
Abstract
S-Adenosyl methionine (SAMe), as a major methyl donor, exerts its influence on central nervous system function through cellular transmethylation pathways, including the methylation of DNA, histones, protein phosphatase 2A, and several catecholamine moieties. Based on available evidence, this review focuses on the lifelong range of severe neuropsychiatric and neurodegenerative diseases and their associated neuropathologies, which have been linked to the deficiency/load of SAMe production or/and the disturbance in transmethylation pathways. Also included in this review are the present-day applications of SAMe in the treatment in these diseases in each age group.
Similar content being viewed by others


Background
Since its discovery in 1951, S-adenosyl-L-methionine (SAMe, also known as AdoMet), has been shown to be second only to adenosine triphosphate when serving as a cofactor [1], and it is involved in a great number of reactions. SAMe is found in every living cell and has been commonly used as a dietary supplement since 1952. The liver plays a central role in the homeostasis of SAMe through both its synthesis and degradation [2].
Three principal pathways are related to SAMe metabolism: transmethylation, trans-sulfuration, and transaminopropylation. The transmethylation pathway is thought to play an especially important role in brain development, reflecting its dominant and ubiquitous effects on cell biology [3].
Most cells contain numerous SAMe-dependent methyltransferases that can transfer the methyl group (–CH3) to all varieties of key substrates, for example nucleic acid, protein, phospholipids, and monoamine neurotransmitters [4]. These molecular targets not only support normal biological development, but can also contribute to pathological states in the central nervous system (CNS) when epigenetically methylated at inappropriate times in the human life span.
The notion that a combination of environment and gene–environment interactions modulates brain development has become an accepted tenet of modern neuroscience, which has led to the definition of a new term, “neuroepigenetics”, for example DNA methylation and histone modification (methylation and acetylation) [5]. SAMe, as the principal methyl donor in cells, takes part in these critical epigenetic mechanisms, thus connecting nurture-based metabolism with brain development, in addition to other direct targets of methyl group-dependent biological functions in cells.
It is generally recognized that brain development is a long-term equilibrium dynamically vibrating between neurogenesis and neurodegeneration (neural cell deletion) throughout the whole mammalian life span. Emerging studies have linked deficient SAMe production and disruption in transmethylation pathways with delayed brain development and severe neuropsychiatric diseases [6].
Besides a broad application in the treatment of hepatic diseases, numerous preclinical and clinical studies over the last 2 decades demonstrated that SAMe showed promise in the treatment of critical neuropsychiatric diseases, including Alzheimer’s disease (AD) and major depression disorder (MDD) [7, 8]. However, despite its increasing popularity as a supplement, SAMe has not been as well studied as more conventional neural psychotherapeutic agents, especially with regards of underlying mechanisms [9]. The purpose of this review is to focus on the mechanistic role of SAMe in transmethylation pathways and its relation to neuropsychiatric diseases, as well as to discuss its efficacy.
SAMe processing and 1-carbon cycle
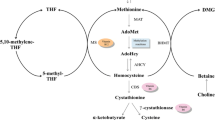
The process of generating SAMe involves a bicyclic cellular pathway consisting of folate and methionine cycles, which both belong to the 1-carbon cycle, a metabolic network that integrates nutrients from the environment to yield multiple epigenetic regulatory and biological functions [10]. A simplified version of this 1-carbon cycle is illustrated in Figure 1.

Three pathways of S = adenosyl-L-methionine (SAMe) metabolism and the 1- carbon cycle. The transmethylation pathway (light blue), trans-sulfuration pathway (brown), and transaminopropylation pathway (purple); 1- carbon cycle: the folate cycle (green) and methionine cycle (dark blue). THF = tetrahydrafolate; 5-MTHF = 5-methyltetrahydrafolate; 5, 10 - methylenetetrahydrofolate; Hcy = homocysteine; Cys = cysteine; GSH = glutathione; Gly - Glycine; Met = Methione; SAH = Sadenosylhomocysteine; dSAM = decarboxylated S - adenosyl - L - methionine
SAMe is a metabolic product of methionine generated by methionine-adenosyltransferase (MAT), which catalyzes the reaction at the expense of the complete dephosphorylation of 1 molecule of adenosine triphosphate in the presence of potassium and magnesium ions [11]. SAMe is then utilized as a donor of its methyl group to methylate many substrates to fulfill diverse biological functions. S-Adenosylhomocysteine (SAH or AdoHC) is produced in this process and then is hydrolyzed to homocysteine (Hcy). This reaction is reversible, during which process increased Hcy levels can lead to the accumulation of SAH, an inhibitor of the transmethylation events related to SAMe. Lower levels of SAMe and higher levels of SAH may result in a reduced methylation capacity, which is represented by the SAM/SAH ratio [3].
Of note, Hcy can be re-methylated to regenerate methionine by donation of a methyl group from 5-methyltetrahydrafolate (5-MTHF) via methionine synthase (MTR). Then the regenerated methionine goes into next cycle to produce SAMe (Fig. 1). B vitamins play important roles in this process, involving vitamin B12 (cobalamin, Cbl) as an essential cofactor and 5-MTHF as an indispensable substrate.
Vitamin B12 is used to mediate the MTR catalyzing process via 2 methyl transfer reactions [12]. Methylcobalamin is the metabolically active form of vitamin B12, and serves as an essential cofactor for the methylation of Hcy to methionine, affecting hundreds of SAMe-dependent methylation reactions [13]. In addition, adenosylcobalamin is another metabolically active form of vitamin B12 to be used as a cofactor for the enzyme methylmalonyl CoA mutase, which converts methylmalonylCoA to succinylCoA [14], Of note, a deficiency in vitamin B12 leads to a functional loss of folate, which results from folate being trapped in the form of methyltetrahydrofolate [15].
5-MTHF is an important product of folate metabolism cycle and this is also the primary circulating form of folate [16]. Dietary folate or dihydrofolate can be reduced to tetrahydrofolate by dihydrofolate reductase. Then tetrahydrofolate is catalyzed by serine hydroxy methyltransferase to produce 5,10-methylenetetrahydrofolate (5,10-MTHF). 5,10-MTHF reductase (5,10-MTHFR) carries out a central reaction in folate metabolism with the production of 5-MTHF, which is required for the biosynthesis of methionine, purine nucleotides, and thymidylate [17].
A failure or abnormality of either of these 2 cycles has been implicated in the pathogenesis of neuropsychiatric diseases. The most common example comes from the deficiency of folate and/or vitamin B12, which would disturb the methylation-dependent pathways by incapacitating a source of methyl groups, a phenomenon that has been linked to mental retardation, psychiatric disorders, myelopathy and neurological diseases [9, 18].
The most common condition associated with folate deficiency is affective disorders, such as MDD. Depressed patients with low folate appeared to have greater severity of depression and poorer response to various antidepressants and augmentation strategies [19, 20]. Furthermore, folate deficiency also correlates with dementia and cognitive impairment [21]. The mechanisms underlying this activity include a reduction in purine/pyrimidine synthesis and genomic and/or specific methylation reactions in brain, leading to decreased DNA stability and altered gene expression resulting in the mental dysfunction [22].
Vitamin B12 deficiency is often manifested in the form of neurological symptoms [22]. During vitamin B12 deficiency, Hcy cannot be converted to methionine, leading to Hcy accumulation; methylmalonic acid (MMA) cannot be converted to succinyl-CoA, resulting in MMA accumulation [23]. Both of these consequences contribute to the damage of myelin, resulting in a condition known as subacute combined degeneration of the spinal cord [24]. In addition, SAMe production is reduced and methylation reactions are impaired, which are subsequently associated with a reduced methylation capacity [25]. Fernàndez-Roig et al. [26] reported that vitamin B12 deficiency in the brain led to DNA hypomethylation in the TCblR/CD320 knockout mouse, which supported that DNA hypomethylation could be a link between vitamin B12 deficiency and neurological pathology [26]. Vitamin B12 can influence cell proliferation and differentiation in brain through protein phosphatase 2 (PP2A), nerve growth factor, and TACE [tumor necrosis factor (TNF)-α ADAM metalloprotease converting enzyme] pathways [13]. Methylcobalamin can facilitate neurite outgrowth and inhibit neural apoptosis through the Erk1/2 and Akt signaling pathways [27], which were implicated in neuropsychiatric diseases. For instance, Zhang et al. [28] found decreased brain levels of vitamin B12 across the lifespan and in autism and schizophrenia. Fortunately, high vitamin B12 levels can help maintain low blood levels of Hcy and MMA, and enhance the synthesis of SAMe [9].
SAMe is a major end product of the “1-carbon cycle”. The dysregulation of SAMe leads to alterations in > 100 methyltransferase reactions, which modulate metabolic pathyways [29]. The aberrant methylation has been implicated as a pathogenic mechanism in psychiatric disorders, such as MDD, as outlined in the section of the review (below), and also in neurological diseases, such as AD, as outlined in later sections. Consequently, this methyl-donor therapy has become a therapeutic strategy to deal with disease progression and/or enhance clinical outcome [30]. The detailed interpretation is described below.
Cellular targets of SAMe transmethylation pathways pertinent to mental/neurological diseases
SAMe and DNA Methylation Status
DNA methylation, as an epigenetic mechanism, plays a pivotal role in the regulation of gene transcription and the establishment and maintenance of cellular identity [31], which is disrupted in many diseases. It allows for sustained adaptability of gene expression in response to developmental or environmental factors [31]. Several DNA methylation marks have been implicated in the dynamics of gene regulation in the CNS. There are 2 kinds of DNA methylation dynamics that include 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC), involving 2 kinds of enzymes as DNA methyltransferases (DNMTs; Dnmt1, Dnmt3a and Dnmt3b) and the demethylases ten–eleven translocation (TET) methylcytosine dioxygenase (Tet1–3), and the methyl-CpG-binding protein 2 (MeCP2). We will interpret these events in the following sections. Tables (1, 2 and 3).
5-mC, derived from the addition of a methyl group from SAMe to the 5’ position of cytosine, has generally been viewed as a stable covalent modification to DNA [32]. There are 2 types of it in brain: 5mCG and 5mCH (H = A, T, or C), respectively according to whether the cytosine methylated predominantly occurs at cytosine–phosphate–guanine dinucleotides (CpGs) or non-CpGs [33, 34].
In fact, in the mammalian brain ~62% of CpGs are found to be methylated [33]. 5mCG is mainly located at intergenic regions, repetitive DNA regions, and silenced genes, directly interfering with the binding of certain transcriptional regulators to their cognate DNA sequences or recruiting repressive methyl-CpG binding domain (MBD) proteins [33, 35]. This results in transcriptional repression in differentiated neurons, whereas 5mCG levels remain unchanged during neurodevelopment [36].
However, 5mCH is the predominant form of DNA methylation in adult neurons [33, 37]. 5mCH methylatation differences occur around neuronal transcription factor binding sites and around transcription start sites, suggesting that it is sufficient to cause transcriptional repression in neurons, as effectively as, but independently of, 5-mCG [38]. Furthermore, neuronal 5mCH is actively maintained by DNMT3a in vivo [38], supporting the accumulation of 5mCH in neurons during postnatal neuronal maturation, which makes it an even better signal than 5mCG in this regard [35, 38].
5-hmC is enzymatically modified from 5-mC by TET [39]. Mouse brain tissues contain high levels of 5-hmC [39]. 5hmC levels in neurons are up to 10-fold higher than other cell types, accounting for ~40% of modified cytosine in the brain [38]. 5-hmC was reported to present at many highly methylated CG sites in mouse adult and fetal frontal cortex [40]. 5hmC is the main modification status for a many CpGs situated at transcriptionally active regions, such as DNaseI-hypersensitive regions and enhancers [35], suggesting its positive correlation with gene expression [41]. Moreover, 5-hmC is also enriched within gene bodies and at intron–exon boundaries of synaptic plasticity-related genes [42]. 5-hmC increases markedly from the fetal to the adult stage and in the adult brain, which implies the importance of 5hmC in neuronal maturation and function [35]. Besides acting as an intermediate in an active DNA demethylation pathway, 5hmC may also serve as a stable neuronal epigenetic mark in its own right [43].
Three DNMTs include 2 de novo methyltransferases, Dnmt3a and Dnmt3b [44], and 1 classical maintenance methyltransferase, Dnmt1 [45]. The former establish methylation patterns on unmethylated DNA [44] and the latter preserves existing methylation patterns by methylating hemi-methylated DNA [46]. Three enzymes were confirmed to catalyze the chemical reaction:
Suetake et al. [47] reported that the methyl transferases Dnmt3a or Dnmt3b showed similar Km values towards poly(dIdC)-poly(dIdC) and poly(dGdC)-poly(dGdC), suggesting little room for regulator activity dependent on the relative abundance of at least these 2 enzymes between tissues. The Km values for SAMe were not affected by the methyl-group acceptors, poly(dIdC)-poly(dIdC) and poly(dG-dC)-poly(dGdC) [47]. The Km values of Dnmt3a for dIdC and dGdC. respectively, were 1.2 ± 0.1 μM and 1.6 ± 0.2 μM; those of Dnmt3b for dIdC and dGdC, respectively, were 0.4 ± 0.1 μM and 0.5 ± 0.1 μM; the Km values of Dnmt3b for SAMe were 0.3 ± 0.1 μM and 0.7 ± 0.3 μM [47]. Gros et al. [48] reported that the Km values of Dnmt1were 4.4± 0.5μM and 0.27 ± 0.03μM for SAMe and DNA, respectively.
In support of developmental selectivity, Dnmt3a and Dnmt1 are expressed in both the embryonic and adult stages of the brain, whereas Dnmt3b is detectable only during early neurogenesis [49]. Furthermore, there is a transition of mouse de novo methyltransferase expression from Dnmt3b to Dnmt3a during neural progenitor cell development [50], suggesting that particular DNMTs play a role in neuronal function at specific times over neuronal development and maturation.
The 3 prominent demethylases are the TETs, which are expressed in the brain, respectively, as Tet1 [39], Tet2, and Tet3 [51]. Tet2 and Tet3 levels are considerably higher than Tet1 postnatally [51]. Tet1 is thought to have a role in neuronal activity-induced, region-specific, active DNA demethylation with concomitant gene expression changes [43]. In addition to its catalytic activity, Tet1 controls DNA methylation levels and thus regulates memory formation [52]. Tet3, the most highly expressed TET in the brain, is activity-dependent in primary cortical neurons [53]. Tet3 activity within the infralimbic prefrontal cortex is essential for the learning-dependent accumulation of 5-hmC and related chromatin modifications, which underpins rapid behavioral adaptations based on epigenetic activation [53]. Moreover, Tet3-KD neurons or Tetrodotoxin-induced decreases in Tet3 expression exhibited increased CpG methylation at the BDNF promoter IV region, whereas bicuculline-induced increases in Tet3 expression were shown to be associated with decreased methylation at the same region, supporting a pivotal role of Tet3 in regulating gene expression in response to global synaptic activity changes [54].
MeCP2 belongs to the MBD protein family, known as “readers” of DNA methylation, which bind methylated DNA to mediate the molecular consequences of this epigenetic mark [55]. MeCP2 is the highest expressed nuclear protein in the brain among various tissues [56], and 7 times higher in neurons than glia [57]. Several studies showed that MeCP2 is important for synapse maturation [57]. MeCP2 has 2 functional domains, transcriptional repression domain, which exhibits a long-range repression and MBD [58]. MBD confers it to bind DNA at CG methylation-dense regions with high levels [55], which also interacts with the nuclear receptor co-repressor/histone deacetylase 3 and Sin3a transcriptional co-repressor complexes, suggesting a role for MeCP2 as a transcriptional repressor [59]. In addition to 5mCG, several studies showed that MeCP2 can also bind to 5mCH in neurons with high affinity [60]. Later, Gabel et al. [61] reported that MeCP2 repressed gene expression by binding to methylated Cornu Ammonis (CA) sites within long genes. Chen et al. [62] also reported that MeCP2 binds to mCH in the mature brain to regulate transcription and the timing of onset for Rett’s syndrome (see below).
The development of brain is a spatial temporally orchestrated process that partly requires appropriate gene regulation through the control of DNA methylation and demethylation [63]. During the differentiation and maturation of CNS, a dynamic process, involving both de novo DNA methylation and demethylation, is required. Several studies have established that Dnmt1 plays a critical role in cellular differentiation as well as in dividing cells [64]. Some other findings support that Dnmt3b is required during early development, whereas Dnmt3a is required for normal cellular differentiation [49]. In the dividing cells, passive DNA demethylation occurs as the depletion of Dnmt1 results in newly incorporated cytosine to remain unmethylated and consequent hypomethylation [65]. The coincidence of the differentiation of the astrocytic lineage with DNA methylation and demethylation occurs on the Gfap gene promoter region [64]. Thus, the coordinated expression of Dnmts, key to the dynamic methylation and demethylation pattern, plays an important role in regulating neuronal development.
Moreover, in several recent studies, Chen et al. [66] found that the mammalian DNMTs (Dnmt3a and Dnmt3b) can also act as Ca2+ ion- and redox state-dependent active DNA demethylases. In the context of extremely low levels of SAMe, Dnmt3a and Dnmt3b might demethylate 5mC via deamination to thymine, which confirmed the dynamic activity of DNMTs with methyltransferases and demethylases according to the levels of SAMe [67].
In addition to acting as the body’s principal methyl donor, SAMe can play an important role in regulating the methylation status in any given cell type. In a recent study, Varela-Rey et al. [68] found that peripheral nerves with enforced elevated levels of SAMe, were specially characterized by DNA hypermethylation at promoter and enhancer regions of several lipid synthesis genes, which contributed to the peripheral myelin defects. Reduced SAMe levels correlate with DNA demethylation in peripheral nerves, which also can play a part in the peripheral myelin defects [68]. In fact, this group demonstrated that Schwann cell myelination was characterized by methylation dynamics, which were dependent on a tight control of SAMe levels [68]. Thus, the levels of SAMe likely also play a key gene regulatory role during development of neuropathies.
In the adult brain, dynamic DNA modifications (active demethylation or de novo methylation), is a general mechanism for rapid modifications in response to external stimuli in vivo [69]. As one of the active demethylation forms, TET enzymes and their corresponding product, 5hmC, partly act as intermediates in DNA demethylation. Several studies have shown that specific promoters of genes, related to neural plasticity and learning and memory, could be modified by neuronal activity through methylation and demethylation [70].
In a recent study, Saunderson et al. [71] reported an intricate balance between DNA methylation, DNA demethylation, and availability of SAMe, which governed the induction of the immediate–early gene (IEG) products in dentate gyrus neurons, as well as to determine behavioral responses after stress. In the context of normal SAMe levels, forced swim (FS) testing resulted in DNA demethylation at the IEG loci; conversely, with increased SAMe levels, DNA methylation at these loci was induced in response to FS [71]. The underlying mechanism may relate to the activity changes of Dnmt3a from DNA demethylase to methyltransferase after raising SAMe levels [71]. Therefore, even though little is known about the control of SAMe levels in the brain, the neuronal concentration of SAMe can be further investigated as a key factor in the molecular and behavioral responses to the stressors [71].
In a study in yeast cells, Sutter et al. [72] proposed an intriguing mechanism for sensing SAMe levels. They found the methylation modification of the catalytic subunit of PP2A, determining metabolic processes underlying growth, was responsive to methionine and SAMe levels [72]. They modeled that PP2A acted as a “sensor” of methionine and SAMe levels in yeast cells [72]. Current investigation seeks to elucidate the underlying mechanisms that are available to monitor SAMe levels in the mammalian brains and to determine the neuronal responses to environmental stressors. Overall, the DNA methylation status of brain neurons may be determined, not only by expression levels of DNA methylation/demethylation enzymes, but also by levels of SAMe.
SAMe, DNA Methylation, and Related Disease
Marked disturbances of DNA methylation in neurodevelopment can occur through the loss of the key enzymes—DNMTs and TETs—thereby leading to deficits in neuronal function. In addition to the examples listed in the last section, embryonic deletion of Dnmt3a results in neuronal dysfunction, such as hypoactivity, motor abnormalities, decreased neuromuscular function, fewer motor neurons, and a shortened lifespan [73]. The deletion of Dnmt1 in excitatory neurons of the dorsal forebrain in mice, although these mice are viable into adulthood, results in hypomethylation, dysregulation of gene expression, defects in dendritic branching, and progressive apoptotic degeneration of dorsal forebrain [74]. Depletion of Tet1 and Tet2, causes a bias towards trophectoderm differentiation in embryonic stem cells [75].
Notably, in a recent study, Shojaei Saadi et al. [76] treated bovine early embryos with SAMe in vitro and found that SAMe caused genome-wide hypermethylation mainly in exonic regions and in CpG islands, imprinting that significantly improved embryo hatching [76]. This study demonstrated that SAMe, as a dietary treatment, may operate in some circumstances to therapeutically boost methylation patterns to improve neurodevelopment. We will list numerous studies that are associated with specific neuropsychiatric diseases to demonstrate that, except for the loss of key enzymes in DNA methylation, nutritional deficiency and/or abnormal levels in the 1-carbon cycle or mutations in genes that control DNA methylation patterns have been linked to hypomethylated CNS neurons with impaired function [77].
Neural Tube Defects, Rett’s Syndrome, and Down’s Syndrome
SAMe metabolism has a critical role in the pathway of nutritional deprivation or mutations in genes is associated with neural tube defects (NTDs) and several developmental disorders, such as Rett’s syndrome and Down’s syndrome (DS).
NTDs are severe birth defects that result from a failure of the neural tube to close during the fourth week of embryogenesis. A group of studies support that an imbalance of SAMe metabolism, caused by the deficiency of B vitamins (folate, vitamin B12, and vitamin B6), leads to decreased availability of donor methyl groups as associated with NTDs [78, 79]. Two studies support that B vitamin supplements can increase the metabolic capacity of SAMe to provide methyl units and ameliorate NTDs [80, 81]. In fact, the literature provides explicit recommendations about pre- and postconception use of oral folic acid with or without a multivitamin/micronutrient supplement for the prevention of NTDs and other congenital anomalies [82]. Furthermore, a dietary supplement of methyl groups—methionine or betaine—plays a unique role in SAMe metabolism. For instance, previous studies showed the importance of methionine in neural tube closure [83], and the degree to which Hcy is re-methylated appears to be dependent on the ingestion of dietary methionine [83]. Different levels of methionine supplementation provide different functionalities in SAMe metabolism. Consistent with this, Dunlevy et al. [84] reported excess methionine supplement (≥ 5 mM) suppressed the methylation cycle with a decreased ratio of SAM/SAH, and unexpectedly caused NTDs in cultured mouse embryos [84]. To further explain this phenomenon, SAH might be increased to a greater extent than SAMe; thus, the ratio of SAM/SAH would be altered consistently with NTDs [84]. In fact, reduction in the SAM/SAH ratio is predicted to inhibit transmethylation reactions and is associated with NTDs [85]. Another study supported the use of betaine, a specific methyl donor, which resulted in a lowering of Hcy and the production of methionine or SAMe in conversion of Hcy to methionine [86]. Regarding drug-induced nutritional changes, methotrexate, an inhibitor of folate metabolism, can cause DNA hypomethylation and NTDs [87]. In a further therapeutic study, folate was found to serve as a supplement to restore the SAM/SAH ratio to benefit patients. For instance, periconceptional supplemental maternal folic acid increased methylation of IGF2, improving intrauterine programming of growth and development [88]. In these cases, dietary supplements, as outlined in Figure 1, help to increase the production of SAMe and restore the SAM/SAH ratio to benefit patients.
Rett’s syndrome, which is caused by a mutation in the gene for MeCP2 [89], is characterized by normal development for 6 to 12 months of life followed by regression and symptomatic presentation of intellectual disability in females [90]. The mouse models of MeCP2-null or MeCP2-mutant display features found in patients with Rett’s syndrome [91]. This mutation influences the interplay of the MeCP2 protein with methylated DNA and consequently changes the repression of transcription associated with a mental retardation [92]. Several studies supported an association between Rett’s syndrome and disturbances of cerebral methyl-group pools, including folate, SAMe, and the SAM/SAH ratio, and speculated on a compromised DNA-methylation [93,94,95,96]. However, there were no objective signs of clinical improvement or upregulation of SAMe and SAH levels in cerebrospinal fluid (CSF) following folic acid supplement [93,94,95,96]; thus, the authors did not recommend folic acid therapy for individuals with Rett’s syndrome. Interestingly, emerging literature has shown that prenatal folic acid supplements reduced risk for autism [97]. In sum, we have outlined evidence for an apparent lack of capacity of patients with Rett’s syndrome to regulate SAMe, SAH levels, and SAM/SAH ratio in the CSF, which leads to disturbances in the donation of methyl groups in defined cell types.
DS is a complex metabolic and genetic disorder that results from nondisjunction of chromosome 21 [98]. Premature aging and disturbed methylation are components of DS [99]. Linking these 2 facts, the gene for cystathionine β-synthase is located on chromosome 21 and is overexpressed in children with DS [91]. The 1-carbon metabolism pathway is greatly affected by overexpression of cystathionine β-synthase, for instance, decreasing levels of Hcy and thus methionine and SAMe, to thereby reduce the ratio of SAM/SAH [98, 100]. Consistent with this observation, Fountoulakis et al. [101] found an overexpression of tetrahydrofolate synthase, a key element in folate metabolism, in the fetal DS brain during the early second trimester to suggest abnormal folate metabolism and folate deficiency. In recent studies, Song et al. [102] showed that folic acid and vitamin B12 levels were decreased with increasing age in patients with DS [102]. Obeid et al. [99] found that methylation of 3 CpGs combined with plasma amyloid-β (Aβ) showed a 3-year accelerated aging in subjects with DS at the age of 12 years. DNA hyper- and hypomethylation were, respectively, observed in patients with DS with vitamin B12 deficiency and hyperhomocysteinemia [102]. Infantino et al. [103] found that the mitochondrial levels of SAM were reduced in DS, suggesting that impairment of the methyl cycle affected mitochondrial methyl availability in DS. We conclude that it will be worth investigating the role of the abnormalities in the 1-carbon cycle and methylation cycle to explain pathoepigenetic modifications in subjects with DS. Perturbation of the 1-carbon cycle result in folate deficiency and abnormal plasma Hcy concentrations [104, 105], which may contribute to the risk that any given mother will or will not have a child with DS. Specific enzymes, MTR, methionine synthase reductase, and MTHFR, can express polymorphisms that result in elevated homocysteinemia, a risk factor for having a child with DS [106].
In a key study, Pogribna et al. [98] supplied specific nutrients, (folinic acid, methyl-B12, methionine) to DS lymphoblastoid cells, resulting in modulation of the metabolites in 1-carbon cycle. It is premature to suggest that the use of 1-carbon cycle supplements (folate acid, vitamin B, SAMe) could ameliorate the progression of DS. However, it will useful to determine whether such supplements could be of benefit for women in order to reduce their risks of giving birth to a child with DS.
Schizophrenia
The hypothesis that there might be a relationship between the disorders of 1-carbon metabolism (and transmethylation) with the genesis of psychiatric disorders was first forwarded by Smythies as early as 1963 [107]. Several studies demonstrated that MAT, the critical enzyme of the 1-carbon cycle to produce SAMe, was significantly underactive in patients with schizophrenia (SZ) [108]. However, other studies showed that the disturbance in folate and/or Hcy metabolism was a risk factor for SZ, such that low plasma folate might be a potential independent factor for SZ [109]. This is also true for high Hcy levels [110]. In addition, several reports showed that low maternal folate linked with high Hcy levels were related to the teratogenic effect at the fetal stage [111] with late-stage risk of the development of SZ [112]. These studies implied that the disorders of 1-carbon cycle, including that of epigenetic DNA methylation pathways, played a role both in the etiology and progression of SZ and also in the predisposition toward SZ in offspring. This idea is also supported by Picker and Coyle [113], and has received additional traction through replicated studies demonstrating a transient doubling of SZ risk 2 decades following fetal exposure to famine conditions [114].
With regard to the disruption of 1-carbon cycle leading to the development of SZ, the abnormality in DNA methylation in many loci has been found to relate to brain development, stress response, and mitochondrial function [115]. Several reports show increased expression of DNA methyltransferases in telencephalic γ-aminobutyric acidergic neurons, for instance increased expression of DNMT1 and DNMT3a [116, 117], which can methylate several SZ-specific candidate genes including reelin, glutamic acid decarboxylase-67, and catechol-O-methyltransferase (COMT). Some examples include the hypermethylation of the reelin gene promoter with its related hypoactivity in SZ [118]; also observed were higher amounts of methylation in the human reelin promoter in postmortem SZ cortices compared with controls [119, 120]. To ascribe the mechanism, the overexpression of Dnmt 1 mRNA presented in the same γ-aminobutyric acidergic neurons in which the levels of the reelin and glutamic acid decarboxylase-67 genes were reduced, suggesting a common negative regulatory mechanism that likely involves the coordinated hypermethylation of these promoters [116, 121]. The DNA promoter for the membrane-bound protein for COMT (MB-COMT) was found frequently to be hypomethylated in patients with SZ and bipolar disorder versus controls, thus contributing to MB-COMT overexpression and, consequently, increased dopamine degradation in the frontal lobe [122, 123]. Problems with attention, cognitive processes, and working memory have been linked to frontal lobe dopamine deficiency in patients with SZ [124].
Our MEDLINE searches have revealed little evidence of clinical studies for SAMe as a treatment for patients with SZ (PubMed, PubMed Central, Google Scholar; keywords: S-adenosylmethionine, schizophrenia). However, earlier studies for this purpose concerned the use of L-methionine, a precursor in the biosynthesis of SAMe. These clinical trials found that excessive oral methionine actually exacerbated the symptoms of SZ (reviewed in Grayson et al. [125]). Tremolizzo et al. [126] fed a protracted methionine diet to heterozygous reeler mice and found that their behavioral startle response worsened, whereas there was a marked decrease of reelin mRNA associated with an increase in the methylation of the reelin promoter. The startle response can be seen as a model of SZ vulnerability [126]. Of note, there is always a higher conversion rate of SAMe into SAH during such protracted L-methionine treatment [126]. Therefore, it seems that there may be a methionine-induced hypermethylation of CpG island-containing promoters in patients with SZ treated with methionine.
Moreover, there have been several randomized controlled trials of supplementation of folateor methylfolate in patients with SZ [127,128,129,130,131]. Levine et al. [127] reported that the symptoms of patients with SZ with hyperhomocysteinemia were improved after addition of folate and vitamin B12 [127]. However, Hill et al. [128] did not report benefits of folate supplementation in patients with SZ with prominent negative symptoms, but did report a significant interaction between treatment and MTHFR genotype. Consistent with this, Roffman et al. [129] reported that folate plus vitamin B12 supplementation can improve negative symptoms of SZ but that the treatment response is influenced by the MTHFR and FOLH1 genotype. Methylfolate is the fully reduced and bioactive form of folate. Several previous studies reported symptom improvement in patients with SZ with low red blood cell folate levels when treated with methyfolate [130, 132]. In a recent study, Roffman et al. [131] reported that methylfolate supplementation was associated with salutary physiological changes and negative symptomatic improvement in patients with SZ, regardless of genotype.
In conclusion, the phenomenon of a higher conversion rate of SAMe into SAH may be related to hypermethylation of reelin promoter and symptoms exacerbation in patients with SZ treated with late-stage dietary intake (methionine). However, folate or methyfolate supplementation in certain SZ genotype may ameliorate the negative symptoms, which may be reflect reverse changes of hypomethylation of the MB-COMT promoter that resulted from decreased SAMe levels or a reduced ratio of SAM/SAH in variants in folate-related enzymes. The normative expression of MB-COMT may help sustain dopamine levels in the frontal lobe associated with negative symptoms in patients with SZ.
AD
Perturbations of DNA methylation pathways can be, at least in part, included as an epigenetic basis of AD pathology. This was supported by several reports of an AD-associated reduction of SAMe levels in various neocortical areas, such as the hippocampus and putamen [133], and this reduction sometimes was found concurrent with an increase in brain SAH levels. As stated above, SAH is a strong DNA methyltransferase inhibitor contributing to DNA hypomethylation [134].
Although it is clear that 5mC is altered in AD, there remains no clear conclusion about global DNA methylation changes in the AD brain. Previous studies referred to both hypo- and hypermethylation, which may arise from the types of techniques used to investigate the DNA modifications [135, 136]. Emerging studies focused on changes in 5mC at specific promoter regions of genes related to AD pathology, such as the amyloid precursor protein (APP); presenilin1 (PS1), an enzyme involved in the processing of APP; β-site amyloid precursor protein-cleaving enzyme (BACE); microtubule-associated protein tau (MAPT); and apolipoprotein 4 in AD, as are described below.
It is well documented and accepted that the Aβ peptide production resulting from APP cleavage is of great relevance in the amyloidogenic etiology of AD [137]. A previous study showed that APP expression is subject to DNA methylation regulation (hypomethylation of the APP gene) [138] and the demethylation of the APP gene with age may have a significant influence on the development of Aβ deposition [139].
The group of Fuso has studied the impact of 1-carbon cycle and DNA methylation pathway on the genes encoding APP processing enzymes underlying AD pathology. SAMe was shown to inhibit PS1 promoter demethylation and thereby downregulated the PS1 mRNA expression in SK-N-SH human neuroblastoma cells, which resulted in a lowered secretion of Aβ [140]. In order to correlate changes in the SAM/SAH cycle and DNA methylation with AD pathology, Fuso et al. [6] studied the consequences of growing SK-N-SH and SK-N-BE neuroblastoma cells in a medium deprived of folate and vitamin B12. They found that an imbalance of SAMe and SAH resulting from these vitamin deficiencies was associated with changes in methylation pattern of PSEN1 and BACE with consequent increases in PS1 and BACE levels, contributing to neurotoxic Aβ deposition [6]. Hypomethylation of the promoter of PSEN1 induced an overexpression of this protein, which is consistent with another report of Scarpa et al. [140]. It is particularly noteworthy that they proposed the regulatory effect of Aβ production is gene-specific based on the model of SAM/SAH cycle and therapeutic intervention to reduce, but not totally inhibit, the slight overproduction of Aβ. Subsequently, the same team also used glioblastoma cells to further confirm that vitamin B deprivation could impair the DNA methylation potential of PSEN1 and BACE, providing the basis for Aβ overproduction [141]. Using in vivo studies, they demonstrated that a B vitamin deprivation diet led to hyperhomocysteinemia, an imbalance of SAM/SAH, PS1, and BACE upregulation, each of which increased Aβ deposition in TgCRND8 mice [142, 143].
Of note, some studies have indicated that Aβ itself could play a role in the pathological mechanisms associated with AD. Hodgson et al. [144] investigated the effects of soluble Aβ oligomers (oAβ) on intracellular thiol metabolite levels, DNA methylation, and the transcription status of redox and methylation-associated genes. They found a reduced ratio of SAM/SAH and decreased global DNA methylation with conversely increased Hcy levels. These authors concluded that the capacity of oAβ to modulate gene expression could generate redox and methylation-dependent epigenetic effects that contributed to the pathology of AD [144]. This prospect has been supported by a recent study [145]. Using primary hippocampal neuronal cells and HT-22 cells, as well as AD transgenic mice, Liu et al. [145] investigated the influence of oAβ on DNA methylation pathway in these cells, including DNMT activity, methylation potential of cells, APP and PS1 expression, cell viability, and the methylation of respective promoters. They found that oAβ caused neuronal toxicity with lowered DNMT activity, increased APP and PS1 expression, and decreased cell viability, each consequence of which could be prevented by folic acid via methylation-dependent pathways [145].
Other than those genes participating in Aβ processing (i.e., APP, PSEN1 and BACE), several target genes show significant interindividual epigenetic variability in late-onset AD [146]. For instance, the methylation pattern of APOE was found to be of a bimodal structure, ranging from a hypomethylated CpG-poor promoter to a fully methylated 3’-CpG-island [146]. By comparison, MTHFR, crucial for SAMe synthesis, was found to be hypermethylated [146]. These authors emphasized that an associated epigenetic drift may affect AD predisposition and course of disease [146].
The most proximal hallmark of AD is intraneuronal neurofibrillary tangles, which are composed of abnormally hyperphosphorylated tau protein. Tau phosphorylation expression is subject to epigenetic regulation mechanisms. For instance, Mastroeni et al. [147] reported that detriments in the DNA methylation pathways, including 2 markers of DNA methylation and 8 methylation maintenance factors, particularly those marked in PHF1/PS396 immunoreactivity, caused an increase in AD-specific neurofibrillary tangle-bearing neurons. In a vitamin B deficiency study, Nicolia et al. [148] reported that B vitamin deficiency promoted tau phosphorylation through upregulation of glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A) genes by inhibiting methylation [148].
Further work by Iwata et al. [138] found aberrant CpG methylation of APP, MAPT, and GSK3B in AD brain. The increased expression of APP and MAPT in both neuronal and non-neuronal cells, and the decreased expression of GSK3B in non-neuronal cells, may be directly linked to AD pathogenesis [138]. The cells that overproduce APP and MAPT are located among normal cells in AD brains, which could serve as seed clones for aggregated protein production, which supports the aggregation propagation hypothesis [138].
In general, there is an age-related increase of 5hmC levels in the brain, pointing towards a role for 5hmc in the development of AD [136, 149, 150]. However, some results differed between these studies, which could be explained by differences in tissue processing and immunostaining. Any dysregulation of 5hmC in AD brain could due to aging or environmental stressors [136]. Bradley-Whitman and Lovell [135] reported that global levels of 5mC and 5hmC were significantly increased, whereas 2 intermediates (5-formylcytosine and 5-carboxylcytosine), in the demethylation steps, were significantly decreased. They proposed that the absence of a mechanism to restore alterations in methylation/demethylation pattern after an environmental insult could result in an accumulation of 5mC and 5hmC during the course of AD [135]. In fact, 5mC and 5hmC both have been linked to neuronal cell death [151, 152]. In a recent study, Bernstein et al. [153] identified 325 genes containing differentially hydroxymethylated loci, which are involved in neuron projection development and neurogenesis. Some of these 5hmC-associated genes modulated tau-induced neurotoxicity in AD [153]. Ellison et al. [154] found significant alterations in 5-mC and 5-hmC in early AD stages across multiple regions of the brain. They suggested that the alterations in 5-mC and 5-hmC may play an early role in the progression of AD [154]. In conclusion, DNA methylation/demethylation patterns play a central role in the connection between amyloidogenic, fibrillogenic pathways in AD, both as a cause and as a consequence of the disease.
SAMe and Histone Methylation Patterns During Related Neuropsychiatric Diseases
Histone methylation refers to the fact that histones can be mono-, di-, or trimethylated at lysines and arginines by histone methyltransferases (HMTs) [155]. It was reported that transcriptionally silent genes correlate with di- and trimethylation of histone H3 at lysine-9 (H3K9) [156], whereas active genes are associated with di- and trimethylation of histone H3 at lysine4 (H3K4) [157]. Of note, several studies reported that these specific histone methylations have been implicated in the regulation of chromatin structure in the nervous system [158]. Two kinds of histone lysine methyltransferase, histone-lysine N-methyltransferase 2A (corresponding gene: MLL1) [159], and histone-lysine N-methyltransferase EHMT2 (corresponding gene: G9a) [160], respectively, catalyze the chemical reaction as below with different products, H3K4 and H3K9:
One recent study reported the kinetic constants for histone-lysine N-methyltransferase EHMT2 (G9a): the Km value of G9a for SAMe was 0.53 ± 0.043 μM; the Km value of G9a for H3K9 was 0.6 ± 0.096 μM [161].
Emerging evidence demonstrates that the dynamic modification of histones through methylation/demethylation can link metabolism with transcription, acting as molecular transducers of metabolic signals to chromatin. For example, Sadhu et al. [162] demonstrated that folate and methionine deficiency, which are both important for the maintenance of the SAMe pool, led to reduced methylation of lysine 4 of histone H3 (H3K4) in Saccharomyces cerevisiae and human cells in culture, suggesting histone methylation marks are susceptible to nutritional limitation [162]. In this report, results with K562 erytholeukemia cells led the authors to hypothesize that the abnormalities of histone methylation, as generated with SAMe, could be tested to impact on the development of NTDs secondary to folate deficiency [162].
This mechanism is also supported by other studies. Mentch et al. [163] showed that modulation of methionine metabolism, via altered nutrient availability, regulated SAMe and SAH levels to drive specific histone methylation events that affect disease-specific gene expression. Using the Drosophila melanogaster model, Liu et al. [164] reported that disruption of methionine metabolism can directly affect histone methylation levels, consistent with the viewpoint of Mench et al. [163], leading to lethality, as well as wing development and cell proliferation defects. In naïve human embryonic stem cells, Sperber et al. [165] recently showed that the knockdown of nicotinamide-N-methyl transferase led to higher SAMe levels and upregulation of H3K27me3. nicotinamide-N-methyl transferase is conferred to catalyze the chemical reaction:
This path therefore appears to make SAMe unavailable for the histone methylation that represses Wnt and activates HIF pathway in primed human embryonic stem cells, thus supporting the hypothesis that the metabolome regulates the epigenetic landscape of the earliest steps in human development [165]. In sum, the specificities of HMTs, and the availability of the cofactor SAMe, can be found in detailed reviews on methionine metabolism as an essential regulator of histone methylation and aforementioned possibility for specific gene regulation and related to biochemical events [10].
Ara et al. [166] found that the consequence of upregulation of TNF-α due to lipopolysaccharide (LPS) stimulation could be blocked by SAMe, MAT, or SAH pretreatment, through increasing the binding of H3K4 to the TNFA promoter. In this case, SAMe displayed no methyl donor role but served as an inhibitor to the trimethylation of H3K4 binding to the promoter region of TNFA. In the future, SAMe can be tested for its capacity to alter the histone methylation in studies when it is to be applied as a therapeutic agent to ameliorate neurodegenerative diseases, such as AD.
SAMe, PP2A Methylation, and Related Diseases
PP2A is a heterotrimeric protein composed of a catalytic subunit, scaffolding subunit, and regulatory subunit [167]. PP2A plays an important role in the biological function of tau protein, which stimulates microtubule assembly and stabilizes microtubules. In the AD brain, tau is abnormally hyperphosphorylated. PP2A has been shown to be methylated by methyltransferase, leucine carboxyl methyltransferases 1 (LCMT-1) [168] and demethylated by methylesterase, a potent phosphatase methylesterase1 [169]. LCMT-1 is conferred to catalyze the chemical reaction:
The kinetic constants for LCMT-1 were reported in 1 experiment: the Km value of LCMT-1 for SAMe was 1.3 μM; the Km value of LCMT-1 for PP2AD was 0.1 μM [170]. Alterations in PP2A-dependent (de)methylation patterns certainly play an important role in the pathogenesis of AD. For instance, Sontag et al. [171] reported that PP2A methyltransferase protein expression and PP2AC methylation levels were quantitatively decreased in AD-affected brain regions, and there was a regional loss of PP2A methyltransferase immunoreactivity that closely paralleled the severity of phosphorylative tau pathology. Nicholls et al. [172] reported that LCMT-1 overexpression protected animals from Aβ-induced impairments, whereas overexpression of phosphatase methylesterase 1 worsened Aβ neurotoxicity [172]. Therefore, PP2A methylation may control sensitivity and resistance to Aβ-induced cognitive and electrophysiological impairments, whereas PP2A demethylation functions in reverse.
The degree of PP2A methylation is subject to changes according to metabolism. For example, incubation of Neuro-2a cells with SAH resulted in reduced methylation of PP2A and detrimentally increased tau phosphorylation [173]. By contrast, incubation of Neuro-2a cells with SAMe enhanced PP2A methylation, led to the beneficial accumulation of dephosphorylated tau, and increased secretion of neuroprotective α-secretase-cleaved APP fragments [173]. This finding provides a new link between metabolism, PP2A methylation, and key CNS proteins involved in AD pathogenesis. In a follow-up study, the same team reported that, in neuroblastoma cells, folic acid deficiency contributed to decreased SAMe levels, and induced downregulation of LCMT-1 expression [174]. This resulted in accumulation of demethylated PP2A and ultimately tau mediated cell death [174]. In another study, Yoon et al. [175] reported that after treatment of rat primary neurons with methotrexate, a folate antagonist, the levels of PP2A methylation decreased and phosphorylated tau, APP, and β-secretase were increased. These findings supported that PP2A methylation correlated with metabolism, which played a corresponding role in pathological progression of AD [175]. In these cases, SAMe, acting as an adaptive responder to nutrition deficiency and also as a methyl group donor, contributed to the dynamic regulation of PP2A by methylation/demethylation pathways.
SAMe, Monoamine Neurotransmitters, and Depression
SAMe is required for the synthesis of monoamine neurotransmitters, such as norepinephrine (NE), dopamine (DA), and serotonin [5-hydroxytryptophan (5-HT)], which play important roles in maintaining normal mood [176]. The neurotransmitters DA and NE are synthesized from the amino acid tyrosine in a series of chemical reactions dependent on tyrosine hydroxylase [177]. Serotonin is synthesized from the amino acid tryptophan in a series of chemical reactions, of which the rate-limiting step is catalyzed by the enzyme tryptophan hydroxylase [178, 179], wherein SAMe functions as a methyl-donating cofactor in the rate-limiting step in these synthetic reactions to increase the levels of this neurotransmitter. In addition, the folate cycle is essential for the synthesis and regeneration of tetrahydrobiopterin [180]. The latter acts as an important cofactor for the enzymes that convert amino acids to monoamine neurotransmitters [181].
We will address SAMe as a treatment option for depression that operates by increasing neurotransmitter levels. Bottiglieri et al. [182] reported that elevated Hcy levels in MDD have been associated with low SAMe levels, and low CSF levels of 5-HT, DA, and NE metabolites. Enhancement of SAMe levels permit it to act as the cofactor of COMT, which prevents catalysis in the first step in a degradation pathway of the catecholamine neurotransmitters, and can therapeutically decrease COMT enzyme activity [183].
In another mode, SAMe has shown efficacy in accelerating the onset of the antidepressant effect of imipramine in depressed patients. Bellido et al. [184] found that SAMe prevented 5-HT1A receptor up-regulation induced by acute imipramine in the frontal cortex, which may contribute to the augmentation effect of SAMe [184]. Mechanistically, there is a structural similarity of serotonin indole ring to the adenosine motif of SAMe. Selective serotonin reuptake inhibitor (SSRIs) can bind to the COMT catalytic site, as a COMT inhibitor, and inhibit SAMe access, thus preventing the methylation of COMT substrates [185]. In these cases, SAMe, either acting as a methyl group donor, or a mediator of specific enzyme activity, has augmentative effects on MMD treatments.
SAMe, Phospholipids, and Depression
Phospholipids play essential roles in the composition of the cellular membrane and membrane dynamic functions, which are pertinent to maintaining healthy neurons and adequate neurotransmission, thus preventing depression. Specifically, a major SAMe-consuming pathway is methylation of phospholipids in mammals [186]. Normal membrane fluidity enhances receptor function and enhances the role of receptor coupling. To promote this function by methylating plasma phospholipids, SAMe may alter the fluidity of the neuronal membrane, affecting the function of proteins that traverse the membrane. This beneficial induction includes that of defined monoamine receptors, monoamine transporters, and other elements of the second messenger system [4].
SAMe Treatment
Numerous preclinical and clinical studies over the past 2 decades have shown that SAMe is effective in the treatment of psychiatric/neurological diseases, with the consequences of inhibiting progression and improving prognosis of these diseases. The mechanism underlying its therapeutic effects partly correlated to its influence on dynamic methylation/demethylation pathways. It has been difficult for clinicians to employ an easy measure the levels of SAMe and SAH in blood or tissue samples. However, research has recently generated antigens and antibodies that will pave the way for immunoassays for SAMe and related molecules [187]. This development of Immunoassays of SAMe and SAH should in the future provide a methylation index as a biomarker for disease and health status and tissue specific and brain region prevalence abundance of SAMe levels. Experimental and clinical studies about the applications of SAMe to specific neuropsychiatric diseases will be discussed below.
SAMe Treatment in Psychiatric Disorders
The FS test monitors inappropriate responses that have been linked with psychiatric disorders, such as major depression and anxiety [188]. Saunderson et al. [71] found that administration of SAMe before the FS challenge resulted in enhanced CpG methylation at IEG loci. The suppression of IEG induction, specifically in the dentate gyrus, was associated with significant changes to behavioral immobility responses 24 h later, suggesting that SAMe may enhance behavioral responses after stress by suppressing the induction of IEG. This finding is consistent with a previous study conducted by Weaver et al. [189]. Those investigators centrally infused the adult offspring with the essential amino acid L-methionine, a precursor to SAMe, to examine whether the epigenetic programming of stress responses established early in life is reversible. They found that the inherent stability of the epigenomic marks is potentially reversible through methyl supplementation [189]. According to these results, we propose a potential role of earlier administration of SAMe to improve depression and also to alter epigenetic markers, which supports its potential capability for inhibiting and reversing neuropathogenesis secondary to stress early in life.
In mood disorders, recent clinical studies supported the use of SAMe as a supplement to standard medications, which effectively ameliorates depressive symptom and cognitive dysfunction in MDD [190]. This clinical trial convincingly demonstrated that the abundance and bioavailability of SAMe increased in the serum of treated depressed patients as associated with clinical improvement in depression [190]. Another systematic review assessed the effects of SAMe in comparison with placebo or antidepressants for the treatment of depression in adults with MDD, with less support for firm conclusions [191]. Both high intracellular levels of SAMe (due to SAMe administration) and low intracellular levels of serotonin (due to SSRI action) prevent binding of serotonin to COMT and its consequent inhibition. Therefore, SAMe has been used as a supplement to treat patients with MDD [8], even in SSRI-resistant patients [192]. Furthermore, despite its good safety profile, SAMe has been contraindicated in patients with bipolar disorder owing to its induction of mania or hypomania [191].
Strous et al. [193] investigated the efficacy of SAMe in managing SZ symptoms in patients with the low-activity COMT polymorphism and found an improvement of aggressive behavior and quality of life following SAMe augmentation in SZ [193]. SAMe can increase COMT enzyme activity, which may help to ameliorate aggressive symptoms in specific patients with SZ.
SAMe Treatment in Neurological Diseases
SAMe was proposed to tilt the epigenetic marking of DNA towards increased methylation and reduced demethylation as a treatment in neurological diseases. In demyelinating diseases, after the dietary restoration of SAMe, substantial clinical improvement has long been associated with apparent remyelination [194].
The deficiency of SAMe may underlie the gradual hypomethylation of DNA that accompanies aging, and this hypomethylation can be alleviated by supplementation with SAMe [195]. The lower availability of SAMe may be related to the altered expression of genes involved in APP metabolism, finally producing the accumulation of Aβ peptide, contributing to the pathological processing of AD [6, 140]. DNA demethylation, when reduced by aging or nutritional deficiencies, may at least be partially avoided or restored by SAMe administration [6, 140]. In addition, BACE expression is modulated by methylation, and SAMe can restore its normal expression pattern [6]. Therefore, there have been several attempts to use SAMe nutritional supplementation in clinical trials in AD [7]. As an example of its mechanism of action, SAMe supplementation was shown to inhibit DNA demethylase, DNMT1 activity, and demethylation of PS1, a prime gene product involved in amyloidogenesis [143], as discovered in transgenic AD mice.
A further study examined whether SAMe could delay neuropathology in 3×Tg-AD mice, which harbor mutant genes for human AβPP, PS-1, and tau, here showing that SAMe supplementation in the diet did, in fact, reduce extracellular Aβ deposits [196]. SAMe treatment should therefore start early to effectively delay onset of AD symptoms. Fuso et al. [197] reported that SAMe reduced amyloid production and increased spatial memory in TgCRND8 mice. Here, SAMe inhibited the effects of B vitamin deficiency to induce PSEN1 and BACE1 expression and tau phosphorylation in TgCRND8 and wild-type mice. Certainly, SAMe treatment also reduced plaque spreading in pathways independent of B vitamin deficiency [197]. Persichilli et al. [198] reported a significant decrease of thiol levels when the B vitamin-deficient diet was supplemented with SAMe + superoxide dismutase and superoxide dismutase alone [198]. In a recent study on the APP transgenic mouse model of AD-like amyloid pathology, Do Carmo et al. [199] found that the early BACE-1 and global DNA demethylation were rescued by chronic administration of SAMe, which contributed to the reduction of amyloid pathology and the improvement in cognition function. In conclusion, these studies in AD support the rationale that it is possible to use SAMe to treat brain disorders with a plausible epigenetic mechanism. The studies are consistent with the hypothesis that SAMe administered orally is bioavailable in an active form or as an active metabolite in the brain.
SAMe Treatment in Chronic Inflammation Associated With Depression, SZ, and AD
It has been reported that inflammation plays a critical role in chronic conditions in the brain, such as depression [200], SZ [201], and AD[202]. Previous studies showed that the anti-inflammatory effects of SAMe in LPS-stimulated monocytes/macrophages were mediated by reducing the expression of the [203], and increasing the expression of the anti-inflammatory cytokine interleukin 10 [204]. Later, Ara et al. [166] reported that the mechanism of the pharmacologic inhibitory effects of SAMe on proinflammatory mediators, are, in part, mediated by altering the methylation capacity and blocking the binding of trimethylated H3K4 to the TNF-α promoter through increasing MTA and SAH levels [166]. In addition, Gobejishvili et al. [205] reported that SAMe pretreatment led to decreased PDE4B expression by increasing the binding of transcriptionally repressive trimethylated H3K9 to its promoter region, thus inhibiting cyclic adenosine monophosphate-mediated TNF-α expression. Pfalzer et al. [206] evaluated the effects of SAMe on global and site-specific DNA methylation and gene expression in the context of LPS-stimulated inflammation in human macrophages, and found a decrease in DNA methylation status of inflammatory genes after SAMe treatment. So it is proposed that the anti-inflammation feature of SAMe through methylation regulation supports its therapeutic effects in treatment of chronic inflammatory conditions.
Prospects
SAMe, as a universal methyl donor, clearly shows promise in the treatment of neuropsychiatric diseases throughout the human life span. The malfunction of methylation in the developing brain due to inherited mutations, compounded by epigenetic stresses, underlies the pathological process of a variety of neurodevelopmental and/or psychiatric disorders, including diseases that manifest at different stages of life, such as MDD and SZ. The critical period for use of SAMe or related agents has been proposed to be as early as possible, thus enhancing therapeutic effects and/or potentially preventing the illness from manifesting in the first place. Disease reversal and the harmless nature of SAMe are projected to counteract the latent risk of pathological diseases in brain.
Relevant to its efficacy in individuals with existing psychiatric disorders, there has been considerable evidence to support the use of SAMe in the treatment of MDD. SAMe is recommended as a second-line treatment option following inadequate treatment response to conventional antidepressants [207]. Owing to its favorable safety profile, SAMe may be specifically suitable for the treatment of MDD in children, adolescents, and pregnant women. However, further studies are required because there is little scientific support for its use in these potentially vulnerable populations. However, as a cautionary note, in patients with SZ, SAMe may at times exacerbate the symptoms of this disease. One study showed that the treatment of heterozygous reeler mice with dietary L-methionine led to a decreased PPI [126] as a model of SZ. However, the treatment of SAMe resulted in an improved neurotransmission and thus attenuated aggressive behavior and ameliorated the quality of life of patients with SZ with a low-activity COMT polymorphism [193]. Therefore, further studies are required to assess the use of SAMe in patients with SZ. Owing to concerns about neurotransmitter hyperactivity, SAMe is not recommended in patients with bipolar disorder because of its history of leading to manic and hypomanic states [191].
Emerging studies have focused on the function of SAMe as a potential disease-modifying agent to treat the pathological process of AD. AD is a progressive and irreversible disease without effective treatments. A number of risk factors exacerbate AD, including folate and vitamin B12 deficiency. SAMe showed a positive effect by preventing the progression of the hallmarks of AD resulting from this dietary deficiency. SAMe reduced amyloid production and prevented phospho-tau accumulation in animal models. At the same time, the clinical manifestations of AD were also ameliorated by SAMe. Thus, recent clinical efficacy trials have shown that SAMe can be used as a part of a nutritional formulation [208], to maintain or improve cognitive performance and mood/behavior in patients with AD [209].
Furthermore, the dosage of SAMe recommended in different situations needs to be carefully studied with additional clinical trials. The usual starting dose of SAMe is 400 mg/day and the maximum reported in the literature is 3200 mg/day (given in 2 doses) [210]. It is clear that this dosing should be increased gradually every 5 to 7-day period [9].
Although SAMe has a favorable safety profile, it is still worth paying attention to the risk that SAMe might cause increased Hcy levels, which is associated with higher risk of cardiovascular disease [211]. Despite evidence that SAMe augmentation of antidepressants did not result in elevation of total Hcy [190], caution with vulnerable populations seems appropriate until additional data emerge. For example, patients lacking the efficient capability of eliminating excessive Hcy might not be advised to use SAMe as a long-term treatment, and if they choose to use it, they should do so under physician supervision.
References
Cantoni GL. Biological methylation: selected aspects. Annu Rev Biochem. 1975;44:435-451.
Mato JM, Alvarez L, Ortiz P, et al. S-Adenosylmethionine synthesis: molecular mechanisms and clinical implications. Pharmacol Ther. 1997;73(3):265-280.
Giulidori P, Galli-kienle M, Catto E, et al. Transmethylation, transsulfuration, and aminopropylation reactions of S-adenosyl-l-methionine in vivo. J Biol Chem. 1984;259(7):4205-4211.
Lu SC. S-Adenosylmethionine. Int J Biochem Cell Biol. 2000;32(4):391-395.
Teh AL, Pan H, Chen L, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014;24(7):1064-1074.
Fuso A, Seminara L, Cavallaro RA, et al. S-Adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and bace and beta-amyloid production. Mol Cell Neurosci. 2005;28(1):195-204.
Panza F, Frisardi V, Capurso C, et al. Polyunsaturated fatty acid and S-adenosylmethionine supplementation in predementia syndromes and Alzheimer's disease: a review. Scientificworldjournal. 2009;9:373-389.
De Berardis D, Orsolini L, Serroni N, et al. A comprehensive review on the efficacy of S-adenosyl-L-methionine in major depressive disorder. Cns Neurol Disord Drug Targets. 2016;15(1):35-44.
Bottiglieri T. Folate, vitamin B(1)(2), and S-adenosylmethionine. Psychiatr Clin North Am. 2013;36(1):1-13.
Mentch SJ, Locasale JW. One-carbon metabolism and epigenetics: understanding the specificity. Ann N Y Acad Sci. 2016;1363:91-98.
Mato JM, Corrales FJ, Lu SC, et al. S-adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16(1):15-26.
Jarrett JT, Huang S, Matthews RG. Methionine synthase exists in two distinct conformations that differ in reactivity toward methyltetrahydrofolate, adenosylmethionine, and flavodoxin. Biochemistry. 1998;37(16):5372-5382.
Gueant JL, Caillerez-Fofou M, Battaglia-Hsu S, et al. Molecular and cellular effects of vitamin B12 in brain, myocardium and liver through its role as co-factor of methionine synthase. Biochimie. 2013;95(5):1033-1040.
Gherasim C, Lofgren M, Banerjee R. Navigating the B(12) road: assimilation, delivery, and disorders of cobalamin. J Biol Chem. 2013;288(19):13186-13193.
Reynolds E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006;5(11):949-960.
Sharp L, Little J. Polymorphisms in genes involved in folate metabolism and colorectal neoplasia: a huge review. Am J Epidemiol. 2004;159(5):423-443.
Nazki FH, Sameer AS, Ganaie BA. Folate: metabolism, genes, polymorphisms and the associated diseases. Gene. 2014;533(1):11-20.
Kim JM, Stewart R, Kim SW, et al. Predictive value of folate, vitamin B12 and homocysteine levels in late-life depression. Br J Psychiatry. 2008;192(4):268-274.
Fava M, Borus JS, Alpert JE, et al. Folate, vitamin B12, and homocysteine in major depressive disorder. Am J Psychiatry. 1997;154(3):426-428.
Papakostas GI, Petersen T, Mischoulon D, et al. Serum folate, vitamin B12, and homocysteine in major depressive disorder, pART 1: predictors of clinical response in fluoxetine-resistant depression. J Clin Psychiatry. 2004;65(8):1090-1095.
Aisen PS, Schneider LS, Sano M, et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: a randomized controlled trial. JAMA. 2008;300(15):1774-1783.
Kennedy DO. B vitamins and the brain: mechanisms, dose and efficacy—a review. Nutrients. 2016;8(2):68.
Ankar A, Bhimji SS. Vitamin, B12 (cobalamin), deficiency. Treasure Island (FL): StatPearls Publishing; 2017.
Kumar N. Neurologic aspects of cobalamin (B12) deficiency. Handb Clin Neurol. 2014;120:915-926.
Yi P, Melnyk S, Pogribna M, et al. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J Biol Chem. 2000;275(38):29318-2923.
Fernandez-Roig S, Lai SC, Murphy MM, et al. Vitamin B12 deficiency in the brain leads to dna hypomethylation in the TCBLR/CD320 knockout mouse. Nutr Metab (Lond). 2012;9:41.
Tanaka H. [Old or new medicine? Vitamin B12 and peripheral nerve neuropathy]. Brain Nerve. 2013;65(9):1077-1082.
Zhang Y, Hodgson NW, Trivedi MS, et al. Decreased brain levels of vitamin B12 in aging, autism and schizophrenia. PLOS ONE. 2016;11(1):E0146797.
Loenen WA. S-Adenosylmethionine: jack of all trades and master of everything? Biochem Soc Trans. 2006;34(PT 2):330-333.
Martinez-Lopez N, Varela-Rey M, Ariz U, et al. S-adenosylmethionine and proliferation: new pathways, new targets. Biochem Soc Trans. 2008;36(PT 5):848-852.
Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(Suppl.):245-254.
Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293(5532):1089-1093.
Xie W, Barr CL, Kim A, ET AL. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148(4):816-831.
Fernando HJ, Mammarella MC, Grandoni G, et al. Forecasting PM10 in metropolitan areas: efficacy of neural networks. Environ Pollut. 2012;163:62-67.
Lister R, Mukamel EA, Nery JR, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905.
Smith ZD, Chan MM, Mikkelsen TS, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484(7394):339-344.
Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23(3):555-567.
Guo JU, Su Y, Shin JH, et al. Distribution, recognition and regulation of non-CPG methylation in the adult mammalian brain. Nat Neurosci. 2014;17(2):215-222.
Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930-935.
Yu M, Hon GC, Szulwach KE, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149(6):1368-1380.
Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29(1):68-72.
Khare T, Pai S, Koncevicius K, et al. 5-HMC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19(10):1037-1043.
Guo JU, Su Y, Zhong C, et al. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423-434.
Okano M, Bell DW, Haber DA, et al. DNA methyltransferases DNMT3A and DNMT3B are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247-257.
Rhee I, Jair KW, Yen RW, et al. CPG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404(6781):1003-1007.
Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395-2402.
Suetake I, Miyazaki J, Murakami C, et al. Distinct enzymatic properties of recombinant mouse DNA methyltransferases DNMT3A and DNMT3B. J Biochem. 2003;133(6):737-744.
Gros C, CHauvigne L, Poulet A, et al. Development of a universal radioactive DNA methyltransferase inhibition test for high-throughput screening and mechanistic studies. Nucleic Acids Res. 2013;41(19):E185.
Feng J, Chang H, Li E, et al. Dynamic expression of de novo DNA methyltransferases DNMT3A and DNMT3B in the central nervous system. J Neurosci Res. 2005;79(6):734-746.
Watanabe D, Uchiyama K, Hanaoka K. Transition of mouse de novo methyltransferases expression from DNMT3B to DNMT3A during neural progenitor cell development. Neuroscience. 2006;142(3):727-737.
Ito S, D'Alessio AC, Taranova OV, et al. Role of TET proteins in 5MC to 5HMC conversion, ES-celL self-renewal and inner cell mass specification. Nature. 2010;466(7310):1129-1133.
Zhang RR, Cui QY, Murai K, et al. TET1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13(2):237-245.
Li X, Wei W, Zhao QY, et al. Neocortical TET3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A. 2014;111(19):7120-7125.
Yu H, Su Y, Shin J, et al. TET3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat Neurosci. 2015;18(6):836-843.
Nan X, Meehan RR, Bird A. Dissection of the methyl-CPG binding domain from the chromosomal protein MECP2. Nucleic Acids Res. 1993;21(21):4886-4892.
Skene PJ, Illingworth RS, Webb S, et al. Neuronal MECP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37(4):457-468.
Shahbazian MD, Antalffy B, Armstrong DL, et al. Insight into Rett syndrome: MECP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11(2):115-124.
Nan X, Campoy FJ, Bird A. MECP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471-481.
Lyst MJ, Ekiert R, Ebert DH, et al. Rett syndrome mutations abolish the interaction of MECP2 with the NCOR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898-902.
Kinde B, Gabel HW, Gilbert CS, et al. Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MECP2. Proc Natl Acad SCI U S A. 2015;112(22):6800-6806.
Gabel HW, Kinde B, Stroud H, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522(7554):89-93.
Chen L, Chen K, Lavery LA, et al. MECP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett Syndrome. Proc Natl Acad Sci U S A. 2015;112(17):5509-5514.
Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23-38.
Fan G, Martinowich K, Chin MH, et al. DNA methylation controls the timing of astrogliogenesis through regulation of Jak-STAT signaling. Development. 2005;132(15):3345-3356.
Oda M, Oxley D, Dean W, et al. Regulation of lineage specific DNA hypomethylation in mouse trophectoderm. PLOS ONE. 2013;8(6):E68846.
Chen CC, Wang KY, Shen CK. DNA 5-Methylcytosine demethylation activities of the mammalian DNA methyltransferases. J Biol Chem. 2013;288(13):9084-9091.
Van Der Wijst MG, Venkiteswaran M, Chen H, et al. Local chromatin microenvironment determines DNMT activity: from DNA methyltransferase to DNA demethylase or DNA dehydroxymethylase. Epigenetics. 2015;10(8):671-676.
Varela-Rey M, Iruarrizaga-Lejarreta M, Lozano JJ, et al. S-adenosylmethionine levels regulate the Schwann cell DNA methylome. Neuron. 2014;81(5):1024-1039.
Guo JU, Ma DK, Mo H, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14(10):1345-1351.
Mcgowan PO, Sasaki A, D'Alessio AC, et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12(3):342-348.
Saunderson EA, Spiers H, Mifsud KR, et al. Stress-induced gene expression and behavior are controlled by DNA methylation and methyl donor availability in the dentate gyrus. Proc Natl Acad Sci U S A. 2016;113(17):4830-4835.
Sutter BM, Wu X, Laxman S, et al. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell. 2013;154(2):403-415.
Nguyen S, Meletis K, Fu D, et al. Ablation of de novo dna methyltransferase DNMT3A in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236(6):1663-1676.
Golshani P, Hutnick L, Schweizer F, et al. Conditional DNMT1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat Syst. 2005;3(3):227-233.
Santiago M, Antunes C, Guedes M, et al. TET enzymes and DNA hydroxymethylation in neural development and function - how critical are they? Genomics. 2014;104(5):334-340.
Shojaei Saadi HA, Gagne D, Fournier E, et al. Responses of bovine early embryos to S-adenosyl methionine supplementation in culture. Epigenomics. 2016;8(8):1039-1060.
Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67-84.
Molloy AM, Kirke PN, Troendle JF, et al. Maternal vitamin B12 status and risk of neural tube defects in a population with high neural tube defect prevalence and no folic acid fortification. Pediatrics. 2009;123(3):917-923.
Ratan SK, Rattan KN, Pandey RM, et al. Evaluation of the levels of folate, vitamin B12, homocysteine and fluoride in the parents and the affected neonates with neural tube defect and their matched controls. Pediatr Surg Int. 2008;24(7):803-808.
Czeizel AE. Periconceptional folic acid and multivitamin supplementation for the prevention of neural tube defects and other congenital abnormalities. Birth Defects Res A Clin Mol Teratol. 2009;85(4):260-268.
Czeizel AE, Dudas I, Paput L, et al. Prevention of neural-tube defects with periconceptional folic acid, methylfolate, or multivitamins? Ann Nutr Metab. 2011;58(4):263-71.
Wilson RD, Audibert F, Brock JA, et al. Pre-conception folic acid and multivitamin supplementation for the primary and secondary prevention of neural tube defects and other folic acid-sensitive congenital anomalies. J Obstet Gynaecol Can. 2015;37(6):534-552.
Coelho CN, Klein NW. Methionine and neural tube closure in cultured rat embryos: morphological and biochemical analyses. Teratology. 1990;42(4):437-451.
Dunlevy LP, Burren KA, Chitty LS, et al. Excess methionine suppresses the methylation cycle and inhibits neural tube closure in mouse embryos. FEBS Lett. 2006;580(11):2803-2807.
Afman LA, Blom HJ, Drittij MJ, et al. Inhibition of transmethylation disturbs neurulation in chick embryos. Brain Res Dev Brain Res. 2005;158(1-2):59-65.
Olthof MR, Van Vliet T, Boelsma E, et al. Low dose betaine supplementation leads to immediate and long term lowering of plasma homocysteine in healthy men and women. J Nutr. 2003;133(12):4135-4138.
Wang X, Guan Z, Chen Y, et al. Genomic DNA hypomethylation is associated with neural tube defects induced by methotrexate inhibition of folate metabolism. PLOS ONE. 2015;10(3):E0121869.
Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, et al. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLOS ONE. 2009;4(11):E7845.
Amir RE, Van Den Veyver IB, Wan M, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185-188.
Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68(6):944-950.
Guy J, Hendrich B, Holmes M, et al. A mouse MECP2-null mutation causes neurological symptoms that mimic rett syndrome. Nat Genet. 2001;27(3):322-326.
Kaludov NK, Wolffe AP. MECP2 driven transcriptional repression in vitro: selectivity for methylated DNA, action at a distance and contacts with the basal transcription machinery. Nucleic Acids Res. 2000;28(9):1921-1928.
Glaze DG, Percy AK, Motil KJ, et al. A study of the treatment of Rett syndrome with folate and betaine. J Child Neurol. 2009;24(5):551-556.
Hagebeuk EE, Koelman JH, Duran M, et al. Clinical and electroencephalographic effects of folinic acid treatment in Rett syndrome patients. J Child Neurol. 2011;26(6):718-723.
Hagebeuk EE, Duran M, Koelman JH, et al. folinic acid supplementation in Rett syndrome patients does not influence the course of the disease: a randomized study. J Child Neurol. 2012;27(3):304-309.
Hagebeuk EE, Duran M, Abeling NG, et al. S-Adenosylmethionine and S-adenosylhomocysteine in plasma and cerebrospinal fluid in Rett syndrome and the effect of folinic acid supplementation. J Inherit Metab Dis. 2013;36(6):967-972.
Yu XF, Li M, Zheng Y. [Association between maternal folate supplementation during pregnancy and the risk of autism spectrum disorder in the offspring: a meta analysis]. Zhongguo Dang Dai Er Ke Za Zhi. 2017;19(3):286-291.
Pogribna M, Melnyk S, Pogribny I, et al. homocysteine metabolism in children with DOWN syndrome: in vitro modulation. Am J Hum Genet. 2001;69(1):88-95.
Obeid R, Hubner U, Bodis M, et al. Plasma amyloid beta 1-42 and DNA methylation pattern predict accelerated aging in young subjects with Down syndrome. Neuromolecular Med. 2016;18(4):593-601.
Obeid R, Hartmuth K, Herrmann W, et al. Blood Biomarkers of methylation in down syndrome and metabolic simulations using a mathematical model. Mol Nutr Food Res. 2012;56(10):1582-1589.
Fountoulakis M, Gulesserian T, Lubec G. Overexpression of c1-tetrahydrofolate synthase in fetal down syndrome brain. J Neural TRansm Suppl. 2003(67):85-93.
Song C, He J, Chen J, et al. Effect of the onecarbon unit cycle on overall dna methylation in children with down's syndrome. Mol Med Rep. 2015;12(6):8209-8214.
Infantino V, Castegna A, Iacobazzi F, et al. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in down's syndrome. Mol Genet Metab. 2011;102(3):378-382.
Beetstra S, Thomas P, Salisbury C, et al. Folic acid deficiency increases chromosomal instability, chromosome 21 aneuploidy and sensitivity to radiation-induced micronuclei. MUTAT RES. 2005;578(1-2):317-326.
Da Silva LR, Vergani N, Galdieri Lde C, et al. Relationship between polymorphisms in genes involved in homocysteine metabolism and maternal risk for down syndrome in brazil. Am J Med Genet A. 2005;135(3):263-267.
Biselli JM, Goloni-Bertollo EM, Zampieri BL, et al. Genetic polymorphisms involved in folate metabolism and elevated plasma concentrations of homocysteine: maternal risk factors for down syndrome in brazil. Genet Mol Res. 2008;7(1):33-42.
Smythies JR. Biochemistry of schizophrenia. Postgrad Med J. 1963;39:26-33.
Kelsoe JR, Tolbert LC, Crews EL, et al. Kinetic evidence for decreased methionine adenosyltransferase activity in erythrocytes from schizophrenics. J Neurosci Res. 1982;8(1):99-103.
Muntjewerff JW, Van Der Put N, Eskes T, et al. Homocysteine metabolism and b-vitamins in schizophrenic patients: low plasma folate as a possible independent risk factor for schizophrenia. Psychiatry Res. 2003;121(1):1-9.
Garcia-Miss Mdel R, Perez-Mutul J, Lopez-Canul B, et al. Folate, homocysteine, interleukin-6, and tumor necrosis factor alfa levels, but not the methylenetetrahydrofolate reductase C677T Polymorphism, are risk factors for schizophrenia. J Psychiatr Res. 2010;44(7):441-446.
Brown AS, Bottiglieri T, Schaefer CA, et al. Elevated prenatal homocysteine levels as a risk factor for schizophrenia. Arch Gen Psychiatry. 2007;64(1):31-39.
Kumar KS, Govindaiah V, Naushad SE, et al. Plasma homocysteine levels correlated to interactions between folate status and methylene tetrahydrofolate reductase gene mutation in women with unexplained recurrent pregnancy loss. J Obstet Gynaecol. 2003;23(1):55-58.
Picker JD, Coyle JT. Do maternal folate and homocysteine levels play a role in neurodevelopmental processes that increase risk for schizophrenia? Harv Rev Psychiatry. 2005;13(4):197-205.
Kirkbride JB, Susser E, Kundakovic M, et al. Prenatal nutrition, epigenetics and schizophrenia risk: can we test causal effects? Epigenomics. 2012;4(3):303-315.
Szyf M. Epigenetics, a key for unlocking complex cns disorders? therapeutic implications. Eur Neuropsychopharmacol. 2015;25(5):682-702.
Veldic M, Guidotti A, Maloku E, et al. In psychosis, cortical interneurons overexpress dna-methyltransferase 1. Proc Natl Acad Sci U S A. 2005;102(6):2152-2157.
Zhubi A, Veldic M, Puri NV, et al. An upregulation of dna-methyltransferase 1 and 3A expressed in telencephalic gabaergic neurons of schizophrenia patients is also detected in peripheral blood Lymphocytes. Schizophr Res. 2009;111(1-3):115-122.
Chen Y, Sharma RP, Costa RH, et al. On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 2002;30(13):2930-2939.
Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (reln) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B(1):60-66.
Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A. 2005;102(26):9341-9346.
Veldic M, Caruncho HJ, Liu WS, et al. DNA-Methyltransferase 1 MRNA is selectively overexpressed in telencephalic gabaergic interneurons of schizophrenia brains. Proc Natl Acad Sci U S A. 2004;101(1):348-353.
Abdolmaleky HM, Cheng KH, Faraone SV, et al. Hypomethylation of mb-comt promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15(21):3132-3145.
Nohesara S, Ghadirivasfi M, Mostafavi S, et al. DNA Hypomethylation of MB-COMT Promoter in the dna derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res. 2011;45(11):1432-1438.
Rosa A, Peralta V, Cuesta MJ, et al. New evidence of association between comt gene and prefrontal neurocognitive function in healthy individuals from sibling pairs discordant for psychosis. Am J Psychiatry. 2004;161(6):1110-1112.
Grayson DR, Chen Y, Dong E, et al. From Trans-Methylation To Cytosine Methylation: Evolution Of The Methylation Hypothesis Of Schizophrenia. Epigenetics. 2009;4(3):144-149.
Tremolizzo L, Carboni G, Ruzicka WB, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci U S A. 2002;99(26):17095-17100.
Levine J, Stahl Z, Sela BA, et al. Homocysteine-reducing strategies improve symptoms in chronic schizophrenic patients with hyperhomocysteinemia. Biol Psychiatry. 2006;60(3):265-269.
Hill M, Shannahan K, Jasinski S, et al. Folate Supplementation in schizophrenia: a possible role for mthfr genotype. Schizophr Res. 2011;127(1-3):41-45.
Roffman JL, Lamberti JS, Achtyes E, et al. Randomized multicenter investigation of folate plus vitamin B12 Supplementation in Schizophrenia. JAMA Psychiatry. 2013;70(5):481-489.
Procter A. Enhancement of recovery from psychiatric illness by methylfolate. BR J Psychiatry. 1991;159:271-272.
Roffman JL, Petruzzi LJ, Tanner AS, et al. Biochemical, physiological and clinical effects of l-methylfolate in schizophrenia: a randomized controlled trial. Mol Psychiatry 2017 MAR 14.
Godfrey PS, Toone BK, Carney MW, et al. Enhancement of recovery from psychiatric illness by methylfolate. Lancet. 1990;336(8712):392-395.
Morrison LD, Smith DD, Kish SJ. Brain S-Adenosylmethionine levels are severely decreased in alzheimer's disease. J Neurochem. 1996;67(3):1328-1331.
Kennedy BP, Bottiglieri T, Arning E, et al. Elevated s-adenosylhomocysteine in alzheimer brain: influence on methyltransferases and cognitive function. J Neural Transm (VIENNA). 2004;111(4):547-567.
Bradley-Whitman MA, Lovell MA. Epigenetic changes in the progression of alzheimer's disease. Mech Ageing Dev. 2013;134(10):486-495.
Coppieters N, Dieriks BV, Lill C, et al. Global changes in dna methylation and hydroxymethylation in alzheimer's disease human brain. Neurobiol Aging. 2014;35(6):1334-1344.
Chaney MO, Baudry J, Esh C, et al. A beta, aging, and alzheimer's disease: a tale, models, and hypotheses. Neurol Res. 2003;25(6):581-589.
Iwata A, Nagata K, Hatsuta H, et al. Altered CPG Methylation in Sporadic alzheimer's disease is associated with APP and MAPT Dysregulation. Hum Mol Genet. 2014;23(3):648-656.
Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region –224 approximately –101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70(2):288-292.
Scarpa S, Fuso A, D'Anselmi F, et al. Presenilin 1 gene silencing by s-adenosylmethionine: a treatment for alzheimer disease? Febs Lett. 2003;541(1-3):145-148.
Fuso A, Cavallaro RA, Zampelli A, et al. Gamma-Secretase is differentially modulated by alterations of homocysteine cycle in neuroblastoma and glioblastoma cells. J Alzheimers Dis. 2007;11(3):275-290.
Fuso A, Nicolia V, Cavallaro RA, et al. B-Vitamin deprivation induces hyperhomocysteinemia and brain s-adenosylhomocysteine, depletes brain s-adenosylmethionine, and enhances PS1 and bace expression and amyloid-beta deposition in mice. Mol Cell Neurosci. 2008;37(4):731-746.
Fuso A, Nicolia V, Pasqualato A, et al. Changes in presenilin 1 Gene methylation pattern in diet-induced b vitamin deficiency. Neurobiol Aging. 2011;32(2):187-199.
Hodgson N, Trivedi M, Muratore C, et al. Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J Alzheimers Dis. 2013;36(1):197-209.
Liu H, Li W, Zhao S, et al. Folic acid attenuates the effects of amyloid beta oligomers on dna methylation in neuronal cells. Eur J Nutr. 2016;55(5):1849-1862.
Wang SC, Oelze B, Schumacher A. Age-specific epigenetic drift in late-onset alzheimer's disease. Plos One. 2008;3(7):E2698.
Mastroeni D, Grover A, Delvaux E, et al. Epigenetic changes in alzheimer's disease: decrements in dna methylation. Neurobiol Aging. 2010;31(12):2025-2037.
Nicolia V, Fuso A, Cavallaro RA, et al. B Vitamin deficiency promotes tau phosphorylation through regulation of GSK3BETA and PP2A. J Alzheimers Dis. 2010;19(3):895-907.
Chen H, Dzitoyeva S, Manev H. Effect of aging on 5-hydroxymethylcytosine in the mouse hippocampus. Restor Neurol Neurosci. 2012;30(3):237-245.
Phipps AJ, Vickers JC, Taberlay PC, et al. Neurofilament-Labeled Pyramidal neurons and astrocytes are deficient in dna methylation marks in alzheimer's disease. Neurobiol Aging. 2016;45:30-42.
Padurariu M, Ciobica A, Mavroudis I, et al. Hippocampal Neuronal Loss in the CA1 and CA3 areas of alzheimer's disease patients. Psychiatr Danub. 2012;24(2):152-158.
Jin SG, Wu X, Li AX, et al. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39(12):5015-5024.
Bernstein AI, Lin Y, Street RC, et al. 5-Hydroxymethylation-Associated epigenetic modifiers of alzheimer's disease modulate tau-induced neurotoxicity. Hum Mol Genet. 2016;25(12):2437-2450.
Ellison EM, Abner EL, Lovell MA. Multiregional Analysis of global 5-methylcytosine and 5-hydroxymethylcytosine throughout the progression of alzheimer's disease. J Neurochem. 2017;140(3):383-394.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074-1080.
Fiszbein A, Kornblihtt AR. Histone methylation, alternative splicing and neuronal differentiation. Neurogenesis (AUSTIN). 2016;3(1):E1204844.
Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6(11):838-849.
Morse SJ, Butler AA, Davis RL, et al. Environmental enrichment reverses histone methylation changes in the aged hippocampus and restores age-related memory deficits. Biology (BASEL). 2015;4(2):298-313.
Patel A, Dharmarajan V, Vought VE, et al. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (mll1) core complex. J Biol Chem. 2009;284(36):24242-24256.
Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 Methyltransferases. Plos One. 2010;5(1):E8570.
Horiuchi KY, Eason MM, Ferry JJ, et al. Assay development for histone methyltransferases. Assay Drug Dev Technol. 2013;11(4):227-236.
Sadhu MJ, Guan Q, Li F, et al. Nutritional control of epigenetic processes in yeast and human cells. Genetics. 2013;195(3):831-844.
Mentch SJ, Mehrmohamadi M, Huang L, et al. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015;22(5):861-873.
Liu M, Barnes VL, Pile LA. Disruption of methionine metabolism in drosophila melanogaster impacts histone methylation and results in loss of viability. G3 (BETHESDA). 2015;6(1):121-132.
Sperber H, Mathieu J, Wang Y, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17(12):1523-1535.
Ara AI, Xia M, Ramani K, et al. S-adenosylmethionine inhibits lipopolysaccharide-induced gene expression via modulation of histone methylation. Hepatology. 2008;47(5):1655-1666.
Van Kanegan MJ, Adams DG, Wadzinski BE, et al. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and AKT. J Biol Chem. 2005;280(43):36029-36036.
Stanevich V, Jiang L, Satyshur KA, et al. The structural basis for tight control of PP2A Methylation and function by LCMT-1. Mol Cell. 2011;41(3):331-342.
Xing Y, Li Z, Chen Y, et al. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133(1):154-163.
De Baere I, Derua R, Janssens V, et al. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry. 1999;38(50):16539-16547.
Sontag E, Hladik C, Montgomery L, et al. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004;63(10):1080-1091.
Nicholls RE, Sontag JM, Zhang H, et al. PP2A Methylation controls sensitivity and resistance to Beta-Amyloid-Induced Cognitive and electrophysiological impairments. Proc Natl Acad Sci U S A. 2016;113(12):3347-3352.
Sontag E, Nunbhakdi-Craig V, Sontag JM, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J Neurosci. 2007;27(11):2751-2759.
Sontag JM, Nunbhakdi-Craig V, Montgomery L, et al. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A B(ALPHA) Subunit expression that correlate with enhanced tau phosphorylation. J Neurosci. 2008;28(45):11477-11487.
Yoon SY, Choi HI, Choi JE, et al. Methotrexate decreases PP2A Methylation and increases tau phosphorylation in neuron. Biochem Biophys Res Commun. 2007;363(3):811-816.
Brummelte S, Mc Glanaghy E, Bonnin A, et al. Developmental changes in serotonin signaling: implications for early brain function, behavior and adaptation. Neuroscience. 2017;342:212-231.
Losada ME, Rubio MC. Acute effects of S-Adenosyl-L-Methionine on catecholaminergic central function. Eur J Pharmacol. 1989;163(2-3):353-356.
Mischoulon D, Fava M. Role of S-Adenosyl-L-Methionine in the treatment of depression: a review of the evidence. Am J Clin Nutr. 2002;76(5):1158S-1161S.
Otero-Losada ME, Rubio MC. Acute changes in 5-HT Metabolism after S-Adenosyl-L-Methionine administration. Gen Pharmacol. 1989;20(4):403-406.
Moat SJ, Clarke ZL, Madhavan AK, et al. Folic acid reverses endothelial dysfunction induced by inhibition of tetrahydrobiopterin biosynthesis. Eur J Pharmacol. 2006;530(3):250-258.
Sumi-Ichinose C, Urano F, Kuroda R, et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. a study from 6-Pyruvoyltetrahydropterin synthase knockout mice. J Biol Chem. 2001;276(44):41150-60.
Bottiglieri T, Laundy M, Crellin R, et al. Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry. 2000;69(2):228-232.
Tsao D, Diatchenko L, Dokholyan NV. Structural mechanism of S-Adenosyl methionine binding to catechol O-methyltransferase. Plos One. 2011;6(8):E24287.
Bellido I, Gomez-Luque A, Plaza A, et al. S-adenosyl-L-Methionine prevents 5-HT(1A) receptors up-regulation induced by acute imipramine in the frontal cortex of the rat. Neurosci Lett. 2002;321(1-2):110-114.
Tsao D, Wieskopf JS, Rashid N, et al. Serotonin-induced hypersensitivity via inhibition of catechol O-Methyltransferase Activity. Mol Pain. 2012;8:25.
Obeid R, Herrmann W. Homocysteine and Lipids: S-Adenosyl Methionine As A Key Intermediate. FEBS LETT. 2009;583(8):1215-1225.
Hao X, Huang Y, Qiu M, et al. Immunoassay of S-adenosylmethionine and S-adenosylhomocysteine: the methylation index as a biomarker for disease and health status. BMC Res Notes 2016; 9:498.
Reul JM. Making memories of stressful events: a journey along epigenetic, gene transcription, and signaling pathways. Front Psychiatry. 2014;5:5.
Weaver IC, Champagne FA, Brown SE, et al. Reversal of maternal programming of stress responses in adult offspring through methyl supplementation: altering epigenetic marking later in life. J Neurosci. 2005;25(47):11045-11054.
Mischoulon D, Alpert JE, Arning E, et al. Bioavailability of S-Adenosyl methionine and impact on response in a randomized, double-blind, placebo-controlled trial in major depressive disorder. J Clin Psychiatry. 2012;73(6):843-848.
Galizia I, Oldani L, Macritchie K, et al. S-adenosyl methionine (SAME) for depression in adults. Cochrane Database Syst Rev. 2016;10:CD011286.
De Berardis D, Marini S, Serroni N, et al. S-adenosyl-L-methionine augmentation in patients with stage ii treatment-resistant major depressive disorder: an open label, fixed dose, single-blind study. Scientific World Journal. 2013;2013:204649.
Strous RD, Ritsner MS, Adler S, et al. Improvement of aggressive behavior and quality of life impairment following s-adenosyl-methionine (sam-e) augmentation in schizophrenia. Eur Neuropsychopharmacol. 2009;19(1):14-22.
Surtees R, Leonard J, Austin S. Association of demyelination with deficiency of cerebrospinal-fluid s-adenosylmethionine in inborn errors of methyl-transfer pathway. Lancet. 1991;338(8782-8783):1550-1554.
Loehrer FM, Schwab R, Angst CP, et al. Influence of oral S-adenosylmethionine on Plasma 5-Methyltetrahydrofolate, S-adenosylhomocysteine, homocysteine and methionine in healthy humans. J Pharmacol Exp Ther. 1997;282(2):845-850.
Lee S, Lemere CA, Frost JL, et al. Dietary supplementation with s-adenosyl methionine delayed amyloid-beta and tau pathology in 3xtg-ad mice. J Alzheimers Dis. 2012;28(2):423-431.
Fuso A, Nicolia V, Ricceri L, et al. S-adenosylmethionine reduces the progress of the alzheimer-like features induced by b-vitamin deficiency in mice. Neurobiol Aging. 2012;33(7):1482 E1-16.
Persichilli S, Gervasoni J, Di Napoli A, et al. Plasma thiols levels in alzheimer's disease mice under diet-induced hyperhomocysteinemia: effect of s-adenosylmethionine and superoxide-dismutase supplementation. J Alzheimers Dis. 2015;44(4):1323-1331.
Do Carmo S, Hanzel CE, Jacobs ML, et al. Rescue of early bace-1 and Global dna demethylation by S-Adenosylmethionine reduces amyloid pathology and improves cognition in an alzheimer's model. Sci Rep. 2016;6:34051.
Bustamante AC, Aiello AE, Galea S, et al. Glucocorticoid receptor DNA Methylation, Childhood Maltreatment And Major Depression. J Affect Disord. 2016;206:181-188.
Bergink V, Gibney SM, Drexhage HA. Autoimmunity, inflammation, and psychosis: a search for peripheral markers. Biol Psychiatry. 2014;75(4):324-331.
Stefano P, Concetta C, Luigi D, et al. Role of neurodevelopment involved genes in psychiatric comorbidities and modulation of inflammatory processes in alzheimer's disease. J Neurol Sci. 2016;370:162-166.
Song Z, Uriarte S, Sahoo R, et al. S-Adenosylmethionine (SAME) modulates interleukin-10 and interleukin-6, but not tnf, production via the adenosine (A2) receptor. Biochim Biophys Acta. 2005;1743(3):205-213.
Gobejishvili L, Avila DV, Barker DF, et al. S-Adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via camp/protein kinase a pathway. J Pharmacol Exp Ther. 2011;337(2):433-443.
Pfalzer AC, Choi SW, Tammen SA, et al. S-Adenosylmethionine mediates inhibition of inflammatory response and changes in dna methylation in human macrophages. Physiol Genomics. 2014;46(17):617-623.
Cleare A, Pariante CM, Young AH, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2008 british association for psychopharmacology guidelines. J Psychopharmacol. 2015;29(5):459-525.
Remington R, Bechtel C, Larsen D, et al. A phase ii randomized clinical trial of a nutritional formulation for cognition and mood in alzheimer's disease. J Alzheimers Dis. 2015;45(2):395-405.
Mischoulon D, Price LH, Carpenter LL, et al. A double-blind, randomized, placebo-controlled clinical trial of S-Adenosyl-L-methionine (SAME) Versus Escitalopram In Major Depressive Disorder. J Clin Psychiatry. 2014;75(4):370-376.
Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT. Selective Translational Control of the Alzheimer’s Amyloid Precursor Protein Transcript by Iron Regulatory Protein-1. J. Biol. Chem. (cover issue), 2010;(285)31217-32.
Lippi G, Mattiuzzi C, Meschi T, et al. Homocysteine and migraine. a narrative review. Clin Chim Acta. 2014;433:5-11.
Hao, X., et al., Novel immunoassays to detect methionine adenosyltransferase activity and quantify S-adenosylmethionine. FEBS Lett, 2017. 591(8): p. 1114-1125.
Acknowledgements
Dr. Jin Gao was supported by the grant from National Natural Science Foundation of China (http://www.nsfc.gov.cn, No:81501170) Dr. Jack Rogers is supported by 5R01MH102279-03 (M. Fava, MGH), and was a recipient of The Zenith Fellows Award of Alzheimer’s Association and of a National Institutes of Health grant (R21NS077079-01A1). Dr. Xudong Huang and JTR are supported by a National Institutes of Health grant (1R01AG056614-01).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
ESM 1
(PDF 1239 kb)
Rights and permissions
About this article

Cite this article
Gao, J., Cahill, C.M., Huang, X. et al. S-Adenosyl Methionine and Transmethylation Pathways in Neuropsychiatric Diseases Throughout Life. Neurotherapeutics 15, 156–175 (2018). https://doi.org/10.1007/s13311-017-0593-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0593-0