
Abstract
Cerebral vasospasm occurs frequently after aneurysmal subarachnoid and contributes to delayed cerebral ischemia. In this article we address systematic problems with the literature on vasospasm and then review both established and experimental treatment options.
Similar content being viewed by others
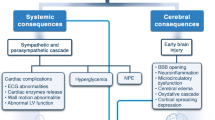
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH), a deadly form of hemorrhagic stroke caused by the rupture of a brain aneurysm, affects 30,000 people annually in the United States [1, 2]. Of the 85% of patients who survive the initial hemorrhage, one third develop further brain injury or “delayed cerebral ischemia” (DCI) [3]. DCI is defined as any neurological deterioration presumed to be related to ischemia that lasts for more than an hour and cannot be attributed to other causes [4]. DCI occurs insidiously within the first 2 weeks after subarachnoid hemorrhage (SAH) and accounts for the majority of morbidity and mortality related to aSAH [5–7].
Following early observations that electrical, chemical, and mechanical stimuli could cause cortical arterial constriction [8–10], Robertson [11] in 1949, speculated that arterial spasm was a possible cause of ischemia after aSAH. Two years later, a small study of patients with aSAH demonstrated angiographic evidence of narrowing of the large arteries at the base of the brain, and the connection between vasospasm and ischemia was established [12]. Subsequent studies reported that the onset of vasospasm occurs at least 3 days after aneurysm rupture and coincides with clinical deterioration [13–15]. Vessel narrowing was found to peak at 5 to 14 days and gradually resolved within 2 to 4 weeks [13–15]. In the ensuing years, arterial narrowing became accepted as the mechanism for DCI, and the term “vasospasm” was used interchangeably to describe angiographic vessel narrowing, elevated cerebral blood flow velocities measured by transcranial Doppler ultrasonography (“TCD-vasospasm”), and clinical deterioration (“clinical vasospasm”). However, there remained an imperfect correlation between angiographic vasospasm, TCD-vasospasm, and clinical deterioration, and it has become apparent that arterial vasospasm, elevated cerebral blood flow velocities, and clinical vasospasm are not synonymous [11, 16–18]. Vasospasm is associated with clinical signs in roughly half of the cases, and half of the symptomatic cases culminate in cerebral infarction [3, 19, 20]. Recent consensus statements recommend that the term “vasospasm” be reserved to describe radiological findings only [4, 21]. Indeed, mechanisms other than arterial narrowing might contribute to DCI [22–25]. Elsewhere, we have reviewed both the evidence against vasospasm as the sole cause of cerebral ischemia and alternate mechanisms of DCI [26]. Unsurprisingly, management of patients with aSAH has lagged behind evolving conceptual changes in the pathogenesis of delayed cerebral ischemia. Despite growing recognition that vasospasm is not the sole cause of DCI, current management remains focused on detection and treatment of vasospasm.
In this article, we review the current state of knowledge regarding the treatment options for cerebral vasospasm, defined as arterial narrowing. In the first section, we discuss established therapies that are either evidence-based or supported by expert guidelines. In the second section, we discuss experimental therapies that have theoretical rationales, but have not yet been established as effective.
Although there are no known methods to prevent vasospasm, numerous treatment strategies have been studied. As these strategies are discussed as follows, it is important to bear in mind that assessment of efficacy is difficult due to significant limitations in the extant literature. One major problem is inconsistent use and definitions of the term “vasospasm.” Anther major problem is that vasospasm may be an inappropriate endpoint for therapeutic studies, as radiological infarction correlates better with functional outcome. An expert panel recently suggested that brain infarction and functional outcome be used as primary endpoints in future therapeutic studies [4, 21], and not vasospasm or clinical deterioration.
Established Therapies
Calcium Channel Antagonists
Multiple clinical studies, including 5 double-blind, placebo-controlled trials, have demonstrated that the dihydropyridine calcium channel antagonist, nimodipine, improves clinical outcome in patients with aSAH [27–31]. However, the mechanism underlying this clinical benefit is unclear, as nimodipine does not influence the incidence of vasospasm [32]. Because nimodipine improves clinical outcome, is cost effective, and safe, oral administration (60 mg every 4 h for 21 days) is recommended as the standard of care [33]. Conversely, 2 randomized controlled trials of intravenous nicardipine, another dihydropyridine calcium channel antagonist, demonstrated a reduction in “symptomatic vasospasm,” but had no impact on 3-month clinical outcomes [34, 35]. Intravenous nicardipine is therefore not recommended for routine use [35].
Hemodynamic Augmentation
The cornerstone of medical management of cerebral vasospasm is hemodynamic augmentation, but the strategy has changed with time. Originally, patients were treated with a combination of hypervolemia, hemodilution, and induced hypertension (“triple-H therapy”) to augment cerebral blood flow by increasing cerebral perfusion pressure and lowering blood viscosity [36–38]. However, as the various components of triple-H therapy were studied, the focus of therapy has shifted to euvolemic-induced hypertension.
Although early retrospective studies of hypervolemia as part of a triple-H therapy suggested benefit, subsequent physiological and prospective observational studies suggest harm [39, 40]. Two randomized trials compared prophylactic hypervolemia to normovolemia [41, 42]. Neither found a significant improvement in cerebral blood flow (CBF), incidence of neurological decline, or functional outcome. Patients randomized to hypervolemia had an excess of bleeding, congestive heart failure, and infections. Hypervolemia was also associated with higher cost. Taken together, these studies suggest that hypervolemia does not improve outcome and is associated with increased cardiopulmonary complications. Therefore, hypervolemia is recommended neither for prophylaxis nor for treatment of vasospasm.
Hemodilution has also fallen out of favor as a treatment strategy for vasospasm, however, controversy exists regarding the “optimal” hemoglobin in SAH patients. Although hemodilution increases CBF through improved rheology, it also reduces oxygen-carrying content and results in no net increase in cerebral oxygen delivery [43]. Furthermore, hemodilution is achieved by volume loading, which itself is associated with complications. Whether transfusion of red blood cells (the opposite of hemodilution) improves outcome is unknown. Blood transfusion increases arterial oxygen content and augments cerebral oxygen delivery. However, whether this translates to increased tissue utilization of oxygen is unclear. Furthermore, it has become apparent in the general critical care population that transfusion is associated with mortality, independent of disease severity [44, 45]. Observational studies in the SAH population suggest that while anemia is associated with increased incidence of cerebral infarction and worse functional outcome [46, 47], transfusion of red blood cells is associated with increased risk of poor outcome, extracerebral complications, and increased intensive care unit and total hospital length of stay [48, 49]. Whether the risks of transfusion outweigh the risks of anemia is unclear. A pilot study suggested that randomizing SAH patients to a restrictive versus a liberal transfusion strategy is feasible [50]. A definitive randomized study of the effects of transfusion strategies on outcome has not been performed. It is likely that the “optimal” hemoglobin level varies from patient to patient depending on individual physiology and that no single hemoglobin threshold is optimal for all patients. Presently, neither hemodilution nor red blood cell transfusion is recommended for routine treatment of vasospasm or DCI [21].
Induced hypertension remains the sole recommended component of triple-H therapy. This strategy does not treat or reverse vasospasm, but aims to ameliorate ischemia, a potential consequence of vasospasm. Induced hypertension increases CBF and brain tissue oxygenation, and reverses neurological deficits believed to be from vasospasm [51–53]. These effects appear to be independent of volume status. Several case series suggest that inotropic agents improve CBF and reverse neurological deficits in patients who do not respond to vasopressors [54, 55]. There is scant data to guide choice of vasopressor or inotropic agent and the choice must be made based on empiricism and theoretical advantages that are based on pharmacology and individual patient physiology.
Endovascular Therapies
Endovascular therapies include percutaneous transluminal angioplasty (PTA), and intra-arterial infusion of vasodilators. Although studies have defined the feasibility and safety profiles of these therapies, none has definitively demonstrated an impact on outcome [56–60].
PTA results in durable dilation of vasospastic vessels [61], augments CBF [61, 62], and may reverse neurological deficits [56]; however, its impact on long-term outcome is unclear. Prophylactic PTA (in the absence of arterial narrowing) reduces the incidence of DCI, but is associated with significant complications, resulting in no net benefit [63]. Complications of PTA include vessel occlusion, dissection, and rupture, dislodging of aneurysm clips, and thrombus formation [64–67]. PTA is typically limited to proximal segments of large cerebral vessels, which are easier to access and presumably less prone to rupture due to thicker muscular walls.
Catheter-based delivery of intra-arterial (IA) vasodilators is a commonly used rescue strategy. Papaverine is the most widely studied IA vasodilator. The administration of papaverine is associated with short-lived (roughly 3 h) effects on both vessel caliber and CBF, and multiple side effects, including brainstem depression, elevated intracranial pressure, seizures, hypotension, and occasionally paradoxical worsening of vasospasm [68, 69]. Intracranial hypertension occurs in ≤42% of cases and may be associated with death and poor outcome [70–73]. Therefore, ICP must be monitored during therapy [71]. Due to the short half-life of papaverine, multiple serial injections are frequently required. However, repeated injections have been associated with worse clinical outcomes in several case series [74–76]. Advancements in microcatheter technology have made the super selective catheterization of third and fourth order cerebral vessels possible, allowing for slow infusion of vasodilators, perhaps reducing side effects [70, 71, 77, 78]. Case series report the use of other IA vasodilators, including verapamil, nicardipine, nimodipine, and milrinone [35, 79–83]. Although these agents appear to be safer than papaverine and provide a more durable response, adequate trials that establish safety and efficacy are lacking.
Endovascular therapies are currently recommended as optional rescue strategies for patients that do not adequately respond to medical therapy [21, 33]. The optimal methods, timing, frequency, and combination of endovascular therapies remain unknown.
Experimental Therapies
Statins
Statins have a favorable safety profile and pleiotropic effects that make them an attractive theoretical treatment for vasospasm and DCI. Levels of several inflammatory cytokines, as well as markers of the central nervous system and endothelial cell injury are increased after SAH, and have been correlated with occurrence of vasospasm [84, 85]. Statins downregulate inflammation and upregulate endothelial nitric oxide synthase, which improve cerebral vasomotor reactivity and CBF [86, 87].
Multiple retrospective studies, and 6 small, prospective, randomized controlled studies (4 of which were published in peer-reviewed journals, 2 of which were published as abstracts) investigated the use of statins in aSAH [88–93]. Study results were inconsistent. A recent meta-analysis of the prospective studies demonstrated a small reduction in DCI and a small reduction in mortality, which was lost when patients from the non-peer-reviewed studies were included [94]. Another meta-analysis that included the 4 high-quality trials reported no benefit with regard to TCD-vasospasm, functional outcome, or mortality [95]. A phase III randomized controlled study, Simvastatin in Aneurysmal Subarachnoid Hemorrhage (STASH), is underway. Currently it is recommended that statins be continued in patients who had been on therapy prior to hospitalization, and that therapy may be considered in statin-naïve patients, pending the outcome of ongoing trials [21].
Magnesium Sulfate
Multiple experimental and clinical observations have justified therapeutic studies of magnesium in patients with aSAH. Magnesium inhibits glutamate release and causes cerebral vasodilation through noncompetitive antagonism of voltage-gated calcium channels [96, 97]. Numerous experimental stroke models have demonstrated that magnesium is neuroprotective. In experimental SAH models, magnesium sulfate reverses cerebral vasospasm and infarct volume [98–100]. Hypomagnesemia occurs in >50% of patients with aSAH and is associated with the occurrence of DCI and poor outcome [101].
Six phase II studies have examined the effects of various magnesium regimens on a variety of endpoints [102–107]. In general, these studies suggest that magnesium therapy is safe. A meta-analysis that included 476 patients suggested that magnesium was associated with a reduced risk of poor outcome (death, vegetative state, or dependency; odds ratio, 0.54 [confidence interval, 0.36-0.81]) and no difference in mortality [108]. A single phase III study, The Intravenous Magnesium Sulfate for Aneurysmal Subararchnoid Hemorrhage (IMASH) trial [109] compared magnesium to a placebo in 327 patients. No difference between groups was detected with respect to 6-month outcomes. A second phase III study, Magnesium in Aneurysmal Subarachnoid Hemorrhage (MASH-II) is ongoing.
Other therapies that have been studied include antiplatelet agents [110–112] endothelin antagonists [113], intracisternal fibrinolysis [114, 115], nitric oxide, and nitric oxide donor compounds [116, 117], anticoagulants [118, 119] corticosteroids [120], antioxidants [121, 122], free radical scavengers [123], and serine protease inhibitors [124]. To date, none have proven efficacious in high quality human studies.
Conclusions
The only therapy currently established for the treatment of cerebral vasospasm after SAH is euvolemic-induced hypertension. Although nimodipine improves outcome and is standard of care, it does not prevent or treat vasospasm per se. Rescue therapy with balloon angioplasty and intra-arterial vasodilator therapy is widely used and supported by expert opinion, but its impact outcome is unclear. As the pathophysiology of aSAH is further elucidated and alternate mechanisms of DCI are uncovered, less emphasis may be placed on the treatment of vasospasm. Future therapeutic trials should use consistent terminology and choose surrogate outcome measures, such as cerebral infarction, and perhaps, in the future, chemical biomarkers that correlate more closely with long-term clinical outcome.
References
Graf CJ, Nibbelink DW. Cooperative Study of Intracranial Aneurysms and Subarachnoid Hemorrhage: report on a randomized treatment study. 3: intracranial surgery. Stroke 1974;5:557-601.
King JT Jr. Epidemiology of aneurysmal subarachnoid hemorrhage. Neuroimaging Clin N Am 1997;7:659-668.
Sundt TM Jr, Kobayashi S, Fode NC, Whisnant JP. Results and complications of surgical management of 809 intracranial aneurysms in 722 cases: related and unrelated to grade of patient, type of aneurysm, and timing of surgery. J Neurosurg 1982;56:753-765.
Vergouwen MDI, Vermeulen M, van Gijn J, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies. Proposal of a multidisciplinary research group. Stroke 2010;41:2391-2395.
Haley EC Jr, Kassell NF, Torner JC. The International Cooperative Study on the Timing of Aneurysm Surgery: the North American experience. Stroke 1992;23:205-214.
Longstreth WT Jr, Nelson LM, Koepsell TD, et al. Clinical course of spontaneous subarachnoid hemorrhage: a population-based study in King County, Washington. Neurology 1993;43:712-718.
Kassell NF, Boarini DJ, Adams HP Jr, et al. Overall management of ruptured aneurysm: comparison of early and late operation. Neurosurgery 1981;9:120-128.
Florey H. Microscopical observations on the circulation of the blood in the cerebral cortex. Brain 1925;48:43-68.
Penfield W. The evidence for a cerebral vascular mechanism in epilepsy. Ann Int Med 1933;5:303-311.
Schultz A. Zur Lehre von der Bluthewegung im Innern des Schaedels. St Petersburg med Ztschr 1866;11:122-144.
Robertson EG. Cerebral lesions due to intracranial aneurysms. Brain 1949;72:150-185.
Ecker A, Riemenschneider PA. Arteriographic demonstration of spasm of the intracranial arteries. With special reference to saccular aneurysms. J Neurosurg 1951;8:660-667
Kagstrom E, Greitz T, Hanson J, Galera R. Changes in cerebral blood flow after subarachnoid hemorrhage, in Proceedings of the Third International Congress of Neurological Surgery, 1965. International Congress Series No. 110. Amsterdam: Excerpta Medica 1966:629-633.
Fisher CM, Roberson GH, Ojemann RG. Cerebral vasospasm with ruptured saccular aneurysm–the clinical manifestations. Neurosurgery 1977;1:245-248.
Graham DI, Macpherson P, Pitts LH. Correlation between angiographic vasospasm, hematoma, and ischemic brain dam- age following SAH. J Neurosurg 1983;59:223-230.
Adams HP Jr. Prevention of brain ischemia after aneurismal subarachnoid hemorrhage. Neurol Clin 1992;10:251-268.
Geraud G, Tremoulet M, Guell A, Bes A. The prognostic value of noninvasive CBF measurement in subarachnoid hemorrhage. Stroke 1984;15:301-305.
Ohkuma H, Manabe H, Tanaka M, Suzuki S. Impact of cerebral microcirculatory changes on cerebral blood flow during cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke 2000;31:1621-1627.
Kassell NF, Kongable GL, Torner JC, Adams HP Jr, Mazuz H. Delay in referral of patients with ruptured aneyrsms to neurosurgical attention. Stroke 1985;16:587-590.
Heros RC, Zervas NT, Varsos V. Cerebral vasospam after subarachnoid hemorrhage: an update. Ann Neurology 1983;14:599-608.
Diringer MN, Bleck TP, Claude Hemphill J 3rd, et al. Critical care management of patients following aneurysmal subarachnoid hemorrhage: recommendations from the Neurocritical Care Society's Multidisciplinary Consensus Conference. Neurocrit Care 2011;15:211-240.
Vergouwen MD, Vermeulen M, Coert BA, Stroes ES, Roos YB. Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J Cereb Blood Flow Metab 2008;28:1761-1770.
Uhl E, Lehmberg J, Steiger HJ, Messmer K. Intraoperative detection of early microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery 2003;52:1307-1315.
Dreier JP, Major S, Manning A, et al. COSBID study group. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 2009;132:1866–1881.
Grubb RL Jr, Raichle ME, Eichling JO, Gado MH. Effects of subarachnoid hemorrhage on cerebral blood volume, blood flow, and oxygen utilization in humans. J Neurosurg 1977;46:446-453.
Sherman SC, Levine JM, Nagpal S, et al. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurg Focus 2006;21:E2.
Allen GS, Ahn HS, Preziosi TJ, et al. Cerebral arterial spasm: A controlled trial of nimodipine in patients with subarachnoid hemorrhage. N Engl J Med 1983;308:619-624.
Phillippon J, Grob R, Dagreou F, et al. Prevention of vasospasm in subarachnoid haemorrhage: A controlled study with nimodipine. Acta Neurochir 1986;82:110-114.
Neil-Dwyer G, Mee E, Dorrance D, Lowe D. Early intervention with nimodipine in subarachnoid hemorrhage. Eur Heart J 1987;8(suppl K):41-47.
Petruck KC, West M, Mohr G, et al. Nimodipine treatment in poor-grade aneurysm patients: results of a multicenter double-blind placebo-controlled trial. J Neurosurg 1988;68:505-517.
Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. Br Med J 1989;298:636-642.
Feigin VL, Rinkel GJ, Algra A, et al. Calcium antagonists in patients with aneurysmal subarachnoid hemorrhage: a systematic review. Neurology 1998;50:876-883.
Bederson JB, Connolly ES Jr, Batjer HH, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke council, American Heart Association. Stroke 2009;40:994-1025.
Haley Jr EC, Kassell NF, Torner JC. A randomized controlled trial of high-dose intravenous nicardipine in aneurysmal subarachnoid hemorrhage. A report of the Cooperative Aneurysm Study. J Neurosurg 1993;78:537-547.
Haley Jr EC, Kassell NF, Torner JC, et al. A randomized trial of two doses of nicardipine in aneurysmal subarachnoid hemorrhage. A report of the Cooperative Aneurysm Study. J Neurosurg 1994;80:788-796.
Keller TS, McGillicuddy JE, LaBond VA, Kindt GW. Modification of focal cerebral ischemia by cardiac output augmentation. J Surg Res 1985;39:420-432.
Tranmer BI, Keller TS, Kindt GW, et al. Loss of cerebral regulation during cardiac output variations in focal cerebral ischemia. J Neurosurg 1992;77:253-259.
Matsui T, Asano T. The hemodynamic effects of prolonged albumin administration in beagle dogs exposed to experimental subarachnoid hemorrhage. Neurosurgery 1993;32:79-84.
Awad IA, Carter LP, Spetzler RF, et al. Clinical vasospasm after subarachnoid hemorrhage: response to hypervolemic hemodilution and arterial hypertension. Stroke 1987;18:365-372.
Kassell NF, Peerless SJ, Durward QJ, et al. Treatment of ischemic deficits from vasospasm with intravascular volume expansion and induced arterial hypertension. Neurosurgery 1982;11:337-343.
Lennihan L, Mayer SA, Fink ME et al. Effect of hypervolemic therapy on cerebral blood flow after subarachnoid hemorrhage: a randomized controlled trial. Stroke 2000;31:383-391.
Egge A, Waterloo K, Sjoholm H, et al. Prophylactic hyperdynamic postoperative fluid therapy after aneurismal subarachnoid hemorrhage: a clinical, prospective, randomized, controlled study. Neurosurgery 2001;49:593-605.
Ekelund A, Reinstrup P, Ryding E et al. Effects of iso- and hypervolemic hemodilution on regional cerebral blood flow and oxygen delivery for patients with vasospasm after aneurysmal subarachnoid hemorrhage. Acta Neurochir (Wien) 2002;144:703-712.
Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. New Engl J Med 1999;340:409-417.
Corwin HL, Gettinger A, Pearl RG, et al. The CRIT Study: Anemia and blood transfusion in the critically ill--current clinical practice in the United States. Crit Care Med 2004;32:39-52.
Naidech AM, Drescher J, Ault AL, et al. Higher hemoglobin is associated with less cerebral infarction, poor outcome, and death after subarachnoid hemorrhage. Neurosurgery 2006;59:775-779.
Naidech AM, Jovanovic B, Wartenberg KE, et al. Higher hemoglobin is associated with improved outcome after subarachnoid hemorrhage. Crit Care Med 2007;35:2383-2389.
Smith MJ, Le Roux PD, Elliott JP, et al. Blood transfusion and increased risk for vasospasm and poor outcome after subarachnoid hemorrhage. J Neurosurgery 2004;101:1-7.
Levine J, Kofke A, Cen L, et al. Red blood cell transfusion is associated with infection and extracerebral complications after subarachnoid hemorrhage. Neurosurgery 2010;66:312-318.
Naidech AM, Shaibani A, Garg RK, et al. Prospective, randomized trial of higher goal hemoglobin after subarachnoid hemorrhage. Neurocrit Care 2010;13:313-320.
Muench E, Horn P, Bauhuf C, et al. Effects of hypervolemia and hypertension on regional cerebral blood flow, intracranial pressure, and brain tissue oxygenation after subarachnoid hemorrhage. Crit Care Med 2007;35:1844-1851.
Raabe A, Beck J, Keller M, et al. Relative importance of hypertension compared with hypervolemia for increasing cerebral oxygenation in patients with cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg 2005;103:974-981.
Otsubo H, Takemae T, Inoue T, et al. Normovolaemic induced hypertension therapy for cerebral vasospasm after subarachnoid haemorrhage. Acta Neurochir 1990;103:18-26.
Levy ML, Rabb CH, Zelman V, Giannotta SL. Cardiac performance enhancement from dobutamine in patients refractory to hypervolemic therapy for cerebral vasospasm. J Neurosurg 1993;79:494-499.
Kim DH, Joseph M, Ziadi S, et al. Increases in cardiac output can reverse flow deficits from vasospasm independent of blood pressure: a study using xenon computed tomographic measurement of cerebral blood flow. Neurosurgery 2003;53:10441051.
Newell DW, Eskridge JM, Mayberg MR, Grady MS, Winn HR. Angioplasty for the treatment of symptomatic vasospasm following subarachnoid hemorrhage. J Neurosurg 1989;71(pt 1):654-660.
Newell DW, Eskridge J, Mayberg M, et al: Endovascular treatment of intracranial aneurysms and cerebral vasospasm. Clin Neurosurg 1992;39:348-360.
Coyne TJ, Montanera WJ, Macdonald RL, et al. Transluminal angioplasty for cerebral vasospasm — the Toronto experience. In: Findlay JM, ed. Cerebral Vasospasm. Amsterdam: Elsevier 1993:333-336.
Fujii Y, Takahashi A, Yoshimoto T. Effect of balloon angioplasty on high grade symptomatic vasospasm after subarachnoid hemorrhage. Neurosurg Rev 1995;18:7-13.
Terada T, Kinoshita Y, Yokote H, et al. The effect of endovascular therapy for cerebral arterial spasm, its limitations and pitfalls. Acta Neurochir 1997;139:227-234.
Firlik AD, Kaufmann AM, Jungreis CA, et al. Effect of transluminal angioplasty on cerebral blood flow in the management of symptomatic vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg 1997;86:830-839.
Le Roux PD, Newell DW, Eskridge J, et al. Severe symptomatic vasospasm: the role of immediate postoperative angioplasty. J Neurosurgery 1994;80:224-229.
Zwienenberg-Lee M, Hartman J, Rudisill N, et al; the Balloon Prophylaxis for Aneurysmal Vasospasm (BPAV) Study Group. Effect of prophylactic transluminal balloon angioplasty on cerebral vasospasm and outcome in patients with Fisher grade III subarachnoid hemorrhage: results of a phase II multicenter, randomized, clinical trial. Stroke 2008;39:1759-1765.
Higashida RT, Halback VV, Dormandy B, Bell JD, Hieshima GB. Endovascular treatment of intracranial aneurysms with a new silicone microballoon device: technical considerations and indications for therapy. Radiology 1990;174(pt 1):687-691.
Higashida RT, Halbach VV, Cahan LD, et al. Transluminal angioplasty for treatment of intracranial arterial vasospasm. J Neurosurg 1989;71(pt 1):648-653.
Higashida RT, Halbach VV, Dowd CF, et al. Intravascular balloon dilatation therapy for intracranial arterial vasospasm: patient selection, technique, and clinical results. Neurosurg Rev 1992;15:89-95.
Terada T, Nakamura Y, Yoshida N, et al. Percutaneous transluminal angioplasty for the M2 portion vasospasm following SAH: development of the new microballoon and report of cases. Surg Neurol 1993;39:13-17.
Mathis JM, Jensen ME, Dion JE. Technical considerations on intra-arterial papaverine hydrochloride for cerebral vasospasm. Neuroradiology 1997;39:90-98.
Clyde BL, Firlik AD, Kaufmann AM, Spearman MP, Yonas H. Paradoxical aggravation of vasospasm with papaverine infusion following aneurysmal subarachnoid hemorrhage: case report. J Neurosurg 1996;84:690-695.
McAuliffe W, Townsend M, Eskridge JM, et al. Intracranial pressure changes induced during papaverine infusion for treatment of vasospasm. J Neurosurg 1995;83:430-434.
Cross DT 3rd, Moran CJ, Angtuaco EE, et al. Intracranial pressure monitoring during intraarterial papaverine infusion for cerebral vasospasm. AJNR Am J Neuroradiol 1998;19:1319-1323.
Zervas NT, Ogilvy CS. Cerebral vasospasm: current clinical management and results. Clin Neurosurg 1999;45:167-76.
Vajkoczy P, Horn P, Bauhuf C, et al. Effect of intraarterial papaverine on regional cerebral blood flow in hemodynamically relevant cerebral vasospasm. Stroke 2001;32:498-505.
Hanel RA, Xavier AR, Mohammad Y, et al. Outcome following intracerebral hemorrhage and subarachnoid hemorrhage. Neurol Res 2002;24(suppl 1):S58-S62.
Qureshi AI, Frankel MR. Recognition and management of subarachnoid hemorrhage. Heart Dis Stroke 1994;3:270-274
Rabinstein AA, Friedman JA, Nichols DA, et al. Predictors of outcome after endovascular treatment of cerebral vasospasm. AJNR Am J Neuroradiol 2004;25:1778-1782.
Milburn JM, Moran CJ, Cross DT 3rd, et al. Effect of intraarterial papaverine on cerebral circulation time. AJNR Am J Neuroradiol 1997;18:1081-1085.
Milburn JM, Moran CJ, Cross DT 3rd, Diringer MN, et al. Increase in diameters of vasospastic intracranial arteries by intraarterial papaverine administration. J Neurosurg 1998;88:38-42.
Kassell NF, Helm G, Simmons N, et al. Treatment of cerebral vasospasm with intra-arterial papaverine. J Neurosurg 1992;77:848-852.
Feng L, Fitzsimmons BF, Young WL, et al. Intraarterially administered verapamil as adjunct therapy for cerebral vasospasm: safety and 2-year experience. AJNR Am J Neuroradiol 2002;23:1284-1290.
Badjatia N, Topcuoglu MA, Pryor JC, et al. Preliminary experience with intra-arterial nicardipine as a treatment for cerebral vasospasm. AJNR Am J Neuroradiol 2004;25:819-826.
Biondi A, Ricciardi GK, Puybasset L, et al. Intra-arterial nimodipine for the treatment of symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage: preliminary results. AJNR Am J Neuroradiol 2004;25:1067-1076.
Fraticelli AT, Cholley BP, Losser MR, et al. Milrinone for the treatment of cerebral vasospasm after aneurismal subarachnoid hemorrhage. Stroke 2008;39:893-898.
Fassbender K, Hodapp B, Rossol S, et al. Endothelin-1 in subarachnoid hemorrhage: an acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke 2000;31:2971-2975.
Fassbender K, Hodapp B, Rossol S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry 2001;70:534-537.
Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005;45:89-118.
McGirt MJ, Lynch JR, Parra A, et al. Simvastatin increases endothelial nitric oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke 2002;33:2950-2956.
Tseng MY, Czosnyka M, Richards H, et al. Effects of acute treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke 2005;36:1627-1632.
Tseng MY, Hutchinson PJ, Czosnyka M, et al. Effects of acute pravastatin treatment on intensity of rescue therapy, length of inpatient stay, and 6-month outcome in patients after aneurysmal subarachnoid hemorrhage. Stroke 2007;38:1545-1550.
Lynch JR, Wang H, McGirt MJ, et al. Simvastatin reduces vaso- spasm after aneurysmal subarachnoid hemorrhage: results of a pilot randomized clinical trial. Stroke. 2005;36:2024–2026.
Chou SH, Smith EE, Badjatia N, et al. A randomized, double- blind, placebo-controlled pilot study of simvastatin in aneurysmal subarachnoid hemorrhage. Stroke 2008;39:2891-2893.
Jaschinski U, Scherer K, Lichtwarck M, Forst H. Impact of treatment with pravastatin on delayed ischemic disease and mortality after aneurysmal subarachnoid hemorrhage. Crit Care 2008;12:P112.
Macedo S, Bello Y, Silva A, et al. Effects of simvastatin in prevention of vasospasm in nontraumatic subarachnoid hemorrhage: preliminary data. Crit Care 2009;13:P103.
Kramer AH, Fletcher JJ. Statins in the management of patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Neurocrit Care 2010;12:285-296.
Sillberg VA, Wells GA, Perry JJ. Do statins improve outcomes and reduce the incidence of vasospasm after aneurysmal subarachnoid hemorrhage: a meta-analysis. Stroke 2008;39:2622-2626.
Johnson JW, Ascher P. Voltage-dependent block by intracellular Mg2+ of N-methyl-D-aspartate-activated channels. Biophys J 1990;57:1085-1090.
Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci 1984;4:1884-1891.
Marinov MB, Harbaugh KS, Hoopes PJ, Pikus HJ, Harbaugh RE. Neuroprotective effects of preischemia intraarterial magnesium sulfate in reversible focal cerebral ischemia. J Neurosurg 1996;85:117-124.
van den Bergh WM, Zuur JK, Kamerling NA, et al. Role of magnesium in the reduction of ischemic depolarization and lesion volume after experimental subarachnoid hemorrhage. J Neurosurg 2002;97:416-422.
Ram Z, Sadeh M, Shacked I, et al. Magnesium sulfate reverses experimental delayed cerebral vasospasm after subarachnoid hemorrhage in rats. Stroke 1991;22:922-927.
Van den Bergh WM, Algra A, van der Sprenkel JW, Tulleken CA, Rinkel GJ. Hypomagnesemia after aneurysmal subarachnoid hemorrhage. Neurosurgery 2003;52:276-282.
van den Bergh WM, Algra A, van Kooten F, et al. Magnesium sulfate in aneurysmal subarachnoid hemorrhage: a randomized controlled trial. Stroke 2005;36:1011-1015.
Westermaier T, Stetter C, Vince GH, et al. Prophylactic intravenous magnesium sulfate for treatment of aneurismal subarachnoid hemorrhage: a randomized, placebo-controlled, clinical study. Crit Care Med 2010;38:1284-1290.
Schmid-Elsaesser R, Kunz M, Zausinger S, Prueckner S, Briegel J, Steiger HJ. Intravenous magnesium versus nimodipine in the treatment of patients with aneurysmal subarachnoid hemorrhage: a randomized study. Neurosurgery 2006;58:1054-1065.
Muroi C, Terzic A, Fortunati M, Yonekawa Y, Keller E. Magnesium sulfate in the management of patients with aneurismal subarachnoid hemorrhage: a randomized, placebo-controlled, dose-adapted trial. Surg Neurol 2008;69:33-39.
Veyna RS, Seyfried D, Burke DG, et al. Magnesium sulfate therapy after aneurysmal subarachnoid hemorrhage. J Neurosurg 2002;96:510-514.
Wong GK, Chan MT, Boet R, Poon WS, Gin T. Intravenous magnesium sulfate after aneurysmal subarachnoid hemorrhage: a prospective randomized pilot study. J Neurosurg Anesthesiol 2006;18:142-148.
Zhao XD, Zhou YT, Zhang X, et al. A meta analysis of treating subarachnoid hemorrhage with magnesium sulfate. J Clin Neurosci 2009;16:1394-1397.
Wong GK, Poon WS, Chan MT, et al. Intravenous magnesium sulphate for aneurysmal subarachnoid hemorrhage (IMASH): a randomized, double-blinded, placebo-controlled, multicenter phase III trial. Stroke 2010;41:921-926.
Juvela S. Aspirin and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. J Neurosurg 1995;82:945-952.
Dorhout Mees SM, Rinkel GJ, Hop JW, Algra A, van Gijn J. Antiplatelet therapy in aneurysmal subarachnoid hemorrhage: a systematic review. Stroke 2003;34:2285-2289.
Walter M, van den Bergh MD; on behalf of the MASH Study Group. Randomized controlled trial of acetylsalicylic acid in aneurysmal subarachnoid hemorrhage. Stroke 2006;37:2326-2330.
Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. J Neurosurg 2005;103:9-17.
Findlay JM, Kassell NF, Weir BK, et al. A randomized trial of intraoperative, intracisternal tissue plasminogen activator for the prevention of vasospasm. Neurosurgery 1995;37:168-178.
Zabramski JM, Spetzler RF, Lee KS, et al. Phase I trial of tissue plasminogen activator for the prevention of vasospasm in patients with aneurysmal subarachnoid hemorrhage. J Neurosurg 1991;75:189-196.
Egemen N, Turker RK, Sanlidilek U, et al. The effect of intrathecal sodium nitroprusside on severe chronic vasospasm. Neurol Res 1993;15:310-315.
Kistler JP, Lees RS, Candia G, et al. Intravenous nitroglycerin in experimental cerebral vasospasm. A preliminary report. Stroke 1979;10:26-29.
Wurm G, Tomancok B, Nussbaumer K, et al. Reduction of ischemic sequelae following spontaneous subarachnoid hemorrhage: a double-blind, randomized comparison of enoxaparin versus placebo. Clin Neurol Neurosurg 2004;106:97-103.
Siironen J, Juvela S, Varis J, et al. No effect of enoxaparin on outcome of aneurysmal subarachnoid hemorrhage: a randomized, double-blind, placebo controlled clinical trial. J Neurosurg 2003;99:953-959
Feigin V, Anderson N, Rinkel G, et al. Corticosteroids for aneurysmal subarachnoid haemorrhage and primary intracerebral haemorrhage. Cochrane Database Syst Rev: CD004583, 2005.
Haley EC Jr, Kassell NF, Alves WM, et al. Phase II trial of tirilazad in aneurysmal subarachnoid hemorrhage A report of the Cooperative Aneurysm Study. J Neurosurg 1995;82:786-790.
Haley EC Jr, Kassell NF, Apperson-Hansen C, et al. A randomized, double-blind, vehicle-controlled trial of tirilazad mesylate in patients with aneurysmal subarachnoid hemorrhage: a cooperative study in North America. J Neurosurg 1997;86:467-474.
Asano T, Takakura K, Sano K, et al. Effects of a hydroxyl radical scavenger on delayed ischemic neurological deficits following aneurysmal subarachnoid hemorrhage: results of a multicenter, placebo-controlled doubleblind trial. J Neurosurg 1996;84:792-803.
Yanamoto H, Kikuchi H, Sato M, et al. Therapeutic trial of cerebral vasospasm with the serine protease inhibitor, FUT-175, administered in the acute stage after subarachnoid hemorrhage. Neurosurgery 1992;30:358-363.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 211 kb)
Rights and permissions
About this article
Cite this article
Athar, M.K., Levine, J.M. Treatment Options for Cerebral Vasospasm in Aneurysmal Subarachnoid Hemorrhage. Neurotherapeutics 9, 37–43 (2012). https://doi.org/10.1007/s13311-011-0098-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-011-0098-1