramidal neurons in monkey prefrontal cortex. Proc. Natl. Acad. Sci. U. S. A. 2007;104:11465–11470.
Kohama SG, Renner L, Landauer N, et al. Effect of Ovarian Hormone Therapy on Cognition in the Aged Female Rhesus Macaque. J. Neurosci. 2016;36:10416–10424.
Gibbs RB. Long-term treatment with estrogen and progesterone enhances acquisition of a spatial memory task by ovariectomized aged rats☆. Neurobiol. Aging. 2000;21:107–116.
Gibbs RB. Estrogen Therapy and Cognition: A Review of the Cholinergic Hypothesis. Endocr. Rev. 2010;31:224–253.
Gonzalez GA, Hofer MP, Syed YA, et al. Tamoxifen accelerates the repair of demyelinated lesions in the central nervous system. Sci. Rep. 2016;6:31599.
Baez-Jurado E, Rincón-Benavides MA, Hidalgo-Lanussa O, et al. Molecular mechanisms involved in the protective actions of Selective Estrogen Receptor Modulators in brain cells. Front. Neuroendocrinol. 2019;52:44–64.
Ishihara Y, Itoh K, Ishida A, et al. Selective estrogen-receptor modulators suppress microglial activation and neuronal cell death via an estrogen receptor-dependent pathway. J. Steroid Biochem. Mol. Biol. 2015;145:85–93.
Aisen PS, Cummings J, Jack CR, et al. On the path to 2025: understanding the Alzheimer’s disease continuum. Alzheimers. Res. Ther. 2017;9:60.
Farrer LA, Cupples LA, Haines JL, et al. Effects of Age, Sex, and Ethnicity on the Association Between Apolipoprotein E Genotype and Alzheimer Disease. JAMA. 1997;278:1349.
Bove R, Secor E, Chibnik LB, et al. Age at surgical menopause influences cognitive decline and Alzheimer pathology in older women. Neurology. 2014;82:222–229.
Zandi PP, Carlson MC, Plassman BL, et al. Hormone Replacement Therapy and Incidence of Alzheimer Disease in Older Women. The Cache County Study. JAMA. 2002;288:2123.
Paganini-Hill A, Henderson VW. Estrogen Replacement Therapy and Risk of Alzheimer Disease. Arch. Intern. Med. 1996;156:2213.
Kantarci K, Lowe VJ, Lesnick TG, et al. Early Postmenopausal Transdermal 17β-Estradiol Therapy and Amyloid-β Deposition. J. Alzheimers. Dis. 2016;53:547–556.
Merlo S, Spampinato SF, Sortino MA. Estrogen and Alzheimer’s disease: Still an attractive topic despite disappointment from early clinical results. Eur. J. Pharmacol. 2017;817:51–58.
Jaffe AB, Toran-Allerand CD, Greengard P, et al. Estrogen regulates metabolism of Alzheimer amyloid β precursor protein. J. Biol. Chem. 1994;269:13065–13068.
Manthey D, Heck S, Engert S, et al. Estrogen induces a rapid secretion of amyloid β precursor protein via the mitogen-activated protein kinase pathway. Eur. J. Biochem. 2001;268:4285–4291.
Goodenough S, Schäfer M, Behl C. Estrogen-induced cell signalling in a cellular model of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2003;84:301–305.
Merlo S, Sortino MA. Estrogen activates matrix metalloproteinases-2 and -9 to increase beta amyloid degradation. Mol. Cell. Neurosci. 2012;49:423–429.
Xiao Z-M, Sun L, Liu Y-M, et al. Estrogen Regulation of the Neprilysin Gene Through A Hormone-Responsive Element. J. Mol. Neurosci. 2009;39:22–26.
Liang K, Yang L, Yin C, et al. Estrogen stimulates degradation of beta-amyloid peptide by up-regulating neprilysin. J. Biol. Chem. 2010;285:935–942.
Alvarez De La Rosa M, Silva I, Nilsen J, et al. Estradiol Prevents Neural Tau Hyperphosphorylation Characteristic of Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2005;1052:210–224.
Zhang Z, Simpkins JW. Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res. 2010;1345:176–181.
Wibowo E, Calich HJ, Currie RW, et al. Prolonged androgen deprivation may influence the autoregulation of estrogen receptors in the brain and pelvic floor muscles of male rats. Behav. Brain Res. 2015;286:128–135.
Matousek RH, Sherwin BB. A randomized controlled trial of add-back estrogen or placebo on cognition in men with prostate cancer receiving an antiandrogen and a gonadotropin-releasing hormone analog. Psychoneuroendocrinology. 2010;35:215–225.
Taxel P, Stevens MC, Trahiotis M, et al. The Effect of Short-Term Estradiol Therapy on Cognitive Function in Older Men Receiving Hormonal Suppression Therapy for Prostate Cancer. J. Am. Geriatr. Soc. 2004;52:269–273.
Wu LM, Diefenbach MA, Gordon WA, et al. Cognitive problems in patients on androgen deprivation therapy: A qualitative pilot study. Urol Oncol. 2013;31.
Hogervorst E, De Jager C, Budge M, et al. Serum levels of estradiol and testosterone and performance in different cognitive domains in healthy elderly men and women. Psychoneuroendocrinology. 2004;29:405–421.
Carlson LE, Sherwin BB. Higher levels of plasma estradiol and testosterone in healthy elderly men compared with age-matched women may protect aspects of explicit memory. Menopause. 7:168–177.
Barrett-Connor E, Goodman-Gruen D, Patay B. Endogenous Sex Hormones and Cognitive Function in Older Men. J. Clin. Endocrinol. Metab. 1999;84:3681–3685.
Shumaker SA, Legault C, Rapp SR, et al. Estrogen Plus Progestin and the Incidence of Dementia and Mild Cognitive Impairment in Postmenopausal Women. JAMA. 2003;289:2651.
Maki PM, Henderson VW. Hormone therapy, dementia, and cognition: the Women’s Health Initiative 10 years on. Climacteric. 2012;15:256–262.
Depypere H, Vierin A, Weyers S, et al. Alzheimer’s disease, apolipoprotein E and hormone replacement therapy. Maturitas. 2016;94:98–105.
Schneider LS, Farlow MR, Henderson VW, et al. Effects of estrogen replacement therapy on response to tacrine in patients with Alzheimer’s disease. Neurology. 1996;46:1580–1584.
Ghoshal A, Rook JM, Dickerson JW, et al. Potentiation of M1 Muscarinic Receptor Reverses Plasticity Deficits and Negative and Cognitive Symptoms in a Schizophrenia Mouse Model. Neuropsychopharmacology. 2016;41:598–610.
Foster DJ, Choi DL, Jeffrey Conn P, et al. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer’s disease and schizophrenia. Neuropsychiatr. Dis. Treat. 2014;10:183–191.
Rook JM, Bertron JL, Cho HP, et al. A novel M1 PAM VU0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem. Neurosci. 2018;19:2274–2285.
Uslaner JM, Kuduk SD, Wittmann M, et al. Preclinical to Human Translational Pharmacology of the Novel M1 Positive Allosteric Modulator MK-7622. J. Pharmacol. Exp. Ther. 2018;365:556–566.
Bubser M, Byun N, Wood MR, et al. Muscarinic receptor pharmacology and circuitry for the modulation of cognition. Handb. Exp. Pharmacol. 2012.
Gould RW, Dencker D, Grannan M, et al. Role for the M1 Muscarinic Acetylcholine Receptor in Top-Down Cognitive Processing Using a Touchscreen Visual Discrimination Task in Mice. ACS Chem. Neurosci. 2015;6:1683–1695.
Grannan MD, Mielnik CA, Moran SP, et al. Prefrontal Cortex-Mediated Impairments in a Genetic Model of NMDA Receptor Hypofunction Are Reversed by the Novel M1 PAM VU6004256. ACS Chem. Neurosci. 2017;7:1706–1716.
Jones CK, Byun N, Bubser M. Muscarinic and Nicotinic Acetylcholine Receptor Agonists and Allosteric Modulators for the Treatment of Schizophrenia. Neuropsychopharmacology. 2012;37:16–42.
Acknowledgments
Preparation of this manuscript was partially supported by the National Institute on Aging R01AG047992, Alzheimer’s Association PCTR-16-383171 to PN, and National Institute on Aging 1R01AG054622 to CJ.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 508 kb)
Rights and permissions
About this article

Cite this article
Russell, J.K., Jones, C.K. & Newhouse, P.A. The Role of Estrogen in Brain and Cognitive Aging. Neurotherapeutics 16, 649–665 (2019). https://doi.org/10.1007/s13311-019-00766-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00766-9