
Abstract
Huntington’s disease is an autosomal dominant disorder caused by a mutation in the gene encoding the protein huntingtin on chromosome 4. The mutation is an expanded CAG repeat in the first exon, encoding a polyglutamine tract. If the polyglutamine tract is >40, penetrance is 100% and death is inevitable. Despite the widespread expression of huntingtin, HD has long been considered primarily as a disease of the striatum. It is characterized by selective vulnerability with dysfunction followed by death of the medium size spiny neuron. Considerable effort is being expended to determine whether striatal damage is cell-autonomous, non-cell-autonomous, requiring cell-cell and region to region communication, or both. We review data supporting both mechanisms. We also attempt to organize the data into common mechanisms that may arise outside the medium, spiny neuron, but ultimately have their greatest impact in the striatum.
Similar content being viewed by others


Huntington’s disease (HD) is an autosomal dominant disorder caused by a mutation in the gene (HTT) encoding the protein huntingtin (htt) on chromosome 4. The mutation is an expanded CAG repeat in the first exon [1]. If the polyglutamine (polyQ) tract is >40, penetrance is 100% and death is inevitable. HD has long been primarily considered as a disease of the striatum, characterized by selective, or perhaps better described as differential [2], vulnerability with degeneration and death of the medium spiny neuron (MSN). The Gamma-aminobutyric acid (GABA)ergic MSN represents 95% of striatal neurons. They are all output neurons, with extensive intra-striatal connections with other MSNs and striatal interneurons. For the last two decades, increased attention has been paid to the pathology in the cortex (particularly in layers V and VI [3]), which contains the corticostriatal projection neurons.
Prior to identification of the HTT gene, the huntingtin protein, and the nature of its mutation, it was assumed that selective neuronal vulnerability in HD would be conferred by the expression pattern of the mutated protein (polyQ-htt). As it is now apparent, htt is expressed throughout the nervous system and periphery, without a preference for, or higher level in, MSNs or cortical projection neurons [4].
In the absence of enriched expression of a mutated protein, neuronal subtype vulnerability was assumed to arise from unique protein interactions. For example, in the study of another polyQ disease, spinocerebellar ataxia 7, the interaction between ataxin-7 and the homeobox protein CRX in the retina was reported to specifically lead to pathology [5]. In a follow-up study, the specific role of CRX was not confirmed [6]. To complicate matters further, the same group recently demonstrated that the interactions between a mutant polyQ protein, Ataxin-1, and 14-3-3epsilon differentially affects disease state in cerebellum and brainstem, both of which are vulnerable regions [7]. The regional distribution of the described huntingtin interacting proteins by and large does not explain MSN vulnerability [8–10]. One exception is the htt interacting protein RASD2/Rhes (Ras homologue enriched in striatum), which is striatal-specific. It is a small G-protein that mediates sumoylation of polyQ-htt, and thereby its neurotoxicity. Although RASD2-null mice exist, the impact on phenotype of a cross with HD models remains to be determined [11, 12].
The absence of protein localization to explain neuronal subtype-specific polyglutamine toxicity has led to a conundrum in the study of polyQ diseases, and non-polyQ neurodegenerative diseases as well (e.g., amyotrophic lateral sclerosis). For all of these, evidence of non-cell-autonomous mechanisms of disease is steadily increasing [13, 14]. Does the mutation in huntingtin act cell-autonomously to cause dysfunction and degeneration of MSNs, or does the mutation require non-cell-autonomous pathogenic cell-cell communication via cortical neurons and non-neuronal cells to induce MSN death? For HD, the pendulum has swung markedly since discovery of the gene, all the way to the notion that dysfunction in the striatum absolutely requires cell-cell interactions, particularly with the cortical projection neurons, and it cannot arise from expression of polyQ-htt in MSNs alone [13, 15–17]. Answers to the questions regarding the contribution of cell types and regions to each pathophysiologic mechanism are required for the development of targeted therapy. In actuality, there is strong evidence to support both cell-autonomous and non-cell-autonomous mechanisms and interactions of many cell types, combining to create MSN dysfunction and death.
What is Unique about the MSN?
Among neuronal subtypes, the MSN receives the most massive combination of glutamatergic input (from the cortex) and dopaminergic input (from the substantia nigra pars compacta). Hypothetically, therefore, the combined dopaminergic and glutamatergic input may result in the preferential vulnerability of MSNs to the toxicity of polyQ-htt, but the convergence, of course, does not identify specific pathophysiologic mechanisms. Although morphologically similar, there are 2 classes of MSNs, divided into direct and indirect pathways. MSNs in the direct pathway project to the dopaminergic neurons of the substantia nigra pars compacta and express dopamine D1 receptors and dynorphin. MSNs in the indirect pathway project to the globus pallidus and express dopamine D2 receptors and enkephalin. More than 95 % of MSNs express dopamine and cyclic AMP-regulated protein (32 kDa, also known as DARPP-32) [18]. In HD, MSNs in the indirect pathway are relatively more vulnerable than those in the direct pathway, and were believed to be the first to be dysfunctional. Recent data surprisingly indicate that the MSNs originating the direct pathway are dysfunctional much earlier in the course of HD than previously described [19]. It remains an enigma as to whether there is something unique about the MSN that increases its vulnerability, or something that is lacking, which is neuroprotective in other neurons [1].
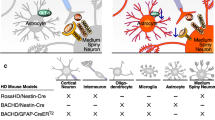
In vitro cell-type specific and in vivo regional “models” of HD serve to identify and distinguish between contributions of cell-autonomous and non-cell-autonomous mechanisms to abnormalities of the MSN. Cell-based systems include: 1) non-striatal neuronal-like cell lines, particularly PC12, expressing htt with various length polyQ expansions [20]; 2) immortalized striatal-like cell lines with pathogenic htt polyQ expansions [21, 22]; and 3) immortalized striatal-like cells derived from polyQ-htt knock-in mice [23]. In vivo models include (Table 1): 1) targeted intrastriatal injections of adenovirus or lentivirus expressing polyQ-htt [24]; and 2) genetically modified mice, using either a cell specific promoter directing expression of polyQ-htt [25], or ROSA26 or BACHD mice combined with cre/lox technology to regulate region-specific expression [15, 16, 26]. In the D9-N171-98Q mouse, we targeted expression of a polyQ-htt fragment (N171-98Q), restricted, within the forebrain, to striatal MSNs via 9Kb of ppp1r1b genomic elements (also known as D9) [25]. In the latter models, region-specific expression of Cre recombinase is used to either “turn on” an exon 1 fragment in the Rosa26 locus, or “turn off” a ubiquitously expressed BAC-97Q transgene containing loxP sites. To date, the majority of the studies addressing the question of cell-autonomous and non-cell-autonomous mechanisms of disease in HD have been largely restricted to fragment models, although preliminary results from a full-length model suggest similarities [26]. Also, complicating interpretation is the fact that the fragment length under study is variable, as are the CAG repeat number and level of transgene expression.
Evidence Supporting Intrinsic Vulnerability of MSNs in HD
Data in support of intrinsic, cell-autonomous vulnerability (“suicide”) may be also derived from pan-neuronal and pan-cellular mouse HD models. These data may be catalogued according to the model or the pathogenic mechanism to be analyzed. Major hypothesized pathogenic mechanisms in HD include abnormal aggregation (inclusions) and clearance of polyQ-htt, bioenergetic deficits, neurotrophin deficiency, transcriptional dysregulation, disorders of axonal transport, and excitotoxicity. Regional models have not been adequately studied yet; therefore, we will also suggest future investigative directions.
These data include:
-
1)
Transcriptional dysregulation induced in MSNs by full-length mutant htt or a fragment thereof (polyQ-htt) are to some extent exclusive to the MSN, even if the same transcripts are expressed outside the striatum (for more detail see Sugars and Rubinsztein [27], Desplats et al. [28]). For example, DARPP-32 is expressed in 97% of the MSNs, several cortical layers, and cerebellar Purkinje cells (PCs) [18, 29]. In the pan cellular R6/2 mice, however, its mRNA is down-regulated in MSNs to a greater degree than in the cortex [30]. In D9-N171-98Q mice, DARPP-32 is decreased only in the striatum, and not in PCs, where the transgene is also expressed. On the other hand, calbindin is decreased in PCs but not in the striatum [25].
-
2)
In several HD mouse models with ubiquitous transgene expression, inclusion formation and/or cell loss is preponderant in the striatum, and sometimes even MSN-subtype specific [31–33]. These data confirm that HD mouse models replicate the human pattern of neuronal disease. Relative to MSNs from nontransgenic or YAC18 mice, isolated MSNs from “knock-in” and YAC128 polyQ-htt mice demonstrate mitochondrial dysfunction, enhanced N-methyl-D-aspartate (NMDA) receptor signaling, and increased sensitivity to excitotoxicity [34–37]. A caveat, however, is that it is unknown whether these isolated neurons have already been compromised in vivo.
-
3)
In vivo, viral-mediated expression of polyQ-htt exclusively in the striatum results in reversible formation of inclusions, transcriptional dysregulation, and MSN death, while sparing interneurons [24, 38, 39]. Conversely, a decrease in striatal polyQ-htt expression mediated by viral siRNA largely corrects many of these deficits both in viral and transgenic HD models [40, 41].
-
4)
In vitro, expression of polyQ-htt in primary striatal neurons is more toxic than in primary cortical neurons, despite accumulation of aggregates in the cortical neurons [42].
-
5)
Emx1-cre-recombinase-mediated expression of Exon1-103Q htt (mixed CAA-CAG) in cortical projection neurons does not lead to striatal disease [15], and Dlx5/6-cre-recombinase-mediated expression of the same fragment in MSNs and a subset of cortical interneurons does not lead to a striatal motor phenotype. In the second model, MSNs develop inclusions and manifest neurophysiological abnormalities, but there is no cell death, and the mice do not develop motor abnormalities [16]. The level of transgene expression was not shown, but it was assumed to be at the same level as in the cortex of the ROSA-HD mouse crossed with Emx1-Cre. The authors concluded that MSN polyQ-htt expression is not sufficient to cause disease, but in fact, cell-autonomous abnormalities of the MSNs were demonstrated.
-
6)
As previously noted, we used DARPP-32 genomic fragments (also known as D9) to direct expression to the MSNs and exclude other forebrain neurons [43]. These mice (D9-N171-98Q) develop MSN inclusions, progressive motor abnormalities, failure to gain weight after 12 months of age, striatal transcriptional abnormalities, decreased striatal mitochondrial complex II activity, and in aged animals, resistance to quinolinic acid-induced excitotoxicity [5, 44, 45], discussed in detail as follows. Expression of the transgene in PCs in this model may contribute to the motor abnormality, but it does not contribute to the other strictly striatal abnormalities. In fact, major differences between the D9-N171-98Q mouse and the RosaHD-Dlx5/6-Cre mouse are PC expression in the D9-N171-98Q and cortical interneuron expression in the latter. Although the cerebellum is usually not a site of major neuropathology in HD, its involvement correlates with the length of the CAG repeat. Moreover, PCs are the primary site of neuropathology in the spinocerebellar ataxias, also caused by CAG repeat expansions. Expression of mutant spinocerebellar ataxia proteins in PCs can result in ataxia and PC degeneration. Microarray analysis of the cerebellum in a knock-in HD mouse reveals that the major alterations in mRNA levels between striatum and cerebellum are quantitative rather than qualitative [46]. Thus, it would be reasonable to determine the phenotype in mice with polyQ-Htt expression restricted to PCs.
-
7)
The expanded CAG repeat is unstable in somatic tissues in mouse HD models. This is particularly true if the mouse is created with a particularly long expansion (e.g., the R6 mouse [47]). Somatic instability may be greater in striatum than in other brain regions and peripheral tissues, and may occur with different dynamics [48, 49]. In another polyglutamine expansion disease (SCA1) somatic instability does not account for selective vulnerability of PCs, but is also more prominent in the striatum [50]. In the D9-N171-98Q mouse, instability is also more pronounced in the striatum than in the cerebellum and tail (Brown and Ehrlich, unpublished data), implicating cell-autonomous mechanisms.
Evidence Suggesting MSN Vulnerability Secondary to Cortical Disease or Other Non-Cell-Autonomous Interactions
Evidence in favor of extrinsic vulnerability ("murder") includes:
-
1)
In human HD, a greater number of cortical neurons contain nuclear inclusions (NIs) relative to striatal neurons [51], and are the first to develop inclusions in some mouse models of HD [52]. As will be discussed, however, inclusion formation and neuronal dysfunction do not always correlate.
-
2)
A major hypothesis regarding the preferential vulnerability of MSNs in HD revolves around increased sensitivity to excitotoxicity, caused by abnormalities in corticostriatal glutamate connectivity [53–58]. Vulnerability to excitotoxicity likely results from aberrations in both the pre-synaptic and post-synaptic compartments, and is therefore a result of cell-autonomous and non-cell-autonomous mechanisms.
-
3)
Similar to preferential transcriptional dysregulation in MSNs, GAD67 expression is down-regulated in the cortex, but is normal in the striatum in R6/2 HD mice [59]. The authors concluded that these data point to the presence of non-cell-autonomous mechanisms.
-
4)
Cortical brain-derived neurotrophic factor (BDNF) synthesis and its transport to the striatum are decreased in human HD and mouse models, likely due to impaired interaction of huntingtin associated protein-1 and p150Glued required for BDNF transport [60, 61]. Moreover, correction of this deficit in mice improves HD-like abnormalities, whereas an engineered decrease in cortical BDNF exacerbates onset of phenotype in mice [62, 63]. The deficit in axonal transport includes mitochondria [64]. Finally, a point mutation in the dynein heavy chain leads to prominent striatal neurodegeneration, implying that MSNs are particularly sensitive to deficits in retrograde axonal transport [65]. Axonal transport has not been studied in regional HD models.
-
5)
Astrocytes are responsible for the regulation of extracellular glutamate level and transport via the excitatory amino acid transporter 2. Glutamate uptake is abnormal in some mouse models of HD [66] and gliosis is a feature of human HD [3] and is observed in some HD mouse models. Transgenic mice with polyQ-htt restricted to glia as driven by the glial fibrillary acidic promoter, develop weight loss, motor abnormalities, and exhibit a shortened life span in the presence of decreased glutamate uptake [67]. When crossed with a primarily pan-neuronal transgenic model (prp-N171-82Q), the phenotype of the double transgenic is more severe than either single transgenic [68]. A caveat is that the glial fibrillary acidic promoter is not always glial-specific. Interestingly, gliosis does not develop in mice with expression of polyQ-htt restricted to MSNs in the forebrain [16, 25]. As MSNs may be more vulnerable to disorders of glutamate uptake than are other neurons, the next step would be to analyze the phenotype in mice with polyQ-htt expression restricted to striatal glia and MSNs. Perhaps, this could be achieved with a combination of viral and transgenic methodology.
-
6)
In addition to astrocytes, a recent study suggests that a decrease in the mitochondrial regulator PGC1α in oligodendrocytes contributes to the dysmyelination that exists in HD mouse models [69]. Interest in the contribution of inflammation to HD pathology has fostered the study of microglia and their receptors [13, 70, 71]. Thus, microglial activation may predict an onset of symptoms, but similar to aggregates, the question is unresolved as to whether microglial activation is destructive or protective [72, 73]. To date, expression of polyQ-htt directed specifically to, or deleted from, microglia has not been studied.
-
7)
Considerable effort has been expended to identify neurophysiologic abnormalities in MSNs secondary to polyQ-htt toxicity, particularly as they may be related to other pathogenic mechanisms, including excitotoxicity, bioenergetics failure, and transcriptional dysregulation. Isolated MSNs from several HD mouse models have an increased response to the glutamate agonist, NMDA, and a reduced sensitivity to Mg2+. These abnormalities are reproduced in the Rosa HD striatal Exon 1 model, implying a cell-autonomous mechanism. The implication of these data is that HD MSNs are activated at more hyperpolarized potentials than normal MSNs, and are therefore more susceptible to injury [16, 74–76]. The presence of an increase in membrane input resistance may also contribute to increased activity, including both an increase in rate of firing (frequency) and burst activity. These abnormalities have also been verified in mouse HD models in vivo [77, 78]. Importantly, the abnormalities evolve as the mouse goes from pre-symptomatic to symptomatic. MSNs from pre-symptomatic mice display increased excitatory postsynaptic currents (EPSCs), whereas MSNs from symptomatic mice display decreased EPSCs [79]. Neurophysiologic abnormalities also exist in cortical neurons, although in the opposite direction as in MSNs. In “cortex only” mice, in which interneurons do not express polyQ-htt, projection neurons do not display abnormalities of inhibitory postsynaptic currents (IPSCs). The authors concluded that within the cortex, cell-cell interactions between interneurons and projection neurons are required for the electrophysiological changes to evolve, and that cell-autonomous alterations do not occur in cortical projection neurons expressing polyQ-htt [15].
Similar neurophysiologic studies were performed in symptomatic D9-N171-98Q mice. The electrophysiological data did not show any decrease of EPSCs in the D9-N171-98Q mice suggesting that glutamate synaptic transmission is not altered in mice in which polyQ-htt expression is restricted to MSNs. IPSCs were not changed either, again in contrast to other studies showing an increase of GABA synaptic activity in several models of HD [79]. Thus, corticostriatal disconnection may not develop in symptomatic D9-N171-98Q mice. In contrast, D9-N171-98Q MSNs displayed increased input resistance and abnormal postsynaptic currents similar to pan-neuronal models of HD, suggesting that these are the changes that are cell autonomous [45]. As noted a major difference between the 2 striatal models is that the D9-N171-98Q mice do not express polyQ-htt in cortical interneurons, as do RosaHD-Dlx5/6-Cre mice. Thus, some of the MSN neurophysiologic alterations noted in the latter model may actually be non-cell-autonomous.
Pathogenic Mechanisms: Autonomous or Non-Cell-Autonomous or Both?
As it is clear from the summary of evidence supporting autonomous and non-cell-autonomous pathology in HD, there are multiple hypotheses as to the etiology of the neurotoxicity in HD. Another approach to synthesize the disparate views is to analyze each mechanism. Not all have been studied in cortical or striatal models in vivo, so in this section we will focus on those that have been analyzed in regional models. It will rapidly become apparent that several of these mechanisms include both autonomous and non-autonomous contributions and potentiate each other.
Protein Aggregation, Cleavage, and Degradation
Huntingtin NIs were identified in the first mouse HD model [52] and in human postmortem tissue [80]. Inclusions were also identified in cytoplasm [81]. Subsequently, multiple cellular and in vivo regional models have demonstrated the cell-autonomous nature of inclusion formation [15, 25, 34, 35]. Mice develop a greater number of NIs than do human HD patients, and the extent of inclusion formation does not correlate with symptoms or with polyQ-htt toxicity in vitro [82, 83]. Inclusion formation occurs later and in smaller numbers in full-length mouse models (e.g., for detail see Hodgson et al. [84]). Thus, the more rapid formation of NIs in fragment models may be due to the propensity to aggregation of N-terminal fragments [85]. In pan-neuronal and pan-cellular models, inclusions can form throughout the brain, but there may be preferential accumulation in striatal nuclei [86]. Aggregate accumulation does not occur in mice with phosphomimetic mutations of serines 13 and 16, in which disease symptoms are also abrogated [87]. The kinases and phosphatases modulating this phosphorylation have not been identified yet. Ser421 is also phosphorylated, and Metzler et al. [88] investigated its function using okadaic acid to inhibit protein phosphatases-1 (PP-1) and phosphatases-2a. Dysregulation of these phosphatases correlates with a reduced level of DARPP-32 and thereby, phospho-DARPP-32. Phospho-DARPP-32 is an inhibitor of PP-1, so the result is increased activity of PP-1, and consequently, decreased phosphorylation of Ser421. This apparent discrepancy between phospho- and dephospho- states may perhaps be resolved via analysis of phenotype following selective up- and down-regulation of DARPP-32 and kinases and their inhibitors in HD models. The phosphorylation state of DARPP-32 and its interacting kinases and phosphatases are regulated in response to neurotransmitters, including glutamate and dopamine [89]. Therefore, it is possible that changes in phosphorylation status are both cell-autonomous and non-cell-autonomous.
Regardless of how the aggregation is regulated, their contribution to toxicity remains highly controversial. Thus, the Rosa HD-Dlx5/6-Cre mouse develops extensive striatal inclusions, but no motor phenotype [16]. Likewise the “short-stop” model develops diffuse inclusions, also in the absence of a motor phenotype, and even the authors were surprised at the lack of phenotypic similarity to other fragment models [90]. As previously noted, full-length mouse HD models develop a phenotype in the absence of NIs. The pathogenic mechanism most strongly linked to polyQ-htt nuclear aggregation is transcriptional dysregulation (see as follows), which has not been studied in either of these models. Such an analysis could serve to answer this outstanding question. Finally, the status of autophagy and the role of ubiquitin-proteasome system and the clearance of aggregated polyQ-htt is of great interest. As these processes have not been examined in any regional HD model to date, we will not discuss them further.
Transcriptional Dysregulation
Three primary mechanisms are proposed for transcriptional dysfunction in HD:
-
1)
Direct interaction of polyQ-htt with transcription factors (TFs) in cytoplasm and nucleus and/or sequestration in NIs, thereby directly interfering with TF function and/or causing depletion of rate-limiting TFs [91–93].
-
2)
Repression of transcription secondary to histone hypoacetylation [20, 94–96].
-
3)
Direct decrease in cortical mRNAs, including Lin7b and anterogradely transported BDNF, leading to transneuronal striatal transcriptional dysregulation [60, 97, 98].
Regarding the first mechanism, polyQ-htt protein interacts with multiple transcription factors, both nuclear and cytoplasmic, and most of which are not cell-specific. These include the adenosine 3′,5′-cyclic monophosphate (cAMP) response element-binding protein (CREB)-binding protein, Specificity protein 1, TATA binding box protein, Bcl11b, repressor element-1 transcription factor/neuron restrictive silencer factor, and widely expressed components of the basal polymerase II transcriptional machinery [94, 99–103]. DARPP-32 is the most widely used marker of mature MSNs, and is highly down-regulated in human HD and in the majority of mouse models [44]. In fact, however, DARPP-32 transcription is not regulated by any of the TFs implicated in HD to date, including CREB [104]. Overall, the genome-wide transcriptome changes induced by polyQ-htt cannot yet be ascribed to a particular transcription factor regulatory network. Part of the difficulty in identifying such a network in the MSN is that little is known about transcriptional regulation of the mature MSN phenotype. As regional-specific transcriptional regulation is highly complex, it is certainly possible that more ubiquitously expressed TFs are involved in a combinatorial fashion [105]. We are successfully using the “D9” striatal regulatory sequences to identify conserved sequences and enhancer marks, followed by biotin-strepavidin column chromatography and mass spectrometry to identify TFs regulating MSN-specific expression (Keilani and Ehrlich, in press).
Microarray has been widely applied to mouse and cellular models of HD. To elucidate the extent of cell-autonomous transcriptional alterations in HD, we performed genome-wide expression profiling of striatal tissue from D9-N171-98Q transgenic mice. We demonstrated a relatively high degree of overlap of changes in gene expression in D9-N171-98Q transgenic mice and 2 transgenic pan-cellular R6 models, human caudate, and Htt N171 fragment-expressing cultured striatal neurons [44]. Eighty-nine of the 90 top-ranked expression differences present in D9-N171-98Q, R6/2-Q150, and R6/2-Q300 that displayed perfect concordance in directionality. Collectively, these data provide strong evidence that intrinsic effects of polyQ-htt are sufficient to result in transcriptional alterations in several pathways relevant to HD pathology. Conversely, these data suggest that expression of polyQ-htt in the cortex is not required for most of the transcriptional dysregulation present in the striatum of mouse models of HD. This conclusion is consistent with that of Runne et al. [106], who performed a similar analysis of mRNA expression in a cellular HD model. A caveat, however, is that the degree of down-regulation overall was less in D9-N171-98Q than in pan-neuronal models, suggesting that both mechanisms are instrumental in MSN transcriptional dysregulation. BDNF and TrkB levels are not altered in D9-N171-98Q mice, implying that a decrease in BDNF is not required for the majority of the observed changes in mRNA levels. The degree of dysregulation, however, may certainly be compounded by the decrease in BDNF noted in most pan-neuronal models, particularly because BDNF has been shown to directly regulate many of the same genes that are down-regulated in D9-N171-98Q [107, 108].
In D9-N171-98Q mice, dysregulated striatal-enriched genes include prodynorphin (Pdyn), protein phosphatase 1, regulatory (inhibitor) subunit 1B (Ppp1r1b, also known as DARPP-32), A2a adenosine receptor (Adora2a), the dopamine D2 receptor (Drd2) and proenkephalin (Penk), all of which were validated by quantitative real time polymerase chain reaction (qPCR). These data are consistent with findings from human subjects with HD [109, 110] and other HD transgenic mouse and cell culture models [28, 111], including a pan-neuronal mouse expressing a similar polyQ-htt fragment (N171-82Q) [112, 113].
It is critically important to determine the cellular localization of expression of genes of interest, changes that may be pathogenic or protective. Whereas changes in gene expression in pan-neuronal models also included decreases in many striatal-enriched, interneuron-selective genes (e.g., Sst, Npy, Pvalb, Cck, and Nos1), parallel changes were absent from D9-N171-98Q striata, but with 1 exception. Thus, the gene expression changes in interneurons also appear to be cell-autonomous, yet these neurons are mostly spared in HD. The exception is level of Pvalb, which is usually decreased; however, in the D9-N171-98Q model, Pvalb expression is increased. Striatal GABAergic-parvalbumin interneurons receive direct, convergent cortical input [114] and are highly sensitive to excitotoxicity, including late in the course of HD [115]. PGC1α mRNA is also paradoxically increased in D9-N171-98Q, and may be driving the Pvalb gene [116]. The recent identification of a phenotype in a “glial-only” polyQ-htt mouse calls attention to the fact that the NF-E2-related factor-2 (Nrf2)/antioxidant response element-mediated protective pathway is largely localized to astrocytes [117]. This pathway appears to protect neurons from oxidative stress in HD mouse models [118], highlighting the importance of mRNA cellular localization in all HD mouse models. Notably, there is an absence of alterations in most dopamine pathway-related genes in the D9-N171-98Q mouse. Assuming these data correlate with the integrity of the nigrostriatal system, they could account for the less severe motor phenotype of the D9-N171-98Q mouse compared with the prp-N171-82Q model [25]. Our underlying assumption is that the changes noted in the D9-N171-98Q mouse striatum are cell autonomous, but can simultaneously be compensating for cell-intrinsic abnormalities [119, 120]. The converse is also true (i.e., compensatory pathways may serve to normalize primary transcriptional alterations). Likewise, in pan-neuronal models (e.g., R6/2), transcriptome alterations that may occur on a cell-autonomous basis may be “masked” by opposing, non-cell-autonomous effects of the mutation on the MSN, resulting in gene alterations that are unique to D9-N171-98Q transgenic mice.
Chromatin acetylation status has not been examined in regional HD models. Transcription factors act, in a large part, by binding to DNA, either directly or as part of a protein complex, followed by recruitment of histone-modifying enzymes that alter chromatin structure and may either induce or repress transcription. Some of the TFs with decreased activity in HD have intrinsic histone acetyltransferase activity (e.g., CREB-binding protein (CBP)). Histone deacetylases (HDACs) are hyperactive in HD, leading to hypoacetylation of histones [95, 121]. Because acetylation usually leads to activation of transcription, hypoacetylation leads to transcriptional repression. Specifically, histones H3/H4 acetylation and/or levels are repressed in HD [94, 122, 123] and HDAC inhibitors, including phenylbutyrate, sodium butyrate, trichostatin A, suberoylanilide hydroxamic acid, and HDACi4b, partially ameliorate polyglutamine toxicity in vitro and in vivo [94, 124–127]. There is a progressive decrease in histone H3 acetylation associated with promoters down-regulated in HD (e.g., the D2 dopamine receptor promoter [123]), but these sequences have not been demonstrated to contribute to striatal-specific expression. For example, the physiologic relevance of selective down-regulation of the DARPP-32 core promoter in striatal, relative to renal, extracts [128] must be cautiously interpreted as this region of the ppp1r1b gene does not direct expression to the striatum and may actually contain repressive elements [129].
The pathogenic nature of only a few of the dysregulated transcripts has been investigated, and it remains possible that many of these alterations may be an epiphenomenon. Down-regulation of the mitochondrial regulator PCG1α results in decreased levels of its targets. Crossing an HD mouse with a PGC1α-null mouse leads to a more severe HD phenotype, whereas increasing PGC1α in the striatum improves phenotype [130, 131]. Importantly, in the mouse model used in these studies, PGC1α was measured in neuronal subtypes, and was decreased in MSNs, but was increased in resistant iNOS interneurons. Overexpression of CBP can rescue HT22 cells polyglutamine toxicity [93], but this has not been attempted in vivo. The decrease in DARPP-32 is hypothetically pathogenic, but it has also not been experimentally tested, as is true for all of the striatal-enriched mRNAs.
Mitochondrial/Energy Dysfunction
Before the creation of transgenic and knockin HD mouse models, chemical insults to the striatum served as useful disease models, and in fact, were the first examples of regional models. Early HD models that recapitulated prominent behavioral and neuropathological features of HD included intra-striatal injection of N-methyl-D-aspartate receptor (NMDAR) agonists (e.g., quinolinic acid [QA]) [76, 132–135], or an irreversible inhibitor of mitochondrial succinate dehydrogenase, 3-nitropropionic acid [136]. The selective degeneration of the striatum in both models directly contributed to the hypotheses that mitochondrial dysfunction and increased vulnerability to excitotoxicity are key pathophysiologic mechanisms contributing to MSN neurodegeneration in HD.
Mitochondrial dysfunction is present in HD patients and in a variety of HD cell and mouse models [45, 137–142]. Although no complex I deficiency was found in the frontal and parietal cortices or the cerebellum of HD patient [143], other clinical studies found down-regulated mRNA levels of 12 subunits of complex I in the brain [130] and decreased complex I activity in platelets and muscle tissues of HD patients [144, 145]. A significant reduction in complex II-III activity was identified in both HD caudate and putamen, and decreased complex IV activity was also observed in HD putamen [143]. Recently, fission and fusion mitochondrial proteins were found to be abnormal in postmortem HD cortex and striatum and polyQ-htt oligomers were localized to the mitochondria, suggesting a cell-autonomous mechanism [146].
The D9-N171-98Q model is the sole regional model in which mitochondrial function has been assayed in vivo [45]. Using a polarographic method, we found that complex I activities were not altered in the striatum of pre-symptomatic and symptomatic D9-N171-98Q mice relative to nontransgenic littermates. States 3 and 4 activities of complex II were significantly reduced in pre-symptomatic D9-N171-98Q mice compared to their age-matched controls. Not surprisingly, state 4 respiration of complex II was significantly reduced in old nontransgenic mice relative to their younger counterparts. Conversely and surprisingly, it was significantly enhanced in symptomatic D9-N171-98Q mice compared to their age-matched controls. Increases in state 3 and carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone (FCCP)-induced maximum respiration were also present in symptomatic D9-N171-98Q mice compared to pre-symptomatic D9-N171-98Q mice. No alterations in complex IV activities were observed in any of the mice. ATP-synthase activities of complex V in symptomatic D9-N171-98Q mice were found to be as efficient as in age-matched controls. Thus, striatal-specific expression of polyQ-htt affected complex II activities of mitochondrial respiration in an age-dependent manner, but did not alter the activities of the other complexes. It is possible that the increased activity in the symptomatic mice is compensatory. Mitochondrial activity assays in R6/2 mice are also biphasic, but further studies are required.
Mitochondrial dysfunction may arise from intrinsic abnormalities of transcription (e.g., a decrease in PGC1α, Ca+2 overload, NMDAR dysfunction, and/or abnormal dopaminergic activity. In primary striatal cultures, dopamine regulates mitochondrial complex II catalytic activity and regulates vulnerability of polyQ-htt-expressing neurons to cell death [147]. As the D9-N171-98Q mouse does not express polyQ-htt in dopaminergic neurons, thereby further differentiating between etiology and impact of complex II abnormalities. Assay of mitochondrial function in a “cortical” model will enable us to distinguish between striatal and cortical contributions to mitochondrial dysfunction in HD.
Excitotoxicity
Intrastriatal injection of an NMDA agonist, particularly quinolinic acid, has served as a model of HD for almost 2 decades [132]. The vulnerability to excitotoxicity, and its origins, has been investigated in primary neurons derived from HD mouse models and 6 pan-cellular or pan-neuronal HD mouse models [148]. Pre-symptomatic YAC128 and R6/1 HD mice exhibit increased vulnerability to QA-induced excitotoxicity and enhanced NMDA responses; conversely, symptomatic, older mice became resistant to an excitotoxic insult [40, 76, 149]. The electrophysiological data in support of enhanced NMDAR activity in early HD has been discussed [40, 74, 150]. NMDA-mediated, and specifically NR2B-dependent, excitotoxicity exacerbates striatal neuronal degeneration in an NR2B-overexpressing HD mouse model [151]. NMDAR is linked to 2 opposing signaling pathways for either cell survival or death, depending on receptor localization. Mislocalization of NMDAR to the peri- and extra-synaptic membrane (non-postsynaptic density fraction) in pre-symptomatic YAC128 striatum leads to a loss of neuroprotection mechanisms and an increase in activity of cell death pathways [58, 152, 153]. In parallel, excitotoxicity triggered by over-activation of NR2B-containing NMDARs results in rapid dissociation of synaptic NMDARs from the postsynaptic scaffolding complex and downstream survival signaling [154]. Thus, 1 possible scenario is that excitotoxic damage occurs in striatal MSNs in the early stage of the disease by dysregulation of NMDAR-linked signaling pathways, which then leads to a cascade of age-dependent pathophysiological events. To examine the contribution of intrinsic, cell-autonomous vulnerability of MSNs to excitotoxicity, we measured the number of degenerating neurons after injection of QA in the striatum of pre-symptomatic and symptomatic D9-N171-98Q transgenic mice [25].
Fluoro Jade-C (FJ) positive neurons were counted after intrastriatal injection of 15 nmol QA in young (pre-symptomatic) and old (symptomatic) D9-N171-98Q mice and age-matched controls. There was no difference in the number of degenerating neurons between young D9-N171-98Q and nontransgenics. Although there was a trend to a greater number of degenerating neurons in young D9-N171-98Q mice (p = 0.09), it was statistically weakened as we increased the number of animals in both groups. These data suggest that non-cell-autonomous factors are responsible for much of the increased sensitivity to excitotoxicity observed at the pre-symptomatic stages in HD pan-cellular and pan-neuronal transgenic models [40, 76, 149]. Our data further imply that cell-autonomous abnormalities are sufficient to cause mitochondrial complex II dysfunction in the absence of a decrease in PGC1α, but that this decrease in complex II function in pre-symptomatic mice is insufficient to increase vulnerability to excitotoxicity.
Contrary to the pre-symptomatic assay and consistent with previous studies in rats and FVB mice [76, 155, 156], both D9-N171-98Q and nontransgenic C57BL/6J mice show a dramatic age-dependent decrease in sensitivity to QA, exhibiting a 10-fold and 2-fold decrease in FJ-positive neurons, respectively, compared to their younger counterparts. Symptomatic D9-N171-98Q mice showed greater resistance to excitotoxicity relative to pre-symptomatic D9-N171-98Q mice (p = 0.0001) and also relative to age-matched nontransgenic mice (p = 0.027), similar to data from pan-cellular HD models [76, 149, 157]. These data imply that striatal-specific expression of polyQ-htt is sufficient to induce resistance to excitotoxicity at the symptomatic stage. The stage-dependent move to resistance to excitotoxicity in the YAC128 model is hypothesized to reflect a corticostriatal disconnection due to cumulative deficits in pre- and post-synaptic elements of glutamate transmission [76]). Thus, going forward, it is critical to identify what mediates cell-autonomous resistance to excitotoxicity in the symptomatic D9-N171-98Q mouse to determine if it can be exploited to decrease sequelae of excitotoxicity in the presymptomatic mouse.
Excitotoxicity, Bioenergetics, and Transcriptional Dysregulation: Are They Related?
It would be particularly satisfying if the pathophysiological mechanisms in HD were intertwined to the extent that correction of 1 abnormality could mitigate more than one detrimental process. As previously discussed, a key connection between transcriptional dysregulation and mitochondrial dysfunction has been identified, initiated with the decreased transcription of PCG1α. The first step in the cascade is hypothesized to occur in the nucleus with decreased expression of PGC1α. This decrease leads to disrupted mitochondrial biogenesis and function, primarily manifested in complex II, and decreased ability to defend against oxidative injury, ultimately leading to vulnerability to excitotoxicity. Mitochondrial release of pro-apoptotic caspases and calcium homeostasis can also be impacted, leading to further vulnerability to oxidative stress (for more detail see Giacomello et al. [158]). Long before it was known that PGC1α is down-regulated in HD, Beal et al. [159] observed that decortication or an NMDAR antagonist ameliorates the striatal lesion induced by malonate, a reversible inhibitor of succinate dehydrogenase. Another interesting observation is that mitochondrial calcium homeostasis, similar to transcriptional dysregulation, can be improved by HDAC inhibitors, which alter both cytoplasmic and nuclear HDAC activity [160].
Striatal vulnerability to excitotoxicity is hypothesized to arise from an increased in extra-synaptic NMDAR (Ex-NMDAR) relative to synaptic NMDAR [161]. Potential mediators include decreased synaptic NMDAR interactions with scaffolding proteins, e.g., membrane-associated guanylate kinases (MAGUKs), or enhanced protein tyrosine phosphatase activation, which dephosphorylates and destabilizes synaptic NMDARs. Increased extrasynaptic Glu-NR2B-PSD-95 binding or protein post-translational modifications may facilitate enhanced Ex-NMDAR stability. Elevated Ex-NMDAR activation facilitates activation of apoptotic signaling and decreased activation of cell survival pathways, such as those involving the pro-survival transcription factor CREB. Increased Ex-NMDAR signaling also facilitates decreased formation of neuroprotective polyQ-htt inclusion bodies, neuronal dysfunction and progressive neurodegeneration [58, 153].
Studies in the D9-N171-98Q mouse may help determine whether the aberrant synaptic localization of NMDAR in HD is regulated in a cell-autonomous manner. Isolated striatal neurons from YAC128 mice are more vulnerable to excitotoxicity in vitro than nontransgenic striatal neurons [40]. Extrapolating, those data would predict that pre-symptomatic D9-N171-98Q mice should be hypersensitive to excitotoxicity. This discrepancy could be explained if similar in vitro experiments were performed with isolated MSNs from the D9-N171-98Q mouse and from a full-length “striatal only” model, and with the analysis of NMDAR subcellular localization in D9-N171-98Q striata.
Dopamine, Glutamate, and Excitotoxicity
Glutamatergic and dopaminergic afferents modulate each other’s activity while converging on MSN dendritic spines, and GABA release from the MSNs further impacts both glutamate and dopamine release [162, 163]. Reciprocal interactions between dopamine and glutamate occur in other forebrain regions compromised in HD, particularly the prefrontal cortex. There is a growing consensus that abnormalities of the nigrostriatal dopamine (DA) pathway contribute to striatal dysfunction and neuronal death in HD, although controversy remains as to whether D1 and D2 receptor transmission equally contribute [164, 165]. Studies in wild-type and polyQ-htt striatal neurons demonstrate that dopamine potentiates glutamate excitotoxicity [164, 166], which could be mediated by the regulation of glutamate receptor cell surface localization by dopamine [167]. Dopamine (DA) also potentiates glutamate-induced calcium signaling in MSNs, potentially further impacting vulnerability to excitotoxicity [164], particularly in the presence of mitochondrial dysfunction. Finally, dopamine D1 receptor agonists are able to induce TrkB phosphorylation and its downstream signal transduction pathways [168]. Thus, the vulnerability to excitotoxicity in the D9-N171-98Q line may be decreased relative to pan-neuronal lines because polyQ-htt is not expressed in dopaminergic neurons.
BDNF Deficit
The cortex is the source of 80 to 90 % plus of the BDNF in the striatum, to where it is anterogradely transported. Anterograde transport of BDNF was first demonstrated by decortication in the adult followed by loss of BDNF protein in the striatum. Striatal cell loss was not evident in these animals, but they were examined at relatively brief times after decortication. MSNs express high levels of TrkB, the major receptor for BDNF [62, 169–172]. Transcriptional dysregulation in human HD patients and mouse HD models includes down-regulation of BDNF in the corticostriatal projection neurons [17, 60, 61, 173], and down-regulation of striatal TrkB [174, 175]. The decrease of BDNF may represent a loss of normal htt function, as wtHtt augments transcription of the BDNF gene [60]. The amount of BDNF reaching the striatum is further reduced secondary to axonal transport deficits [61]. We, along with others, have demonstrated the important role for BDNF in the maturation of the MSN, and there is also a requirement for BDNF in the maintenance of adult striatum. Contrary to what is frequently written, MSNs probably do not require BDNF for survival [107, 108]. In the study by Baquet et al. [176], in which cortical BDNF is prenatally knocked-out, volume loss is complete by P35, but striatal neuron loss does not occur until after 12 months of age. More recently, Rauskolb et al. [177] used a conditional forebrain BDNF deletion to demonstrate that the main effect was on dendritic and spine growth, and not survival. In addition, we have demonstrated that BDNF developmentally regulates transcription of several of the molecules down-regulated in HD, including DARPP-32 [107, 108]. Moreover, as previously noted, the microarray profile of conditional cortical BDNF-knockout mice resembles the human HD profile more than some HD mouse models [97]. This latter study used Emx1-Cre to conditionally delete BDNF in the cortex, and recombination occurs as early as embryonic day 11, long before BDNF is down-regulated in HD. Regulation of transcription by BDNF requires signal transduction systems altered in HD (e.g., phosphatidylinositol-3-kinase and cdk5) [43, 178]. Increased BDNF, either by intraventricular injection or prenatal transgenic overexpression, ameliorates the severity of the phenotype in R6 and YAC128 mice, whereas crossing with BDNF heterozygote-null mice exacerbates the phenotype [62, 63, 179].
In the D9-N171-98Q mouse, levels of cortical BDNF mRNA and striatal TrkB mRNA are equal to nontransgenic littermates, and absence of polyQ-htt in cortical neurons precludes its interference with BDNF transport [25]. Nevertheless, decreased BDNF is almost certainly directly detrimental and has been hypothesized to increase vulnerability of HD MSNs to other insults (e.g., excitotoxicity) [60, 62]. Thus, similar to the presence of an intact nigrostriatal dopamine system, the presence of normal BDNF and TrkB levels may mitigate the D9-N171-98Q phenotype.
How Might Abnormal BDNF/TrkB Neurotransmission Intersect with Other Pathogenic Mechanisms in HD?
Far more attention has been given to the role of BDNF in HD, than to its major striatal receptor, TrkB. MSNs in both the direct and indirect pathways express TrkB, and interneurons express even higher levels of TrkB [180]. There are 3 systems in which BDNF/TrkB neurotransmission has been linked to excitotoxicity. These include cortical neurons in vitro, motor neurons in vitro, and HD R6/1 mice on a BDNF heterozygous-null background in vivo. In all 3, TrkB activation increases sensitivity to excitotoxic insults:
-
1)
NMDA toxicity is increased in cortical neurons cultured in the presence of BDNF [181].
-
2)
Motor neurons cultured in BDNF are also more sensitive to excitotoxicity. This process requires transcription and translation, and can be inhibited by blocking activation of TrkB [182–184]. The effects of TrkB antagonism on rescue from excitotoxicity are attributed to inhibition of downstream signaling cascades from Src-family kinases, mitogen-activated protein kinase, and particularly phosphatidylinositol-3-kinase. The latter 2 pathways are strongly activated in striatal neurons following treatment with BDNF [104] and are likely protective in HD. Also relevant to HD, adenosine A2a receptors transactivate TrkB, and their blockade reduces sensitivity to excitotoxicity in several models, which thereby also implicate TrkB activation [184–186].
-
3)
R6/1;BDNF+/- mice have 50 % of the normal BDNF level, and are resistant to QA injection relative to R6/1;BDNF+/+ mice, at the pre-symptomatic stage [187, 188]. The resistance was ascribed to decreased expression of NMDAR scaffolding proteins and a reduced level of αCaMKII and calcineurin. Thus, the BDNF/TrkB system may exacerbate vulnerability to excitotoxicity in the pre-symptomatic stage, while protecting from other insults. Therefore, it is possible that some of the inconsistencies in the excitotoxicity hypothesis may be dependent on the status of the BDNF/TrkB system at the time of testing, as well as the age of the mouse [157]. Vulnerability to excitotoxicity has not been examined in pre-symptomatic HD mice with overexpression of BDNF, and conversely, has not been assayed in pre- or post-symptomatic HD mice with either over- or under-expression of striatal TrkB. These studies would be most informative if performed in pan-neuronal and regional models.
Conclusion
There has been enormous progress in the identification of pathophysiological mechanisms in HD, and it is now recognized that multiple neuronal subtypes and non-neuronal cells contribute to disease. At this time, it appears that polyQ-htt in neither the cortex nor the striatum is sufficient alone to cause a severe mouse HD phenotype, and that interaction with each other and non-neuronal cells further complicate the development of disease. Several in vitro and in vivo models provide evidence for “striatal-only” pathophysiology, but further study of the available models and the development of additional cell-selective and regional-selective models, with different combinations, is required to determine if regional-specific therapy will be efficacious. Within each region, it will be critical to determine the temporal relationship between pathways and disease stage to efficiently time the delivery of targeted therapeutics.
References
Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993;72:971-983.
Han I, You Y, Kordower JH, Brady ST, Morfini GA. Differential vulnerability of neurons in Huntington's disease: the role of cell type-specific features. J Neurochem 2010;113:1073-1091.
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 1985;44:559-577.
Trottier Y, Devys D, Imbert G, Saudou F, An I, Lutz Y, et al. Cellular localization of the Huntington’s disease protein and discrimination of the normal and mutated form. Nat Genet 1995;10:104-110.
La Spada AR, Fu YH, Sopher BL, et al. Polyglutamine¬expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 2001;31: 913-927.
Yoo SY, Pennesi ME, Weeber EJ, et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 2003;37:383-401
Jafar-Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploin sufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci USA 2011;108:2142-2147.
Harjes P, Wanker EE. The hunt for huntingtin function; interaction partners tell many different stories. Trends Biochem Sci 2003;28:425-433.
Marcora E, Gowan K, Lee JE. Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2. Proc Natl Acad Sci USA 2003;100:9578-9583.
Yanai A, Huang K, Kang R, et al. Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 2006;9:824-831.
Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009;324:1327-1330.
Subramaniam S, Snyder SH. Huntington's disease is a disorder of the corpus striatum: focus on Rhes (Ras homologue enriched in the striatum). Neuropharmacology 2011;60:1187-1192.
Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 2009;187:761-772.
Sambataro F, Pennuto M. Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog Neurobiol 2011. doi:10.1016/j.pneurobio.2011.10.003.
Gu X, Li C, Wei W, et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron 2005;46:433-444.
Gu X, Andre VM, Cepeda C, et al. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener 2007;2:8.
Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron 2011;69:423-435.
Ouimet CC, Langley-Guillon KC, Greengard P. Quantitative immunocytochemistry of DARPP-32-expressing neurons in the rat caudatoputamen. Brain Res 1998;808:8-12.
Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience 2011;198:252-273.
Igarashi S, Morita H, Bennett KM, et al. Inducible PC12 cell model of Huntington’s disease shows toxicity and decreased histone acetylation. Neuroreport 2003;14:565-568.
Ehrlich ME, Conti L, Toselli M, et al. ST14A cells have properties of a medium-size spiny neuron. Exp Neurol 2001;167:215-226.
Sipione S, Rigamonti D, Valenza M, et al. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 2002;11:1953-1965.
Trettel F, Rigamonti D, Hilditch-Maguire P, et al. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000;9:2799-2809.
Ruiz M, Déglon N.Viral-mediated overexpression of mutant huntingtin to model HD in various species. Neurobiol Dis 2011. doi:10.1016/j.nbd.2011.08.023.
Brown TB, Bogush AI, Ehrlich ME. Neocortical expression of mutant huntingtin is not required for alterations in striatal gene expression or motor dysfunction in a transgenic mouse. Hum Mol Genet 2008;17:3095-3104.
Cantle JP, Wang N, Gray M, et al. Genetic and molecular analyses of non-cell-autonomous disease pathogenesis in a conditional BAC transgenic mouse model of Huntington’s disease (BACHD). SFN Annual Meeting 2011:148.21.
Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet 2003;19:233-238.
Desplats PA, Kass KE, Gilmartin T, et al. Selective deficits in the expression of striatal-enriched mRNAs in Huntington’s disease. J Neurochem 2006;96:743-757.
Ouimet CC, Miller PE, Hemmings HC Jr, Walaas SI, Greengard P. DARPP-32, a dopamine- and adenosine 3’: 5’-monophosphate- regulated phosphoprotein enriched in dopamine-innervated brain regions. III. Immunocytochemical localization. J Neurosci 1984;4:111-124.
Bibb JA, Yan Z, Svenningsson P, et al. Severe deficiencies in dopamine signaling in presymptomatic Huntington‘s disease mice. Proc Natl Acad Sci USA 2000;97:6809-6814.
Menalled LB, Sison JD, Wu Y, et al. Early motor dysfunction and striosomal distribution of huntingtin microaggregates in Huntington's disease knock-in mice. J Neurosci 2002;22:8266-8276.
Van Raamsdonk JM, Pearson J, Rogers DA, et al. Loss of wild-type huntingtin influences motor dysfunction and survival in the YAC128 mouse model of Huntington disease. Hum Mol Genet 2005;14:1379-1392.
Tallaksen-Greene SJ, Crouse AB, Hunter JM, Detloff PJ, Albin RL. Neuronal intranuclear inclusions and neuropil aggregates in HdhCAG(150) knockin mice. Neuroscience 2005;131:843-852.
Zeron MM, Hansson O, Chen N, et al. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 2002;33:849-860.
Gines S, Ivanova E, Seong IS, Saura CA, MacDonald ME. Enhanced Akt signaling is an early pro-survival response that reflects N-Methyl-D-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem 2003;278:50514-50522.
Gines S, Seong IS, Fossale E, et al. Specific progressive cAMP reduction implicates energy deficit in presymptomatic Huntington’s disease knock-in mice. Hum Mol Genet 2003;12:497-508.
Cowan CM, Fan MM, Fan J, et al. Polyglutamine-modulated striatal calpain activity in YAC transgenic huntington disease mouse model: impact on NMDA receptor function and toxicity. J Neurosci 2008;28:12725-12735.
de Almeida LP, Ross CA, Zala D, Aebischer P, Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglytamine repeat size, huntingtin expression levels, and protein length. J Neurosci 2002;22:3473-3483.
Regulier E, Trottier Y, Perrin V, Aebischer P, Deglon N. Early and reversible neuropathology induced by tetracycline-regulated lentiviral overexpression of mutant huntingtin in rat striatum. Hum Mol Genet 2003;12:2827-2836.
DiFiglia M, Sena-Esteves M, Chase K, et al. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci USA 2007;104:17204-17209.
Drouet V, Perrin V, Hassig R, et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann Neurol 2009;65:276-285.
Zala D, Benchoua A, Brouillet E, et al. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis 2005;20:785-798.
Bogush AI, McCarthy LE, Tian C, et al. DARPP-32 genomic fragments drive Cre expression in postnatal striatum. Genesis 2005;42:37-46.
Thomas EA, Coppola G, Tang B, et al. In vivo cell-autonomous transcriptional abnormalities revealed in mice expressing mutant huntingtin in striatal but not cortical neurons. Hum Mol Genetics 2011;20:1049-1060.
Kim SH, Thomas CA, André VM, et al. Forebrain striatal-specific expression of mutant huntingtin protein in vivo induces cell-autonomous age-dependent alterations in sensitivity to excitotoxicity and mitochondrial function. ASN Neuro 2011;3:e00060.
Fossale E, Seong IS, Coser KR, et al. Differential effects of the Huntington's disease CAG mutation in striatum and cerebellum are quantitative not qualitative. Hum Mol Genet 2011;20:4258-4267.
Mangiarini L, Sathasivam K, Mahal A, Mott R, Seller M, Bates GP. Instability of highly expanded CAG repeats in mice transgenic for the Huntington's disease mutation. Nat Genet 1997;15:197-200.
Goula AV, Berquist BR, Wilson DM 3 rd, Wheeler VC, Trottier Y, Merienne K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington's disease transgenic mice. PLoS Genet 2009;5:e1000749.
Lee JM, Pinto RM, Gillis T, St Claire JC, Wheeler VC. Quantification of age-dependent somatic CAG repeat instability in Hdh CAG knock-in mice reveals different expansion dynamics in striatum and liver. PLoS One 2011;6:e23647.
Watase K, Venken KJ, Sun Y, Orr HT, Zoghbi HY. Regional differences of somatic CAG repeat instability do not account for selective neuronal vulnerability in a knock-in mouse model of SCA1. Hum Mol Genet 2003;12:2789-2795.
Sieradzan KA Mann DM. The selective vulnerability of nerve cells in Huntington’s disease. Neuropathol Appl Neurobiol 2001;27:1-21.
Davies SW, Turmaine M, Cozens BA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997;90:537-548.
Beal MF. Huntington’s disease, energy, and excitotoxicity. Neurobiol Aging 1994;15:275-276.
Levine MS, Klapstein GJ, Koppel A, et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res 1999;58:515-532.
Grunewald T, Beal MF. Bioenergetics in Huntington’s disease. Ann NY Acad Sci 1999;893:203-213.
Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med 2003;3:65-94.
Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington’s disease. Curr Top Dev Biol 2006;75:25-71.
Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington’s disease. Trends Neurosci 2010;33:513-523.
Gourfinkel-An I, Parain K, Hartmann A, et al. Changes in GAD67 mRNA expression evidenced by in situ hybridization in the brain of R6/2 transgenic mice. J Neurochem 2003; 86:1369-1378.
Zuccato C, Ciammola A, Rigamonti D, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 2001;293:493-498.
Gauthier LR, Charrin BC, Borrell-Pages M, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004;118:127-138.
Canals JM, Pineda JR, Torres-Peraza JF, et al. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J Neurosci 2004;24:7727-7739.
Xie Y, Hayden MR, Xu B. BDNF overexpression in the forebrain rescues Huntington’s disease phenotypes in YAC128 mice. J Neurosci 2010;30:14708-14718.
Trushina E, Dyer RB, Badger JD 2nd, et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 2004;24:8195-8209.
Braunstein KE, Eschbach J, Ròna-Vörös K, et al. A point mutation in the dynein heavy chain gene leads to striatal atrophy and compromises neurite outgrowth of striatal neurons. Hum Mol Genet 2010;19:4385-4398.
Liévens JC, Woodman B, Mahal A, et al. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 2001;8:807-821.
Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci USA 2009;106:22480-22485.
Bradford J, Shin JY, Roberts M, et al. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem 2010;285:10653-10661.
Xiang Z, Valenza M, Cui L, et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington's disease. J Neurosci 2011;31:9544-9553.
Lobsiger CS, Cleveland DW. Glial cells as intrinsic components of non-cell-autonomous neurodegenerative disease. Nat Neurosci 2007;10:1355-1360.
Palazuelos J, Aguado T, Pazos MR, et al. Microglial CB2 cannabinoid receptors are neuroprotective in Huntington's disease excitotoxicity. Brain 2009;132:3152-3164.
Politis M, Pavese N, Tai YF, et al. Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp 2011;32:258-270.
Kraft AD, Kaltenbach LS, Lo DC, Harry GJ. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol Aging 2012;33:621.e17-33.
Cepeda C, Itri JN, Flores-Hernández J, Hurst RS, Calvert CR, Levine MS. Differential sensitivity of medium- and large-sized striatal neurons to NMDA but not kainate receptor activation in the rat. Eur J Neurosci 2001;14:1577-1589.
Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 2006;137:327-336.
Graham RK, Pouladi MA, Joshi P, et al. Differential susceptibility to excitotoxic stress in YAC128 mouse models of Huntington disease between initiation and progression of disease. J Neurosci 2009;29:2193-2204.
Ariano MA, Wagle N, Grissell AE. Neuronal vulnerability in mouse models of Huntington's disease: membrane channel protein changes. J Neurosci Res 2005;80:634-645.
Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience 2006;137:327-336.
Cepeda C, Cummings DM, André VM, Holley SM, Levine MS. Genetic mouse models of Huntington’s disease: focus on electrophysiological mechanisms. ASN Neuro 2010;2:e00033.
DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997;277:1990-1993.
Hackam AS, Singaraja R, Zhang T, Gan L, Hayden MR. In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington's disease. Hum Mol Genet 1999;8:25-33.
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004;431:805-810.
Perrin V, Dufour N, Raoul C, et al. Implication of the JNK pathway in a rat model of Huntington's disease. Exp Neurol 2009;215:191-200.
Hodgson JG, Agopyan N, Gutekunst CA, et al. A YAC mouse model for Huntington‘s disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron 1999;23:181-192.
Schilling G, Becher MW, Sharp AH, et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet 1999;8:397-407. [Erratum in: Hum Mol Genet 1999;8:943].
Havel LS, Wang CE, Wade B, Huang B, Li S, Li XJ. Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum Mol Genet 2011;20:1424-1437.
Gu X, Greiner ER, Mishra R, et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009;64:828-840.
Metzler M, Gan L, Mazarei G, et al. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J Neurosci 2010;30:14318-14329.
Fienberg AA, Hiroi N, Mermelstein PG, et al. DARPP-32:regulator of the efficacy of dopaminergic neurotransmission. Science 1998;281:838-843.
Slow EJ, Graham RK, Osmand AP, et al. Absence of behavioral abnormalities and neurodegeneration in vivo despite widespread neuronal huntingtin inclusions. Proc Natl Acad Sci U S A 2005;102:11402-11407.
Nucifora FC Jr, Sasaki M, Peters MF, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001;291:2423-2428.
Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell 2005;123:1241-1253.
Jiang H, Poirier MA, Liang Y, et al. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol Dis 2006;23:543-551.
Steffan JS, Bodai L, Pallos J, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001;413:739-743.
Sadri-Vakili G, Cha JH. Mechanisms of disease: Histone modifications in Huntington's disease. Nat Clin Pract Neurol 2006;2:330-338.
Sadri-Vakili G, Bouzou B, Benn CL, et al. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet 2007;16:1293-1306.
Strand AD, Baquet ZC, Aragaki AK, et al. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci 2007;27:11758-11768.
Zucker B, Kama JA, Kuhn A, et al. Decreased Lin7b expression in layer 5 pyramidal neurons may contribute to impaired corticostriatal connectivity in huntington disease. J Neuropathol Exp Neurol 2010;69:880-895.
Chai Y, Wu L, Griffin JD, Paulson HL. The role of protein composition in specifying nuclear inclusion formation in polyglutamine disease. J Biol Chem 2001;276:44889-44897.
Dunah AW, Jeong H, Griffin A, et al. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 2002;296:2238-22343.
Li SH, Cheng AL, Zhou H, et al. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 2002;22:1277-1287.
Zuccato C, Belyaev N, Conforti P, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J Neurosci 2007;27:6972-6983.
Desplats PA, Lambert JR, Thomas EA. Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington's disease. Neurobiol Dis 2008;31:298-308.
Stroppolo A, Guinea B, Tian C, Sommer J, Ehrlich ME. Role of phosphatidylinositide 3-kinase in brain-derived neurotrophic factor-induced DARPP-32 expression in medium size spiny neurons in vitro. J Neurochem 2001;79:1027-1032.
Charron F, Nemer M. GATA transcription factors and cardiac development. Semin Cell Dev Biol 1999 Feb;10:85-91.
Runne H, Régulier E, Kuhn A, et al. Dysregulation of gene expression in primary neuron models of Huntington’s disease shows that polyglutamine-related effects on the striatal transcriptome may not be dependent on brain circuitry. J Neurosci 2008;28:9723-9731.
Ivkovic S, Polonskaia O, Farinas I, Ehrlich ME. Brain-derived neurotrophic factor regulates maturation of the DARPP-32 phenotype in striatal medium size spiny neurons: Studies in vivo and in vitro. Neuroscience 1997;79:509-516.
Ivkovic S, Ehrlich ME. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J Neuroscience 1999;19:5409-5419.
Hodges A, Strand AD, Aragaki AK, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet 2006;15:965–977.
Shao J, Diamond MI. Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet 2007;16 Spec No. 2:R115-R123.
Kuhn A, Goldstein DR, Hodges A, et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet 2007;6:1845-1861.
Luthi-Carter R, Strand A, Peters NL, et al. Decreased expression of striatal signaling genes in a mouse model of Huntington‘s disease. Hum Mol Genet 2000;9:1259-1271.
Luthi-Carter R, Strand AD, Hanson SA, et al. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington’s disease mouse models reveal context-independent effects. Hum Mol Genet 2002;11:1927-1937.
Ramanathan S, Hanley JJ, Deniau JM, Bolam JP. Synaptic convergence of motor and somatosensory cortical afferents onto GABAergic interneurons in the rat striatum. J Neurosci 2002:22:8158-8169.
Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington’s disease. Brain Res Rev 2000;34:80-101.
Lucas EK, Markwardt SJ, Gupta S, et al. Parvalbumin deficiency and GABAergic dysfunction in mice lacking PGC-1alpha. J Neurosci 2010;30:7227-7735.
Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med 2009;11:e17.
Stack C, Ho D, Wille E, et al. Triterpenoids CDDO-ethyl amide and CDDO-trifluoroethyl amide improve the behavioral phenotype and brain pathology in a transgenic mouse model of Huntington’s disease. Free Radic Biol Med 2010;49:147-158.
Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington’s disease transgenic mice. J Neurosci 2004;28:791-796.
Qiu Z, Norflus F, Singh B, et al. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J Biol Chem 2006;281:16672-16680.
Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci 2006;7:784-796.
Yazawa I, Hazeki N, Nakase H, Kanazawa I, Tanaka M. Histone H3 is aberrantly phosphorylated in glutamine-repeat diseases. Biochem Biophys Res Commun 2003;302:144-149.
Chen-Plotkin AS, Sadri-Vakili G, Yohrling GJ, et al. Decreased association of the transcription factor Sp1 with genes downregulated in Huntington's disease. Neurobiol Dis 2006;22:233-241.
Hockly E, Richon VM, Woodman B, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 2003;100:2041-2046.
Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem 2005;280:556-563.
Thomas EA, Coppola G, Desplats PA, et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc Natl Acad Sci USA 2008;105:15564-15569.
Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 2003;23:9418-9427.
Gomez GT, Hu H, McCaw EA, Denovan-Wright EM. Brain-specific factors in combination with mutant huntingtin induce gene-specific transcriptional dysregulation. Mol Cell Neurosci 2006;31:661-675.
Blau S, Daly L, Fienberg A, Teitelman G, Ehrlich ME. DARPP-32 promoter directs transgene expression to renal thick ascending limb of loop of Henle. Am J Physiol 1995;269:F564-F570.
Weydt P, Pineda VV, Torrence AE, et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab 2006;4:349-362.
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006;127:59-69.
Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature 1986;321:168-171.
Ellison DW, Beal MF, Mazurek MF, Malloy JR, Bird ED, Martin JB. Amino acid neurotransmitter abnormalities in Huntington's disease and the quinolinic acid animal model of Huntington's disease. Brain 1987;110(pt 6):1657-1673.
DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci 1990;13:286-289.
Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington's disease. Neurobiol Dis 2004;17:455-461.
Brouillet E, Hantraye P, Ferrante RJ, et al. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA 1995;92:7105-7109.
Stahl WL, Swanson PD. Biochemical abnormalities in Huntington’s chorea brains. Neurology 1974;24:813-819.
Brennan WA Jr, Bird ED, Aprille JR. Regional mitochondrial respiratory activity in Huntington’s disease brain. J Neurochem 1985;44:1948-1950.
Sawa A, Wiegand GW, Cooper J, et al. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length dependent mitochondrial depolarization. Nat Med 1999;5:1194-1198.
Tabrizi SJ, Workman J, Hart PE, et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol 2000;47:80-86.
Panov AV, Gutekunst CA, Leavitt BR, et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 2002;5:731-736.
Benchoua A, Trioulier Y, Zala D, et al. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell 2006;17:1652-1663.
Browne SE, Bowling AC, MacGarvey U, et al. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 1997;41:646-653.
Parker WD Jr., Boyson SJ, Luder AS, Parks JK. Evidence for a defect in NADH: ubiquinone oxidoreductase (complex I) in Huntington’s disease. Neurology 1990;40:1231-1234
Arenas J, Campos Y, Ribacoba R, et al. Complex I defect in muscle from patients with Huntington’s disease. Ann Neurol 1998;43:397-400.
Shirendeb U, Reddy AP, Manczak M, et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet 2011;20:1438-1455.
Benchoua A, Trioulier Y, Diguet E, et al. Dopamine determines the vulnerability of striatal neurons to the N-terminal fragment of mutant huntingtin through the regulation of mitochondrial complex II. Hum Mol Genet 2008;17:1446-1456.
Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta 2009;1787:1416-1424.
Hansson O, Guatteo E, Mercuri NB, et al. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the Huntington gene. Eur J Neurosci 2001;14:1492-1504.
Zeron MM, Fernandes HB, Krebs C, et al. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci 2004;25:469-479.
Heng MY, Detloff PJ, Wang PL, Tsien JZ, Albin RL. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J Neurosci 2009;29:3200-3205.
Okamoto S, Pouladi MA, Talantova M, et al. Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 2009;15:1407-1413.
Milnerwood AJ, Gladding CM, Pouladi MA, et al. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington’s disease mice. Neuron 2010;65:178-190.
Gascón S, Sobrado M, Roda JM, Rodríguez-Peña A, Díaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol Psychiatry 2008;13:99-114.
Finn SF, Hyman BT, Storey E, Miller JM, Beal MF. Effects of aging on quinolinic acid lesions in rat striatum. Brain Res 1991;562:276-280.
Cepeda C, Li Z, Levine MS. Aging reduces neostriatal responsiveness to N-methyl-D-aspartate and dopamine: an in vitro electrophysiological study. Neuroscience 1996;73:733-750.
Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington’s disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA 1999;96:8727-8732.
Giacomello M, Hudec R, Lopreiato R. Huntington’s disease, calcium, and mitochondria. Biofactors 2011;37:206-218.
Beal MF, Brouillet E, Jenkins BG, et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 1993;13:4181-4192.
Oliveira JM, Chen S, Almeida S, et al. Mitochondrial-dependent Ca2+ handling in Huntington’s disease striatal cells: effect of histone deacetylase inhibitors. J Neurosci 2006;26:11174-11186.
Hardingham GE. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem Soc Trans 2009;37:1147-1160.
Missale C, Fiorentini C, Busi C, Collo G, Spano PF. The NMDA/D1 receptor complex as a new target in drug development. Curr Top Med Chem 2006;6:801-808.
Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology 2010;58:951-961.
Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci 2007;227:7899-7910.
Deyts C, Galan-Rodriguez B, Martin E, et al. Dopamine D2 receptor stimulation potentiates PolyQ-Huntingtin-induced mouse striatal neuron dysfunctions via Rho/ROCK-II activation. PLoS One 2009;4:e8287.
Cepeda C, Colwell CS, Itri JN, Gruen E, Levine MS. Dopaminergic modulation of early signs of excitotoxicity in visualized rat neostriatal neurons. Eur J Neurosci 1998;10:3491-3497.
Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci 2011;48:308-320.
Iwakura Y, Nawa H, Sora I, Chao MV. Dopamine D1 receptor-induced signaling through TrkB receptors in striatal neurons. J Biol Chem 2008;283:15799-15806.
Altar CA, Cai N, Bliven T, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 1997;389:856-860.
Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci 1997;17:2295-2313.
Yurek DM, Hipkens SB, Wiegand SJ, Altar CA. Optimal effectiveness of BDNF for fetal nigral transplants coincides with the ontogenic appearance of BDNF in the striatum. J Neurosci 1998;18:6040-6047.
Fusco FR, Zuccato C, Tartari M, et al. Co-localization of brain-derived neurotrophic factor (BDNF) and wild-type huntingtin in normal and quinolinic acid-lesioned rat brain. Eur J Neurosci 2003;18:1093-1102.
Hermel E, Gafni J, Propp SS, et al. Specific caspase interactions and amplification are involved in selective neuronal vulnerability in Huntington's disease. Cell Death Differ 2004;11:424-438.
Gines S, Bosch M, Marco S, et al. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur J Neurosci 2006;23:649-658.
Zuccato C, Marullo M, Conforti P, MacDonald ME, Tartari M, Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol 2008;18:225-238.
Baquet ZC, Gorski JA, Jones KR. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci 2004;24:4250-4258.
Rauskolb S, Zagrebelsky M, Dreznjak A, et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci 2010;30:1739-1749.
Bogush A, Pedrini S, Pelta-Heller J, et al. AKT and CDK5/p35 mediate brain-derived neurotrophic factor induction of DARPP-32 in medium size spiny neurons in vitro. J Biol Chem 2007;282:7352-7359.
Gharami K, Xie Y, An JJ, Tonegawa S, Xu B. Brain-derived neurotrophic factor over-expression in the forebrain ameliorates Huntington's disease phenotypes in mice. J Neurochem 2008;105:369-379.
Costantini LC, Feinstein SC, Radeke MJ, Snyder-Keller A. Compartmental expression of trkB receptor protein in the developing striatum. Neuroscience 1999;89:505-513.
Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science 1995;268:573-575.
Fryer HJ, Wolf DH, Knox RJ, et al. Brain-derived neurotrophic factor induces excitotoxic sensitivity in cultures embryonic rat spinal motor neurons through activation of the phosphatidylinositol 3-kinase pathway. J Neurochem 2000;74:582-595.
Hu P, Kalb RG. BDNF heightens the sensitivity of motor neurons to excitotoxic insults through activation of TrkB. J Neurochem 2003;84:1421-1430.
Mojsilovic-Petrovic J, Jeong GB, Crocker A, et al. Protecting motor neurons from toxic insult by antagonism of adenosine A2a and Trk receptors. J Neurosci 2006;26:9250-9263.
Domenici MR, Scattoni ML, Martire A, et al. Behavioral and electrophysiological effects of the adenosine A2A receptor antagonist SCH58261 in R6/2 Huntington’s disease mice. Neurobiol Dis 2007;28:197-205
Popoli P, Blum D, Martire A, Ledent C, Ceruti S, Abbracchio MP. Functions, dysfunctions and possible therapeutic relevance of adenosine A2A receptors in Huntington’s disease. Prog Neurobiol 2007;81:331-348.
Torres-Peraza JF, Giralt A, García-Martínez JM, Pedrosa E, Canals JM, Alberch J. Disruption of striatal glutamatergic transmission induced by mutant huntingtin involves remodeling of both postsynaptic density and NMDA receptor signaling. Neurobiol Dis 2008;29:409-421.
Xifró X, Giralt A, Saavedra A, et al. Reduced calcineurin protein levels and activity in exon-1 mouse models of Huntington’s disease: role in excitotoxicity. Neurobiol Dis 2009;36:461-469.
Acknowledgment
This work was supported by NIH R01NS059936 and CHDI.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
ESM 1
(PDF 510 kb)
Rights and permissions
About this article
Cite this article
Ehrlich, M.E. Huntington’s Disease and the Striatal Medium Spiny Neuron: Cell-Autonomous and Non-Cell-Autonomous Mechanisms of Disease. Neurotherapeutics 9, 270–284 (2012). https://doi.org/10.1007/s13311-012-0112-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-012-0112-2