
Abstract
The efficacy and safety of lamotrigine extended-release tablets (LTG XR) as monotherapy for partial seizures were evaluated using the conversion-to-monotherapy design, and historical data as the control. This methodology was recently approved by the United States Food and Drug Administration, and this study is the first historical control design in epilepsy to complete enrollment. Patients ≥13 years old with uncontrolled partial epilepsy receiving monotherapy with valproate or a noninducing antiepileptic drug were converted to once-daily LTG XR (250 mg or 300 mg) as monotherapy and were followed up for 12 additional weeks. Efficacy was measured by the proportion of patients meeting predefined escape criteria for seizure worsening compared with aggregated pseudoplacebo control data from 8 previously conducted conversion-to-monotherapy trials. Nonoverlap of the 95% confidence limit for LTG XR and the 95% prediction interval of the historical control denotes efficacy. Of 226 randomized patients, 174 (93 in 300 mg/day group and 81 in 250 mg/day group) started withdrawal of the background AED and were evaluated for escape. In the historical control analysis population, the lower 95% prediction interval of the historical control (65.3%) was not overlapped by the upper 95% confidence limit of either LTG XR (300 mg/day; 37.2%) or LTG XR (250 mg/day; 43.4%). Adverse events were reported in 53% and 61% of patients receiving LTG XR (300 mg/day and 250 mg/day, respectively). LTG XR (250 mg or 300 mg once daily) is effective for conversion-to-monotherapy treatment of partial seizures in patients ≥13 years old.
Similar content being viewed by others

Introduction
Once a new antiepileptic drug (AED) is demonstrated to be safe and effective as adjunctive therapy, it is also desirable to determine its efficacy as monotherapy, a common and important setting for its use. Monotherapy trials of AEDs have been fraught with difficulty [1–3]. A key consideration for a monotherapy study is the choice of control. The options for demonstration of a monotherapy effect are 1) demonstration of equivalence or noninferiority using a known effective agent as the control group, and 2) demonstration of superiority to a comparator. Use of an active comparator would be technically acceptable if it had been established that the active comparator in a similar trial design and patient population had regularly demonstrated superiority to another therapy, and thereby confirmed that the trial methodology could separate an effective drug from an ineffective one (a concept known as assay sensitivity). However, this situation does not exist for AEDs. Thus, a demonstration of noninferiority in an active control trial may not be an acceptable proof of efficacy. The standard method of demonstrating superiority is comparison to a placebo control. However, a placebo control is not an appropriate alternative in a monotherapy study of a life-threatening condition such as epilepsy. In fact, Robert Temple of the United States (U.S.) Food and Drug Administration (FDA) suggested that if a placebo control is to be used in a life-threatening condition, an adjunctive design would be more appropriate [4].
Because of these considerations, a conversion-to-monotherapy design was developed in the 1990s [5]. The conversion-to-monotherapy design used an active but presumably inferior medication consisting either of low-dose valproic acid (VPA) or a low dose of the drug under investigation (a “pseudoplacebo”) as a control. Such use of a pseudoplacebo raises ethical concerns as it appears not to fully comply with the principle of the Helsinki Declaration that patients in clinical studies should receive the best proven therapy [6]. In fact, in the pseudoplacebo-controlled withdrawal to monotherapy studies performed to date, 74.9% to 95.9% of patients worsened, as defined by predetermined exit criteria, with half of the studies having an estimated percent exiting of between 77.2 and 87.5% [7]. An alternative approach that avoids these ethical concerns is the use of a historical control, in which meta-analysis of multiple placebo-controlled or pseudoplacebo-controlled studies is performed to “model” the behavior of a placebo or pseudoplacebo in a randomized trial [7, 8]. This approach avoids exposure of patients with a potentially life-threatening illness to a potentially inferior treatment. Moreover, this approach has recently been accepted by the FDA as a pathway to monotherapy approval of new AEDs with the condition that any study using the historical control as a comparator be similar, as much as possible, in design, patient population, and endpoint analysis to the studies that comprise the historical control. Such a historical control was recently compiled [7] and showed a consistently high escape rate (the primary outcome measure), thereby allowing it to be used as an alternate to a concurrent placebo comparison. The study described herein, evaluating the efficacy of LTG XR as conversion-to-monotherapy for the treatment of partial seizures, used this novel historical control comparator, consisting of data from the pseudoplacebo arms of 8 similarly designed and conducted conversion-to-monotherapy studies [7, 9], and it is the first such study to complete enrollment. This study exemplifies the issues and difficulties of using this approach.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The study was approved by all required ethics committees, and written informed consent was obtained from all study participants. Study details are publicly available (see www.clinicaltrials.gov, No. NCT00355082).
Patients
Eligible patients were ≥13 years of age, experienced ≥4 partial seizures during the 8-week baseline phase (with ≥1 seizure in each 4-week period), and they were receiving a stable monotherapy regimen with a nonenzyme-inducing AED. The patients were not experiencing any primary generalized seizures. The first 4 weeks of the baseline phase could be retrospective if proper seizure documentation was available. Patients taking estrogen-based hormonal therapy were excluded.
Procedures
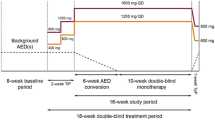
Figure 1 shows the study design. This randomized, double-blind study conducted in 7 countries (57 sites in Argentina, Chile, Costa Rica, Korea, Russia, the Ukraine, and the U.S.) between May 2006 and May 2008 was comprised of a screening phase (<2 weeks), a noninterventional baseline phase (8 weeks), and a double-blind treatment (DBT) phase (22 to 23 weeks). Patients who met the baseline seizure frequency criteria entered the DBT phase and were randomized (1:1) to receive LTG XR (250 mg or 300 mg once daily). The use of 2 randomized treatment groups (250 mg and 300 mg once daily) was implemented to be consistent with the designs of the studies from which the historical control group was drawn, all of which had 2 randomized and blinded arms. The target test dose was 300 mg; the alternative dose was chosen to be 250 mg. These doses were chosen because they both are in the range of effective adjunctive doses of immediate-release (IR) or extended-release (XR) lamotrigine and both doses were expected to be effective as monotherapy.

Schematic representation of study design. AED = antiepileptic drug
The DBT phase consisted of a 10- to 11-week conversion phase and a 12-week maintenance (monotherapy) phase. During conversion, the LTG XR dose was escalated based on the background AED and in accordance with the product label to the target dose, and the background AED was withdrawn. The withdrawal of the background AED occurred during a 4- to 6-week timeframe, according to schedules depending on the specific AED. After background AED withdrawal was complete, patients entered the maintenance (monotherapy) phase and received 12 weeks of LTG XR monotherapy. Patients who completed or were discontinued from the blinded study drug treatment during the DBT phase (and who did not elect to enroll in an optional open-label continuation phase) were converted to another AED or were returned to their prior AED in consultation with the investigator. Seizure type and frequency were recorded on a daily basis throughout the study.
Endpoints and Statistical Analyses
The requirement for use of the historical control is that the study using this type of control should match, in inclusion/exclusion criteria, trial design, trial conduct, and analysis, the studies contributing to the historical control dataset to the greatest extent possible. The 8 trials contributing to the historical control dataset were conducted in the mid-1990s and compared a test medication to a pseudoplacebo that was either low-dose valproate or a low dose of the test medication. Similarities among the 8 studies in patient population, design, conduct, and endpoints allowed aggregation and use of the data as a historical control [7].
The current study was conducted to determine whether LTG XR is superior to a pseudoplacebo historical control in the ability to prevent worsening when patients are converted from their background AED to LTG XR monotherapy. Worsening in the historical control studies was defined as the proportions of patients meeting pre-defined escape criteria, which were very similar among the historical control studies. Efficacy in the current study was initially evaluated as the proportion of patients not meeting ≥1 pre-defined escape criteria, and as the proportion of patients who did not prematurely discontinue from the study for any reason (including escape and adverse events). It was eventually determined that the designation of the discontinuation endpoint as primary had resulted from confusion as to the historical control analysis, and that the secondary endpoint of the proportion of patients meeting predefined escape criteria was consistent with the methodology used in the historical control. Both endpoints were calculated and are reported herein. The measure evaluating efficacy as the proportion of patients who prematurely discontinued for any reason is more conservative than limiting endpoint evaluation only to the pre-defined escape criteria.
Escape criteria, calculated relative to the baseline phase, included: 1) doubling of average monthly seizure frequency, calculated as the sum of countable partial seizures starting the day prior to the study visit and extending back 28 days; 2) doubling of the highest consecutive 2-day seizure frequency; 3) emergence of a new, more severe seizure type; or 4) clinically significant prolongation of generalized tonic-clonic seizures or worsening of seizure considered by the investigator to require intervention.
As data became available during the study, a sample was evaluated for correct application of the escape criteria. A number of errors were detected, in which a patient met an escape criterion but was not discontinued. As a result, study site personnel and sponsor monitors were reinstructed regarding the methodology for evaluating escape. After completion of the DBT phase and prior to unblinding, seizure data were evaluated against the escape criteria and compared with escapes reported by investigators. In addition, adverse events and concurrent use of benzodiazepines for seizure events were investigated for indications of seizure worsening, and these were included in the calculated endpoints (i.e., escape criterion number 4). Results indicated under-reporting of escape by investigators. Therefore, analysis of escape and discontinuation, based on both investigator-reported endpoints and on those calculated from the seizure data, were summarized and reported herein. As the historical control is based on the escape endpoint, statistical comparisons with the historical control group are only reported for the calculated analyses.
The main efficacy analysis compared the upper 95% confidence limit of the proportion of patients in the LTG XR (300 mg/day) group who met escape criteria with the lower 95% prediction interval of the historical pseudoplacebo control data (65.3% [7]). The proportion of patients who prematurely withdrew from the study for any reason was also evaluated. Nonoverlap of the limits was interpreted as denoting efficacy (Fig. 2).

Evaluation using the historical control. CL = confidence limit
In the planned analysis, the proportion of patients in each dose group meeting escape criteria (beginning at the start of the withdrawal of background AED through the end of the DBT phase) was calculated, and the 95% confidence limit was determined. The analysis based on the historical control method used the Kaplan-Meier estimate of the proportion of patients in the pseudoplacebo control arms of the historical control studies meeting escape criteria beginning at the start of withdrawal of background AED through 112 days. From this estimate, a prediction interval for escape was calculated against which a future study could be compared [7]. This prediction interval is more conservative than the standard confidence interval.
In the planned analyses, the primary efficacy population was the per protocol population, which included all randomized patients who received the study drug and began withdrawal of the background AED, excluding those with major protocol violations (study per-protocol analysis). As the analysis of the historical data did not exclude any patients with major protocol violations (i.e., excluded only patients who did not begin withdrawal of the background AED), the endpoint in the current study was also analyzed in a similar fashion (historical control analysis). The intent-to-treat (ITT)/safety population, defined as all patients who received at least 1 dose of study medication, was used for demographic data and to assess safety.
For each patient, the seizure frequencies during the entire treatment phase were compared to the seizure frequency during baseline. Based on these comparisons, each patient was classified as having a reduction in seizure frequency of at least 25%, at least 50%, at least 75%, 100%, or having at least a 25% increase in seizure frequency. The number of patients falling into each of these categories are displayed for each dose of LTG-XR using the ITT population.
Safety was assessed by the recording of adverse events (AEs), serious adverse events (SAEs), and premature withdrawal because of AEs. The AEs were defined as any untoward medical occurrences, regardless of their suspected cause. SAEs were defined as AEs that were fatal, life-threatening, or permanently disabling, or that were a congenital anomaly or cancer, or that required inpatient hospitalization.
Results
Patients
The number of patients randomized to treatment was 226, and 223 took study medication (ITT population). The per protocol population was comprised of 174 patients (93 in the LTG XR [300 mg/day] group and 81 in the LTG XR [250 mg/day] group). Among the patients excluded from the per protocol population were 18 patients (4 in the 300 mg/day group and 14 in the 250 mg/day group) who did not begin withdrawal of the background AED. These patients were the only patients excluded from the historical control analysis. Patient disposition and the derivation of analysis populations are shown in Fig. 3. Demographics and baseline clinical characteristics are shown in Table 1.

Patient disposition. AED = antiepileptic drug; ITT = intent-to-treat; LTG = lamotrigine
Efficacy
Table 2 presents both the investigator reported and the calculated efficacy endpoints. For the proportion of patients who met escape criteria, the lower 95% prediction interval of the historical control (65.3%) was not overlapped by the upper 95% confidence limit of either the 300 mg/day group (37.2% for the historical control population) or the 250 mg/day group (43.4%), which is a result that reflects efficacy of LTG XR as monotherapy. Results were similar, regardless of whether major protocol violators were included in the analysis (study per-protocol analysis or historical control per-protocol analysis). Results remained positive even when the more conservative endpoint of discontinuation for any reason was evaluated. The most frequent endpoint for escape was doubling of the highest 2-day seizure frequency followed by doubling of the highest monthly seizure frequency (Table 3).
Change in seizure frequency for the entire treatment period is presented in Table 4. Approximately 50% of patients experienced at least a 50% reduction in seizure frequency compared to baseline. Unlike usual efficacy studies in epilepsy in which the primary endpoint is based on reduction in seizure frequency, the endpoint in the present study was driven by seizure frequency, but was more broadly based on pre-defined escape criteria and was therefore a measure of “lack of worsening.”
Safety and Tolerability
Adverse events were reported in 53% and 61% of the groups receiving LTG (300 mg/day and 250 mg/day), respectively. Headache and dizziness were the most common AEs (Table 5). Except for rash and insomnia, AE frequency was similar between treatment groups. No deaths occurred. Treatment-emergent serious AEs (SAEs) were reported in 3 patients (3%) in the LTG XR (300 mg/day) group and 5 patients (5%) in the LTG XR (250 mg/day) group. No SAE was reported in >1 patient in either treatment group. The only SAEs judged by the investigator to be reasonably attributable to the study drug were pyrexia and rash, which were reported concurrently for a patient in the LTG XR (250 mg/day) group. AEs leading to discontinuation were reported for 4 patients (4%) in the LTG XR (300 mg/day) group and 11 patients (10%) in the LTG XR (250 mg/day) group. The only AE leading to discontinuation of more than 1 patient was rash (1 patient [300 mg/day] and 7 patients [250 mg/day]).
Discussion
This study constitutes the first clinical trial of AED monotherapy to use a historical control as comparator. Because this was the first epilepsy study to be completed using a historical control, and because the analysis plan was developed before publication of the methodology article using comparison to a historical control [7], the current study had some initial errors in methodology (e.g., selection of a primary endpoint that did not match the historical control and initial failure to compute escape based on seizure diary data). Fortunately, the desired endpoint had been included as a secondary endpoint, and all of the initial errors could be addressed post hoc. The challenges notwithstanding, the data did provide support for the efficacy of LTG XR in monotherapy, and the results can be instructive to others contemplating the use of historical control methodology. In the present study, once daily treatment with LTG XR (250 mg/day or 300 mg/day) met the regulatory criterion for demonstration of efficacy (i.e., nonoverlap of the 95% prediction interval of the historical data and the 95% confidence limits of the test data for escape). It is of interest to compare the results of this study with those of the previously conducted conversion to monotherapy trial with the immediate-release formulation of lamotrigine, which was one of the historical control trials [10]. Because the older study only enrolled patients taking enzyme-inducing AEDs, the dose of lamotrigine was higher in that study (500 mg/day) making a direct comparison with the current study difficult. Due to these enzyme-inducing effects, the patients in the older study were exposed to the equivalent of 250 mg per day of lamotrigine during the monotherapy conversion period. Therefore, the most appropriate comparison with the older study would use only the subset of patients in the current study who were in the 250 mg arm taking neutral AEDs at entry. This comparison reveals similar escape rates: 38% for LTG XR and 44% for LTG IR. In this study and in the historical control studies, efficacy was defined as lack of worsening rather than as seizure improvement, which is the manner of defining efficacy in a typical add-on, placebo-controlled trial.
The driver for the use of historical data as a control is the ethical concern for using a known inferior treatment in the study of an illness with significant morbidity and mortality. The danger of known inferior treatment in epilepsy efficacy studies is discussed in recent publications [11, 12]. In a review of recent epilepsy clinical studies, the authors noted that fluctuation in seizure frequency is a normal occurrence in epilepsy, but it is almost always worse in the placebo arm of placebo-controlled trials of adjunctive therapy [11]. In addition, in a review of data from more than 100 adjunctive treatment trials involving more than 17,000 patients with epilepsy, the incidence of sudden unexplained death in epilepsy among patients randomized to placebo (0.69%/year) was almost fivefold higher than the incidence in patients receiving an efficacious dose of the test drug (0.09%/year), even in the setting of adjunctive treatment [12]. In the context of observations such as these, it is reasonable to believe that conversion to an active drug (in this case LTG XR) would be substantially safer than conversion to a pseudoplacebo.
Historical control use requires that a number of design and conduct criteria be met [7]. Specifically, it is crucial that the study incorporating the historical control be consistent with the studies contributing to the historical control dataset, with respect to study design, patient population, evaluation criteria, and analysis plan. The present study generally met these key criteria (Table 6); however, there were some substantive differences between the control population and the population enrolled in this study, including characteristics such as background AEDs and study site location. Five of the historical studies allowed patients to enter on 2 AEDs if the second AED was at no more than half the recognized minimal effective dose or serum concentration. In all studies, the second AED was discontinued at the start of dosing with the study drug. Approximately 17% of the historical pseudoplacebo control patients used a second AED. Because of the added complexity, and the relatively small contribution of a second background AED to the historical control, the present study allowed only 1 background AED. The present study excluded patients taking enzyme-inducing AEDs (primarily carbamazepine), as these AEDs induce the clearance of LTG approximately twofold. In the 6 historical control studies for which data are available, approximately 64% of patients were converted from the enzyme-inducing AED carbamazepine. An analysis by French et al. showed no significant impact on escape of withdrawal from carbamazepine [7]. Although the majority of patients in the current study were taking VPA, LTG XR was dosed according to current recommendations that result in comparable LTG serum concentrations to patients taking neutral AEDs. Several of the historical control trials also enrolled patients taking VPA. However, this represents a difference from the historical control population.
There was also an impact of study site location. Unlike the historical control studies, which were conducted virtually entirely in the U.S., the present study was conducted in 7 countries with 25% of patients coming from the U.S. Post hoc analyses by region (U.S./non-U.S.) showed higher escape in the U.S. region, but even in this small group, the upper 95% confidence limit did not overlap the lower 95% prediction interval of the historical data (data not shown). It must be noted that the study was not powered for this analysis. Study site location has been specified as an important factor for the FDA when assessing historical control trials, and in future studies the effect of location should be carefully monitored.
It would have been useful to be able to compare other characteristics of the current trial population relative to the historical control population. Unfortunately, characteristics such as etiology, site of focality of partial epilepsy, and number of previous drugs failed are not available (typical for epilepsy drug trials).
There has been recent discussion of rising placebo response in epilepsy trials [13]. Rising placebo response would be of more concern in a study with no placebo arm, as one would not be able to determine the proportion of patients in the active arms that might be placebo responders. This would also be a problem for active control equivalence trials, which are the alternative design for assessment of monotherapy. One reassuring point is that in the studies that comprised the historical control, which were conducted over more than a decade, there was no increase in placebo responder rate over time [7].
As with the studies that contributed to the historical control, the present study consisted of an 8-week baseline phase followed by introduction of the test agent (LTG XR), withdrawal of the background AED, and monotherapy treatment with LTG XR. In the present study, both average age and baseline seizure frequency were consistent with those in the studies contributing to the historical control. Both the current study and the historical control studies enrolled control patients with a diagnosis of partial epilepsy, including seizure types of simple partial, complex partial, and secondarily generalized tonic-clonic. Although the entry criteria pertaining to seizure type were consistent among the present study and the studies contributing to the historical control, the distribution of seizure types, available from 4 of the 8 historical studies, showed a higher percentage of patients (83% to 95%) with complex partial seizures at entry compared with the present study (approximately 62%). The potential implications of these differences cannot be definitively elucidated on the basis of the study data.
In the present study, all patients were aware that they would receive either 1 of the 2 doses of LTG XR, a drug known to be effective as an adjunctive therapy. The expectation would be that at least 1 of the doses would be effective. In the historical control studies, the pseudoplacebo arm, although intended to be suboptimal, was also intended to be minimally effective to protect against more serious generalized tonic-clonic seizures and status epilepticus, and none of the consent forms available for review by French et al. stated that 1 of the study treatments would be suboptimal [7]. It is not known what may have been communicated to the patients by investigators. How these differences may have resulted in bias in the present study is unknown, as is the magnitude of any potential bias from the lack of an internal control; however, the objective nature of seizure counts, on which the endpoint is based, provides some hedge against bias. This limitation should be borne in mind when interpreting the results of the study.
Historical control use in epilepsy was recently a subject of review by a FDA Advisory Committee [14], which endorsed the validity of the historical control study under limited conditions (conversion-to-monotherapy design and proof-of-effectiveness as adjunctive therapy). As part of this review, the FDA performed many “worst-case scenario” evaluations that did not contradict the conclusions of the study as presented here.
The results of the current study demonstrate that a monotherapy study using a historical control is indeed feasible. Future studies might do well to calculate escape in an automated manner in real time at clinic visits. A conversion-to-monotherapy study incorporating the historical control does indeed provide important information to clinicians who often wish to convert their patients from one AED to another. It is important to demonstrate that such conversion does not put the patient at undue risk of seizure worsening, which routinely occurred in the pseudoplacebo control arms of the studies that make up the historical control. However, it does not provide information on whether individual patients will improve with the conversion nor is it intended to do so. This study does not address whether LTG is as effective as other monotherapies, although this has been addressed in other studies in which LTG appeared to be as effective as standard or new AEDs as initial monotherapy [15]. The results of the present study demonstrate that it is possible to convert patients from another drug to LTG XR (250 mg once daily or 300 mg once daily), and that this conversion is well-tolerated, but the results do not address the appropriateness of LTG XR as initial monotherapy.
References
Perucca E. Designing clinical trials to assess antiepileptic drugs as monotherapy: difficulties and solutions. CNS Drugs 2008;22:917–938.
Sachdeo R. Monotherapy clinical trial design. Neurology 2007;69(24 suppl 3):S23-S27.
Gilliam FG. Limitations of monotherapy trials in epilepsy. Neurology 2003;60(suppl 4):S26-S30.
Temple R, Ellenberg SS. Placebo-controlled trials and active-control trials in the evaluation of new treatments. Part 1: ethical and scientific issues. Ann Intern Med 2000;133:455–463.
Pledger GW, Kramer LD. Clinical trials of investigational antiepileptic drugs: monotherapy designs. Epilepsy 1991;32:716–721.
Declaration of Helsinki. Tokyo: World Medical Association, 2004, October 9.
French JA, Wang S, Warnock B, Temkin N. Historical control monotherapy design in the treatment of epilepsy. Epilepsia 2010;51:1936–1943.
Baulac M. Historical data in the design and interpretation of trials with newly diagnosed patients. Epilepsy Res 2006;68:77–81.
Beydoun A, Kutluay E. Conversion to monotherapy: clinical trials in patients with refractory partial seizures. Neurology 2003;60(suppl 4):S13-S25.
Gilliam F, Vazquez B, Sackellares et al. An active-control trial of lamotrigine monotherapy for partial seizures. Neurology 1998;51:1018–1025.
Somerville ER. Aggravation of partial seizures by antiepileptic drugs: is there evidence from clinical trials? Neurology 2002;59:79–83.
Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol 2011, Sept 19 [Epub ahead of print].
Rheims S, Perucca E, Cucherat M, Ryvlin P. Factors determining response to antiepileptic drugs in randomized controlled trials. A systematic review and meta-analysis. Epilepsia 2011;52:219–233.
Food and Drug Administration Center for Drug Evaluation and Research. Summary minutes of the Peripheral and Central Nervous System Drugs Advisory Committee meeting. March 10, 2011. Available at: http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/PeripheralandCentralNervousSystemDrugsAdvisoryCommittee/UCM250489.pdf. Accessed May 5, 2011.
Marson AG, Al-Kharusi AM et al.; SANAD Study group. The SANAD study of effectiveness of valproate, lamotrigine, or topiramate for generalized and unclassifiable epilepsy: an unblinded randomised controlled trial. Lancet 2007;369:1016–1026.
Acknowledgments
We thank Jane Saiers, Ph.D. (The Write Medicine, Inc.) for assistance with editing the manuscript. This work was supported by (and conducted by) GlaxoSmithKline. The team from the sponsor (A.E.H., P.T.C., J.A.M.) in collaboration with the coauthors (J.A.F., N.R.T., and B.F.S.) led the design of the study, and the analysis and interpretation of the data, as well as the preparation, review, and approval of this article. One author (B.F.S.) was also an investigator in the study. All authors had full access to the data. Some authors (J.A.F., N.R.T., and B.F.S.) have consulted for (or conducted research funded by) GlaxoSmithKline and other pharmaceutical companies. One author (A.E.H.) is an employee of GlaxoSmithKline; and 2 other authors (P.T.C. and J.A.M.) were employed by GlaxoSmithKline at the time the study was conducted. GlaxoSmithKline funded the research described in this article. This protocol was posted on the Clinicaltrials.gov database (No. NCT00355082).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 340 kb)
Rights and permissions
About this article
Cite this article
French, J.A., Temkin, N.R., Shneker, B.F. et al. Lamotrigine XR Conversion to Monotherapy: First Study Using a Historical Control Group. Neurotherapeutics 9, 176–184 (2012). https://doi.org/10.1007/s13311-011-0088-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-011-0088-3