
Abstract
Rett syndrome (RTT) is a syndromic autism spectrum disorder caused by loss-of-function mutations in MECP2. The methyl CpG binding protein 2 binds methylcytosine and 5-hydroxymethycytosine at CpG sites in promoter regions of target genes, controlling their transcription by recruiting co-repressors and co-activators. Several preclinical studies in mouse models have identified rational molecular targets for drug therapies aimed at correcting the underlying neural dysfunction. These targeted therapies are increasingly translating into human clinical trials. In this review, we present an overview of RTT and describe the current state of preclinical studies in methyl CpG binding protein 2-based mouse models, as well as current clinical trials in individuals with RTT.
Similar content being viewed by others

Background
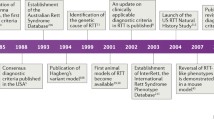
Rett syndrome (RTT) was first noted by Andreas Rett in Vienna, Austria, and virtually simultaneously by Bengt Hagberg in Göteborg, Sweden, about 50 years ago [1]. However, it did not become widely known until the report of Hagberg et al. in 1983 [2]. Thereafter, general awareness expanded rapidly through the 1980s, spurred by symposia in Vienna under the guidance of Andreas Rett, and meetings organized through the International Rett Syndrome Association in the USA. Diagnostic criteria were generated and continuously updated [3–7], clinical studies were organized, and a search for possible causation was initiated.
Neuropathologic examinations confirmed the clinical finding of reduced brain growth, and identified specific cellular abnormalities, including reduced neuronal size and simplified dendritic arborizations. These also failed to identify any evidence of progressive neural pathologies that would indicate a neurodegenerative condition [8–10]. Instead, RTT had all the features of a neurodevelopmental disorder. Owing to the overwhelming predominance of RTT among females, a genetic etiology was proposed, directed at the X chromosome [11]. Eventually, attention was focused on the Xq28 locus [12]. In 1999, Amir et al. [13] described mutations in MECP2, the gene encoding methyl CpG binding protein 2 (MeCP2). The MeCP2 protein binds methylcytosine and 5-hydroxymethycytosine at CpG sites in promoter regions of target genes, controlling their transcription by recruiting co-repressors and co-activators. Mecp2 knockout mouse models demonstrated that MeCP2 is critical for the maturation and maintenance of neurons and glial cells [14, 15]. Thereafter, Mecp2 knockout and knockin mice carrying mutations observed in RTT individuals allowed fundamental research to advance our understanding of the molecular, cellular, and systems-level pathology [16], and to begin addressing possible therapeutic interventions. These strategies have proved disease modifying, and have even resulted in full reversal of the underlying abnormalities [17]. At the same time, human clinical studies expanded rapidly worldwide to obtain natural history information that is essential for the design of appropriate clinical trials [18–21] (see also https://www.clinicaltrials.gov/ct2/show/NCT00299312). This combination of fundamental and preclinical research in mouse models with human clinical research has fostered a vibrant level of translational and clinical trial planning and execution that is described in detail in the following sections.
Clinical Features
RTT is a unique neurodevelopmental disorder with an incidence of about 1 : 10,000 female births [22], and multiple systemic issues appearing first in young females after a period of apparently normal postnatal development. Detailed evaluation of the first 6 months of life established that developmental skills do not follow a typical timeline when viewed critically [23–25]. These findings of delayed or arrested developmental are sufficiently subtle that specific delays are not often suspected until the second year of life when frank regression, accompanied in some by autistic-like social avoidance, is noted. Other clues may also be present, including abnormal deceleration of head circumference increase (a barometer of appropriate brain growth; [26–28]), reduction in muscle tone, and an overall appearance of being too quiet or “good”. This is punctuated in some individuals by periods of inappropriate and unexplained screaming spells. The most critical feature is the appearance of stereotypic movements during wakefulness, most prominent in the hands, but also seen in oro-motor regions and the lower extremities.
The initial period of regression is followed by a period of stabilization beginning by 30 months with improved eye contact and socialization that remains throughout life [29]. However, motor development may transition as muscle tone begins to increase, first with a period of apparently normal tone transitioning ultimately to an increase in tone or rigidity. This may be coupled with the appearance of dystonic positions, particularly at the ankles and wrists. Multiple neurologic or systemic issues often arise: 1) epilepsy beginning typically by the third year of life [30–32]; 2) periodic breathing consisting of breath-holding or hyperventilation or both [33–35]; 3) prominent gastrointestinal (GI) issues from top to bottom, including poor chewing and swallowing, gastroesophageal reflux, delayed stomach emptying, and constipation [36–39]; 4) decline in growth parameters in most, including height, weight, hands, and feet [28, 40]; 5) scoliosis in most with up to 13% requiring surgical correction of the deformity [41]; 6) intolerance of warm temperatures; and 7) cool hands and feet. Ambulation is achieved initially in as many as 80% but is typically broad-based and nonpurposeful. It is often accompanied by retropulsion (first steps are backwards), and may include prominent toe walking. Approximately one-third of this group will stop walking. Overall, about 50% maintain independent gait and another 20% ambulate with some level of assistance.
Genetics
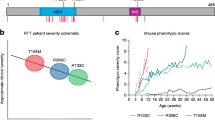
MECP2 mutations were identified in 1999 as causative for RTT [13]. Presently, up to 96% of individuals in the US Natural History Study (NHS) have such mutations [42, 43]. Nevertheless, a MECP2 mutation does not indicate the clinical diagnosis of RTT. Individuals with mutations may not have features of RTT syndrome. Conversely, up to 4% have RTT but do not have a MECP2 mutation [43]. More than 200 pathologic mutations have been identified. However, 8 point mutations (4 missense and 4 nonsense) account for nearly 60% of all affected individuals. Deletions and insertions account for nearly 20%, such that a large number of the remaining mutations occur privately or in only a few individuals. Among the most common mutations, definite phenotype–genotype correlations have been established [42–44]. Based on the Clinical Severity Score employed in the NHS, individuals with R133C, R294X, R306C, and 3’-truncations are significantly less involved than those with the 5 other common point mutations and with deletions of whole exons. However, those with exactly the same mutation may have widely differing clinical abilities. This is thought to represent differences in X-chromosome inactivation (XCI) and overall genetic background. One factor that has not received major attention is the clonal distribution of normal and mutant X chromosomes in brain as differences in expression patterns may lead to markedly different clinical phenotypes. LaSalle et al. noted this in 2001 [45], but scarce mention has been made in recent years. Attempts at identifying variations in XCI in blood have indicated skewing in > 25% of RTT individuals, which is approximately double the occurrence in the general population. Highly skewed (>90%) XCI was noted in > 10%, more than twice that in the general population [unpublished data from the US NHS (NCT00299312)]; in this group, the skewing was roughly equal in favor of the mutant or normal chromosome. These data should be interpreted cautiously, as XCI in brain may differ from that in blood. In addition, environmental background may be critically important to the overall level of clinical involvement.
As an X-linked dominant disorder, RTT is thought to arise from spontaneous or de novo mutations in rapidly dividing germinal cells. As such, the majority of de novo mutations occur in the sperm [46, 47]. In some instances, the mutation is transmitted by a female carrying a specific mutation but, owing to favorable XCI, she is unaffected or has mild learning disability or cognitive impairment [48, 49]. In offspring resulting from affected women, a girl may have classic or typical RTT, but boys are likely to have much greater involvement resulting in infantile encephalopathy and a very much shortened life expectancy (~1 year) owing mainly to failure to maintain normal respiratory function [50]. In other instances, mutations in the 3’-truncation region in boys produce a progressive dystonia against a background of severe cognitive delay [49]. However, in families that do not have an affected female, recognition of such features in a male may not raise suspicion of a MECP2 mutation and hence go undetected.
Males with seemingly typical RTT have been reported in a small number under 2 scenarios: one being co-occurrence of Klinefelter syndrome (47 XXY) [51–54] and the other being somatic mosaicism [55]. In each instance, 2 populations of cells exist: 1 with a normal and 1 with a mutant X chromosome.
More recently, mutations in other genes have been associated with atypical RTT. These include children with mutations in CDKL5, FOXG1, and other associated genes [56–60].
Therapeutic Targets for Clinical Trials
At present, clinical trials are limited to RTT individuals with MECP2 mutations. In addition, careful consideration is given to establish inclusion and exclusion criteria in order to ensure similar groups in the design of placebo-controlled, double-blind studies. Suitable end points are also essential. RTT offers several highly relevant and important clinical end points for which suitable outcome measures exist [61]. These include communication, epilepsy, motor function, GI parameters, and periodic breathing, either breath holding or hyperventilation. Current studies using eye-tracking technology coupled with evoked response potentials are leading the way to assess communication. Video-electroencephalography recordings can provide both spectral analysis of the various waveforms and time-locked assessments of epileptiform features. Coupled with plethysmographic recordings, periodic breathing can also be captured to provide evidence of altered breathing patterns. For those individuals who are ambulatory or have improvement in walking capabilities, gait analysis paradigms provide objective evidence of motor performance. Improvements in muscle tone or dystonia can also be assessed objectively. Actigraphy provides an excellent measure of both purposeful and stereotypic movements. Evaluation of GI function is more varied. Potentially useful targets are objective improvements in chewing and swallowing and better GI motility in terms of improved gastric emptying and reduced constipation.
In terms of effective clinical outcome measures, efforts are ongoing to improve the acute responsiveness of clinical severity measures, communication assessment, and neurophysiologic parameters. What is lacking is an appropriate behavioral measure. This deficiency is actively being pursued. Quality of life (QoL) using standardized instruments (not specific to RTT) has been addressed in both those with RTT [62] and their caregivers (work in progress). QoL for those with RTT has a distinct pattern of involvement. Individuals with greater clinical severity demonstrate more severe motor impairment, and those with lower clinical severity have fewer motor impairments. However, those with greater clinical severity have better behavioral function than those with lower clinical severity. This raises the possibility that modest improvement in motor function could have an adverse effect on behavior. QoL has also been assessed in the primary caregiver, generally the mother, and is currently undergoing analysis. In addition, a separate caregiver inventory is being collected and standardized specifically among families managing an individual with RTT (data from NCT00299312).
At present, no relevant laboratory biomarker is known. A metabolomics pilot study has started with the current NHS; however, the results are not anticipated before 2016.
Repertoire of Clinical Trials
Therapeutic targets for RTT are either aimed at reversing the loss-of-function mutation in MECP2 or modifying its downstream pathways, including neurotransmitter receptor systems, neurotrophins, and their intracellular signaling pathways (Tables 1 and 2) [63–65]. We follow the standard convention: “preclinical” is used for experiments done in experimental animal models, and “clinical” for tests performed in humans.
MECP2 Gene Therapy and “Read-through” Compounds
Preclinical studies have demonstrated that restoring Mecp2 gene expression in mice improves RTT-like neurological symptoms [17, 66]. However, translating this gene therapy approach to clinical trials requires overcoming multiple challenges, including developing safe vectors that allow effective in vivo gene delivery and avoiding overexpression of exogenous MECP2 in wild-type cells in the mosaic brain of individuals with RTT. Among different gene vectors, the serotype 9 of adeno-associated virus is the most promising owing to its ability to cross the blood–brain barrier (BBB) and to infect neurons efficiently, yielding long-term transgene expression in male Mecp2 knockout mice and in female Mecp2 heterozygous mice [67, 68]. It should be noted that no deleterious consequences of the overexpression of Mecp2 in wild-type cells were noted in the mosaic female brain [69]. At present, there are no clinical trials of MECP2 gene therapy in individuals with RTT.
Owing to a premature STOP codon, a nonsense mutation (e.g., R168X, R255X) observed in approximately one-third of RTT individuals causes the termination of translation and may destabilize mRNA molecules [70, 71]. These individuals may have a more severe clinical presentation than those with missense MECP2 mutations that result in single amino acid substitutions, except, perhaps, for the R294X allele. Aminoglycoside antibiotics allow ribosomal read-through of premature STOP codons during translation, yielding a full-length functional protein. Such aminoglycosides and novel nonaminoglycoside “read-through” compounds have been tested for therapeutic efficacy in Duchenne muscular dystrophy and cystic fibrosis [72]. Gentamycin or geneticin allowed translation of full-length MeCP2 protein in a lymphocyte cell line derived from an individual with a R255X nonsense mutation [73], and in fibroblasts from knock-in mice expressing an R168X nonsense mutation [69]. More recently, embryonic fibroblasts from a mouse model with the R255X nonsense mutation were treated with gentamycin and were able to express full-length MeCP2 [74]. Gentamycin also increased dendritic spine density in neurons derived from induced pluripotent stem cells obtained from a RTT individual with a Q244X nonsense mutation [75]. However, the renal and auditory toxicity, as well as poor central nervous system penetration of aminoglycoside antibiotics, limits their applicability. New “read-through” compounds with better safety profiles are currently being developed.
Neurotrophins and Growth Factors: Brain-derived Neurotrophic Factor and Insulin-like Growth Factor-1
One of the most prominent targets of MeCP2 transcriptional regulation is the gene encoding brain-derived neurotrophic factor (BDNF) [76, 77], a neurotrophin well known for its critical role in neuronal growth, synapse formation, and activity-dependent plasticity through activation of its selective tropomyosin receptor kinase B (TrkB) receptors [78]. MeCP2 controls BDNF expression through complex interactions [79, 80]. Knockout of Bdnf results in neurologic phenotypes reminiscent of the deficits observed in Mecp2-deficient mice, suggesting that impaired Bdnf transcription contributes to RTT pathophysiology. Furthermore, conditional overexpression of BDNF in excitatory forebrain neurons of Mecp2-deficient mice—achieved by crossing BDNFSTOP mice with Mecp2;cre mice—led to the improvement of some of their RTT-like neurologic phenotypes [81]. In addition, in vitro expression of green fluorescent protein-tagged BDNF from a cytomegalovirus-driven plasmid rescued the dendritic atrophy caused by short hairpin RNA-mediated knockdown of Mecp2 in cultured rat hippocampal neurons [82], and the dendritic atrophy and lower spine density in cultured hippocampal neurons from Mecp2 knockout mice (Xu and LP-M, in preparation). Unfortunately, BDNF has very low BBB permeability, which limits the bioavailability of peripherally administered BDNF as a potential therapy. Hence, BDNF “boosters” or mimetics with sufficient bioavailability in brain are being developed for therapy.
Currently, there are 2 clinical trials in RTT individuals, testing compounds that boost BDNF levels: a Phase I open-label trial of fingolimod and a Phase II open-label trial of glatiramer acetate (Copaxone; Teva Pharmaceuticals, Petah Tikva, Israel), both Food and Drug Administration (FDA)-approved drugs for the treatment of multiple sclerosis. Fingolimod modulates the sphingosine-1 phosphate receptor and increases BDNF levels in neurons in an activity-dependent fashion through the mitogen-activated protein kinase pathway [83]. Glatiramer acetate is an immunomodulatory agent based on the amino acid structure of myelin basic protein that is currently used for the treatment of relapsing–remitting multiple sclerosis. One of the proposed mechanisms of action of Copaxone (Teva Pharmaceuticals) is the increased expression and release of BDNF by autoreactive T cells [84].
Additional compounds have been described that increase BDNF levels and improve RTT-like phenotypes in Mecp2-deficient cells and mice. Ampakines are fast-acting molecules that prevent the desensitization of α-amino-3-hydroxy-5-methyl-4-isoxalepropionic acid-type glutamate receptors, which results in higher activity-dependent expression of BDNF, both in vivo and in vitro [85]. Peripheral treatment with ampakines significantly improved respiratory function in male Mecp2 knockout mice [86], which phenocopies the recurrent apneas suffered by individuals with RTT. BDNF loop domain mimetics are small molecules designed in silico to interact with the BDNF binding pocket in the TrkB receptor [87]. One of them, LM22A-4, restored respiratory regularity in female Mecp2 heterozygous mice [88, 89], increased dendritic spine density in CA1 pyramidal neurons in organotypic slice cultures of male Mecp2 knockout mice (Miller, Longo, and LP-M, in preparation), and restored long-term potentiation at CA3->CA1 excitatory synapses in hippocampal slices from symptomatic female Mecp2 heterozygous mice (Li, Longo, and LP-M, in preparation). Finally, cysteamine (and its FDA-approved dimer cystamine) were shown to increase BDNF release by increasing heat shock DnaJ-containing protein 1b levels and inhibiting transglutaminase [90], which supports the Phase II/III clinical trial of delayed-release cysteamine (RP103) for Huntington disease. None of these compounds has reached human clinical trials for RTT.
Unlike BDNF, insulin-like growth factor-1 (IGF-1) and its active peptide (1–3)IGF-1 cross the BBB and activate intracellular signaling cascades similar to those triggered by BDNF activation of TrkB receptors, which includes phosphatidylinositol-3 kinase–Akt and mitogen-activated protein kinase [91]. Intraperitoneal injection of (1–3)IGF-1 (glypromate) in male Mecp2 knockout mice improved survival and locomotor activity, as well as social and anxiety behaviors [92]. Recombinant full-length IGF-1 (mecasermin) is already approved by the FDA for the treatment of growth failure in children, and improves survival and RTT-like phenotypes in male Mecp2 knockout mice and in female Mecp2 heterozygous mice [93]. The fact that full-length IGF-1 worsened the metabolic syndrome of Mecp2-deficient mice should be considered [94]. The effects of full-length IGF-1 are owing to the direct activation of IGF-1 receptors and its downstream signaling cascades. However, the (1–3)IGF-1 tripeptide may increase expression of IGF-1 [95], although its molecular mechanism of action is currently unknown. Based on these promising leads, a Phase II double-blind, placebo-controlled clinical trial involving children aged 3–10 years is underway to treat patients with RTT with full-length IGF-1 [96]. More recently, a Phase II double-blind, placebo-controlled clinical trial involving older teenagers and adults aged 16–45 years has been initiated to test the efficacy of NNZ-2566, a protease-resistant analogue of (1–3)IGF-1. The rsults of this short-term trial were sufficiently positive to promote consideration of further study of this agent.
Neurotransmitter Systems: Monoamines, Glutamate and γ-Aminobutyric Acid
Monoamine neurotransmitters like dopamine, serotonin, and noradrenaline have been found to be reduced in autopsy of RTT brains and in Mecp2-deficient mice [97–100]. As these monoamines are associated with the regulation of breathing patterns in the brainstem, augmenting their levels may be therapeutic for breathing dysfunction in RTT. Desipramine is an antidepressant that blocks the uptake of noradrenaline, and has been shown to reverse the breathing dysregulation in male Mecp2 knockout mice [101, 102]. Desipramine is currently in a Phase II double-blind, placebo-controlled clinical trial for RTT. The atypical tricyclic antidepressant tianeptine (REV-003) also improved respiratory activity in Mecp2-deficient mice, although this effect may reflect modulation of monoamine levels, glutamate receptor function, or BDNF levels [103]. Sarizotan and NLX-101 are selective agonists of the 5-HT1α receptor that significantly improved breathing patterns and reduced the frequency of apneas in Mecp2-deficient mice [104].
γ-Aminobutyric acid (GABA)-ergic and glutamatergic systems maintain the fine balance between excitation and inhibition during neuronal maturation. Dysfunctions in these neurotransmitter systems have been described in preclinical models of RTT [105–107]. Dextromethorphan is a N-methyl-D-aspartate (NMDA) receptor antagonist currently in a double-blind, placebo-controlled trial in individuals with RTT; to date, its efficacy has not been noted. The NMDA receptor antagonist ketamine improved some RTT-like phenotypes in Mecp2 knockout mice [108]; based on these encouraging results, a clinical trial of low-dose ketamine is currently planned. A delay of the developmental switch in the expression of GluN2 subunits of the NMDA receptors in the visual cortex contributes to visual acuity deficits in Mecp2-deficient mice, which were improved by genetic deletion of the GluN2A subunit [109]; a negative allosteric modulator selective for GluN2A-containing NMDA receptors is currently in preclinical trials in Mecp2-deficient mice. Selective deletion of Mecp2 in GABAergic neurons caused impaired GABAergic transmission, cortical hyperexcitability, and several neurologic features of RTT and autism spectrum disorders [110]. Vigabatrin is an antiepileptic drug that irreversibly inhibits GABA transaminase, inhibits GABA catabolism, and thereby increases GABA levels [111]. The drug is already FDA-approved for use in epilepsy syndromes. Planning for a clinical trial in RTT is underway. However, retinal toxicity may limit the chronic use of this medication.
Mitochondrial Function: EPI-743
RTT is associated with high levels of systemic oxidative stress and alteration in mitochondrial morphology, while plasma levels of oxidative stress biomarkers correlate with disease severity and progression [112]. Based on these observations, augmenting glutathione synthesis is thought to be potentially beneficial. The structure of the small molecule EPI-743 is based on vitamin E and its proposed mechanisms of action include augmenting glutathione synthesis and acting at the mitochondrial level to regulate electron transport. An exploratory Phase II placebo-controlled trial of EPI-743 in individuals with RTT revealed improvement in head growth but not in other core features of RTT.
Conclusion
The discovery of loss-of-function MECP2 mutations in individuals with RTT, and the generation of experimental cell culture and animal models has provided detailed information of the underlying pathophysiology at the molecular, cellular, and systems levels. In turn, this information prompted preclinical studies of rationally designed therapies aimed at identified molecular targets, some with known mechanism of action. Thus, exciting new opportunities are emerging for the discovery and development of new pharmacotherapies for RTT and other related austism spectrum disorders. However, the complexity of these disorders requires more extensive interdisciplinary collaborations following the highest standards for the validation of animal models, outcome measures, and study design to yield robust and reproducible preclinical information relevant to the human condition. Only then can the preclinical studies provide the strong foundation needed for the success of human clinical trials.
References
Rett A. [On a unusual brain atrophy syndrome in hyperammonemia in childhood]. Wien Med Wochenschr 1966;116:723-726.
Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett's syndrome: report of 35 cases. Ann Neurol 1983;14:471-479.
Hagberg B, Goutieres F, Hanefeld F, Rett A, Wilson J. Rett syndrome: criteria for inclusion and exclusion. Brain Dev 1985;7:372-373.
Diagnostic criteria for Rett syndrome. The Rett Syndrome Diagnostic Criteria Work Group. Ann Neurol 1988;23:425-428.
Hagberg B, Hanefeld F, Percy A, Skjeldal O. An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric Neurology Society Meeting, Baden Baden, Germany, 11 September 2001. Eur J Paediatr Neurol 2002;6:293-297.
Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 2010;68:944-950.
Percy AK, Neul JL, Glaze DG, et al. Rett syndrome diagnostic criteria: lessons from the Natural History Study. Ann Neurol 2010;68:951-955.
Jellinger K, Armstrong D, Zoghbi HY, Percy AK. Neuropathology of Rett syndrome. Acta Neuropathol 1988;76:142-158.
Armstrong DD. The neuropathology of Rett syndrome—overview 1994. Neuropediatrics 1995;26:100-104.
Armstrong DD. Neuropathology of Rett syndrome. J Child Neurol 2005;20:747-753.
Zoghbi HY, Percy AK, Schultz RJ, Fill C. Patterns of X chromosome inactivation in the Rett syndrome. Brain Dev 1990;12:131-135.
Schanen NC, Dahle EJ, Capozzoli F, Holm VA, Zoghbi HY, Francke U. A new Rett syndrome family consistent with X-linked inheritance expands the X chromosome exclusion map. Am J Hum Genet 1997;61:634-641.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999;23:185-188.
Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet 2001;27:327-331.
Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet 2001;27:322-326.
Shahbazian M, Young J, Yuva-Paylor L, et al. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 2002;35:243-254.
Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007;315:1143-1147.
Witt-Engerstrom I, Hagberg B. The Rett syndrome: gross motor disability and neural impairment in adults. Brain Dev 1990;12:23-26.
Engerstrom IW. Rett syndrome: the late infantile regression period--a retrospective analysis of 91 cases. Acta Pediatr 1992;81:167-172.
Clarke A. Rett syndrome. J Med Genet 1996;33:693-699.
Leonard H. Rett syndrome in Australia. University of Western Australia, Perth, Western Australia, 1996.
Laurvick CL, de Klerk N, Bower C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr 2006;148:347-352.
Einspieler C, Kerr AM, Prechtl HF. Is the early development of girls with Rett disorder really normal? Pediatr Res 2005;57:696-700.
Marschik PB, Kaufmann WE, Sigafoos J, et al. Changing the perspective on early development of Rett syndrome. Res Develop Disabil 2013;34:1236-1239.
Neul JL, Lane JB, Lee HS, et al. Developmental delay in Rett syndrome: data from the natural history study. J Neurodev Disord 2014;6:20.
Rett A. Rett syndrome. History and general overview. Am J Med Genet Suppl 1986;1:21-25.
Schultz RJ, Glaze DG, Motil KJ, et al. The pattern of growth failure in Rett syndrome. Am J Dis Child 1993;147:633-637.
Tarquinio DC, Motil KJ, Hou W, et al. Growth failure and outcome in Rett syndrome: Specific growth references. Neurology 2012;79:1653-1661.
Percy AK. Rett syndrome: exploring the autism link. Arch Neurol 2011;68:985-989.
Glaze DG, Schultz RJ, Frost JD. Rett syndrome: characterization of seizures versus non-seizures. Electroencephalogr Clin Neurophysiol 1998;106:79-83.
Glaze DG. Neurophysiology of Rett syndrome. J Child Neurol 2005;20:740-746.
Glaze DG, Percy AK, Skinner S, et al. Epilepsy and the natural history of Rett syndrome. Neurology 2010;74:909-912.
Lugaresi E, Cirignotta F, Montagna P. Abnormal breathing in the Rett syndrome. Brain Dev 1985;7:329-333.
Glaze DG, Frost JD, Jr., Zoghbi HY, Percy AK. Rett's syndrome. Correlation of electroencephalographic characteristics with clinical staging. Arch Neurol 1987;44:1053-1056.
Julu PO, Kerr AM, Apartopoulos F, et al. Characterisation of breathing and associated central autonomic dysfunction in the Rett disorder. Arch Dis Child 2001;85:29-37.
Motil KJ, Schultz RJ, Browning K, Trautwein L, Glaze DG. Oropharyngeal dysfunction and gastroesophageal dysmotility are present in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr 1999;29:31-37.
Motil KJ, Morrissey M, Caeg E, Barrish JO, Glaze DG. Gastrostomy placement improves height and weight gain in girls with Rett syndrome. J Pediatr Gastroenterol Nutr 2009;49:237-242.
Motil KJ, Caeg E, Barrish JO, et al. Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J Pediatr Gastroenterol Nutr 2012;55:292-298.
Leonard H, Ravikumara M, Baikie G, et al. Assessment and management of nutrition and growth in Rett syndrome. J Pediatr Gastroenterol Nutr 2013;57:451-460.
Schultz R, Glaze D, Motil K, Hebert D, Percy A. Hand and foot growth failure in Rett syndrome. J Child Neurol 1998;13:71-74.
Percy AK, Lee HS, Neul JL, et al. Profiling scoliosis in Rett syndrome. Pediatr Res 2010;67:435-439.
Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 2008;70:1313-1321.
Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet 2014;51:152-158.
Bebbington A, Anderson A, Ravine D, et al. Investigating genotype-phenotype relationships in Rett syndrome using an international data set. Neurology 2008;70:868-875.
LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet 2001;10:1729-1740.
Girard M, Couvert P, Carrie A, et al. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet 2001;9:231-236.
Trappe R, Laccone F, Cobilanschi J, et al. Mecp2 mutations in sporadic cases of rett syndrome are almost exclusively of paternal origin. Am J Hum Genet 2001;68:1093-1101.
Schanen C, Francke U. A severely affected male born into a Rett syndrome kindred supports X-linked inheritance and allows extension of the exclusion map. Am J Hum Genet 1998;63:267-269.
Augenstein K, Lane JB, Horton A, Schanen C, Percy AK. Variable phenotypic expression of a MECP2 mutation in a family. J Neurodev Disord 2009;1:313.
Kankirawatana P, Leonard H, Ellaway C, et al. Early progressive encephalopathy in boys and MECP2 mutations. Neurology 2006;67:164-166.
Salomao Schwartzman J, Zatz M, dos Reis Vasquez L, et al. Rett syndrome in a boy with a 47,XXY karyotype. Am J Hum Genet 1999;64:1781-1785.
Hoffbuhr K, Devaney JM, LaFleur B, et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 2001;56:1486-1495.
Leonard H, Silberstein J, Falk R, et al. Occurrence of Rett syndrome in boys. J Child Neurol 2001;16:333-338.
Vorsanova SG, Yurov YB, Ulas VY, et al. Cytogenetic and molecular-cytogenetic studies of Rett syndrome (RTT): a retrospective analysis of a Russian cohort of RTT patients (the investigation of 57 girls and three boys). Brain Dev 2001;23(Suppl. 1):S196-S201.
Clayton-Smith J, Watson P, Ramsden S, Black GC. Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet 2000;356:830-832.
Borg I, Freude K, Kubart S, et al. Disruption of Netrin G1 by a balanced chromosome translocation in a girl with Rett syndrome. Eur J Hum Genet 2005;13:921-927.
Archer HL, Evans JC, Millar DS, et al. NTNG1 mutations are a rare cause of Rett syndrome. Am J Med Genet A 2006;140:691-694.
White R, Ho G, Schmidt S, et al. Cyclin-dependent kinase-like 5 (CDKL5) mutation screening in Rett syndrome and related disorders. Twin Res Hum Genet 2010;13:168-178.
Pini G, Bigoni S, Engerstrom IW, et al. Variant of Rett syndrome and CDKL5 gene: clinical and autonomic description of 10 cases. Neuropediatrics 2012;43:37-43.
Hagebeuk EE, van den Bossche RA, de Weerd AW. Respiratory and sleep disorders in female children with atypical Rett syndrome caused by mutations in the CDKL5 gene. Dev Med Child Neurol 2013;55:480-484.
Katz DM, Berger-Sweeney JE, Eubanks JH, et al. Preclinical research in Rett syndrome: setting the foundation for translational success. Dis Model Mech 2012;5:733-745.
Lane JB, Lee HS, Smith LW, et al. Clinical severity and quality of life in children and adolescents with Rett syndrome. Neurology 2011;77:1812-1818.
Cobb S, Guy J, Bird A. Reversibility of functional deficits in experimental models of Rett syndrome. Biochem Soc Trans 2010;38:498-506.
Gadalla KK, Bailey ME, Cobb SR. MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J 2011;439:1-14.
Chapleau CA, Lane J, Larimore J, Li W, Pozzo-Miller L, Percy AK. Recent progress in Rett syndrome and MeCP2 dysfunction: assessment of potential treatment options. Future Neurol 2013;8.
Giacometti E, Luikenhuis S, Beard C, Jaenisch R. Partial rescue of MeCP2 deficiency by postnatal activation of MeCP2. Proc Natl Acad Sci U S A 2007;104:1931-1936.
Gadalla KK, Bailey ME, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther 2013;21:18-30.
Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci 2013;33:13612-13620.
Brendel C, Belakhov V, Werner H, et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med 2011;89:389-398.
Schanen C, Houwink EJ, Dorrani N, et al. Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am J Med Genet A 2004;126A:129-140.
Percy AK, Lane JB, Childers J, et al. Rett syndrome: North American database. J Child Neurol 2007;22:1338-1341.
Zingman LV, Park S, Olson TM, Alekseev AE, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther 2007;81:99-103.
Popescu AC, Sidorova E, Zhang G, Eubanks JH. Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. J Neurosci Res 2010;88:2316-2324.
Pitcher MR, Herrera JA, Buffington SA, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet 2015 Jan 29 [Epub ahead of print].
Marchetto MC, Carromeu C, Acab A, et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010;143:527-539.
Chen WG, Chang Q, Lin Y, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 2003;302:885-889.
Martinowich K, Hattori D, Wu H, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003;302:890-893.
Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci 2013;14:7-23.
Katz DM. Brain-derived neurotrophic factor and Rett syndrome. Handb Exp Pharmacol 2014;220:481-495.
Li W, Pozzo-Miller L. BDNF deregulation in Rett syndrome. Neuropharmacology 2014;76:737-746.
Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of mecp2 mutant mice is affected by the level of BDNF expression. Neuron 2006;49:341-348.
Larimore JL, Chapleau CA, Kudo S, Theibert A, Percy AK, Pozzo-Miller L. Bdnf overexpression in hippocampal neurons prevents dendritic atrophy caused by Rett-associated MECP2 mutations. Neurobiol Dis 2009;34:199-211.
Deogracias R, Yazdani M, Dekkers MP, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. Proc Natl Acad Sci U S A 2012;109:14230-14235.
Ziemssen T, Kumpfel T, Klinkert WE, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain-derived neurotrophic factor. Brain 2002;125:2381-2391.
Lauterborn JC, Lynch G, Vanderklish P, Arai A, Gall CM. Positive modulation of AMPA receptors increases neurotrophin expression by hippocampal and cortical neurons. J Neurosci 2000;20:8-21.
Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of Rett syndrome. J Neurosci 2007;27:10912-10917.
Massa SM, Yang T, Xie Y, et al. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest 2010;120:1774-1785.
Schmid DA, Yang T, Ogier M, et al. A TrkB small molecule partial agonist rescues TrkB phosphorylation deficits and improves respiratory function in a mouse model of Rett syndrome. J Neurosci 2012;32:1803-1810.
Kron M, Lang M, Adams IT, Sceniak M, Longo F, Katz DM. A BDNF loop-domain mimetic acutely reverses spontaneous apneas and respiratory abnormalities during behavioral arousal in a mouse model of Rett syndrome. Dis Model Mech 2014;7:1047-1055.
Borrell-Pages M, Canals JM, Cordelieres FP, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest 2006;116:1410-1424.
Zheng WH, Quirion R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem 2004;89:844-852.
Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci U S A 2009;106:2029-2034.
Castro J, Garcia RI, Kwok S, et al. Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett Syndrome. Proc Natl Acad Sci U S A 2014;111:9941-9946.
Pitcher MR, Ward CS, Arvide EM, et al. Insulinotropic treatments exacerbate metabolic syndrome in mice lacking MeCP2 function. Hum Mol Genet 2013;22:2626-2633.
Corvin AP, Molinos I, Little G, et al. Insulin-like growth factor 1 (IGF1) and its active peptide (1-3)IGF1 enhance the expression of synaptic markers in neuronal circuits through different cellular mechanisms. Neurosci Lett 2012;520:51-56.
Khwaja OS, Ho E, Barnes KV, et al. Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for the treatment of Rett syndrome. Proc Natl Acad Sci U S A 2014;111:4596-4601.
Brucke T, Sofic E, Killian W, Rett A, Riederer P. Reduced concentrations and increased metabolism of biogenic amines in a single case of Rett-syndrome: a postmortem brain study. J Neural Transm 1987;68:315-324.
Lekman A, Witt-Engerstrom I, Gottfries J, Hagberg BA, Percy AK, Svennerholm L. Rett syndrome: biogenic amines and metabolites in postmortem brain. Pediatr Neurol 1989;5:357-362.
Roux JC, Dura E, Villard L. Tyrosine hydroxylase deficit in the chemoafferent and the sympathoadrenergic pathways of the Mecp2 deficient mouse. Neurosci Lett 2008;447:82-86.
Samaco RC, Mandel-Brehm C, Chao HT, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. Proc Natl Acad Sci U S A 2009;106:21966-21971.
Roux JC, Dura E, Moncla A, Mancini J, Villard L. Treatment with desipramine improves breathing and survival in a mouse model for Rett syndrome. Eur J Neurosci 2007;25:1915-1922.
Zanella S, Mebarek S, Lajard AM, Picard N, Dutschmann M, Hilaire G. Oral treatment with desipramine improves breathing and life span in Rett syndrome mouse model. Respir Physiol Neurobiol 2008;160:116-121.
McEwen BS, Chattarji S, Diamond DM, et al. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol Psychiatry 2010;15:237-249.
Abdala AP, Lioy DT, Garg SK, Knopp SJ, Paton JF, Bissonnette JM. Effect of Sarizotan, a 5-HT1a and D2-like receptor agonist, on respiration in three mouse models of Rett syndrome. Am J Respir Cell Mol Biol 2014;50:1031-1039.
Maliszewska-Cyna E, Bawa D, Eubanks JH. Diminished prevalence but preserved synaptic distribution of N-methyl-d-aspartate receptor subunits in the methyl CpG binding protein 2(MeCP2)-null mouse brain. Neuroscience 2010;168:624-632.
Zhang ZW, Zak JD, Liu H. MeCP2 is required for normal development of GABAergic circuits in the thalamus. J Neurophysiol 2010;103:2470-2481.
Blue ME, Kaufmann WE, Bressler J, et al. Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat Rec 2011;294:1624-1634.
Kron M, Howell CJ, Adams IT, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci 2012;32:13860-13872.
Durand S, Patrizi A, Quast KB, et al. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron 2012;76:1078-1090.
Chao HT, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010;468:263-269.
Connelly JF. Vigabatrin. Ann Pharmacother 1993;27:197-204.
De Felice C, Signorini C, Leoncini S, et al. The role of oxidative stress in Rett syndrome: an overview. Ann N Y Acad Sci 2012;1259:121-135.
Acknowledgments
LP-M was supported by National Institutes of Health (NIH) grants NS-065027 and HD-074418, Rettsyndrome.org (former International Rett Syndrome Foundation), and the Rett Syndrome Research Trust. AKP was supported by NIH grant HD-061222, Rettsyndrome.org, and the Civitan International Research Center.
Protection of Human Subjects Research
The US Natural History Study (NCT00299312) has received Human Subject approval from The University of Alabama at Birmingham IRB.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1.19 mb)
Rights and permissions
About this article

Cite this article
Pozzo-Miller, L., Pati, S. & Percy, A.K. Rett Syndrome: Reaching for Clinical Trials. Neurotherapeutics 12, 631–640 (2015). https://doi.org/10.1007/s13311-015-0353-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-015-0353-y