
Abstract
Mammalian fertilization begins with the fusion of two specialized gametes, followed by major epigenetic remodeling leading to the formation of a totipotent embryo. During the development of the pre-implantation embryo, precise reprogramming progress is a prerequisite for avoiding developmental defects or embryonic lethality, but the underlying molecular mechanisms remain elusive. For the past few years, unprecedented breakthroughs have been made in mapping the regulatory network of dynamic epigenomes during mammalian early embryo development, taking advantage of multiple advances and innovations in low-input genome-wide chromatin analysis technologies. The aim of this review is to highlight the most recent progress in understanding the mechanisms of epigenetic remodeling during early embryogenesis in mammals, including DNA methylation, histone modifications, chromatin accessibility and 3D chromatin organization.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Fertilization is regarded as one of the greatest feats of nature, beginning with a sperm combining with an oocyte, during which these two terminally differentiated germ cells are converted into a totipotent zygote (Rivera and Ross, 2013; Canovas and Ross, 2016; Xu and Xie, 2018). Dramatic changes occur to ensure that a series of pivotal biological events proceed, including oocyte activation(Amdani et al., 2015; Yeste et al., 2017; Bonte et al., 2018) and maternal-to-zygotic transition (MZT) coordinated with zygotic gene activation (ZGA) (Minami et al., 2007; Tadros and Lipshitz, 2009; Jukam et al., 2017; Eckersley-Maslin et al., 2018; Schulz and Harrison, 2019), followed by the first cell-fate decision and lineage-specific differentiation (Mihajlovic and Bruce, 2017; Zhang et al., 2018; Yao et al., 2019). A precise regulatory network must function appropriately to support such a major transition in a brief period. Epigenetic information plays a major role in the maintenance of cell identity and the control of gene expression. Epigenetic modifications in terminally differentiated gametes, including DNA methylation (Guo et al., 2014b; Iurlaro et al., 2017; Zhu et al., 2018; Zeng and Chen, 2019), histone modifications (Dahl et al., 2016; Liu et al., 2016b; Zhang et al., 2016; Inoue et al., 2017a; Wang et al., 2018a), modifications affecting chromatin accessibility (Wu et al., 2016; Jachowicz et al., 2017; Gao et al., 2018a) and 3D chromatin organization (Du et al., 2017; Ke et al., 2017; Kragesteen et al., 2018), can be reset to a basal state after fertilization to achieve totipotency and support development into a new individual. Unexpected changes in the external environment may lead to irreversible damage to proper growth by altered epigenetic patterns that may interfere with gene expression (Legault et al., 2018; Risal et al., 2019; Yu et al., 2019).
Recently, methodological advances such as low-input chromatin analysis technologies (Xu and Xie, 2018) have provided approaches to overcome the inaccessibility of early-stage embryos and elucidate the epigenetic remodeling mechanism at the whole-genome level. In this review, we will discuss the dynamic processes of epigenetic re-establishment during mouse and human early embryonic development, focusing on how these epigenetic mechanisms may promote the acquisition of totipotency and the differences between mice and humans in these reprogramming events.
DNA METHYLATION
Large-scale asymmetric demethylation upon mouse fertilization
DNA methylation (5mC) is an inheritable type of epigenetic mark that provides molecular memory to preserve the transcriptional order during mammalian early embryo development (Li and Zhang, 2014; Messerschmidt et al., 2014; Okae et al., 2014a; Zeng and Chen, 2019). The methylation of cytosine residues is catalyzed by the de novo DNA methyltransferases (DNMT3A/B) and maintenance DNA methyltransferase (DNMT1), while TET family enzymes act in a multistep process to achieve DNA demethylation (Okano et al., 1999; Kohli and Zhang, 2013; Wang et al., 2014; Wu and Zhang, 2014, 2017; Verma et al., 2018). Before the first cleavage, both the maternal and paternal genomes undergo widespread active and passive demethylation, except in imprinting control regions (ICRs) and some retrotransposons (Smith et al., 2012; Guo et al., 2014a; Messerschmidt et al., 2014; Shen et al., 2014; SanMiguel and Bartolomei, 2018). The paternal genome is more rapidly and actively demethylated, along with the exchange of protamines for maternal histones, including HIRA-mediated H3.3 deposition (Loppin et al., 2005; Skene and Henikoff, 2013; Inoue and Zhang, 2014).
Immunofluorescence data show that the paternal genome loses the 5mC signal before the first DNA replication of the zygote at pronuclear stage 3 (PN3) (Oswald et al., 2000). Further investigation suggests that paternal 5mC is converted to 5hmC by TET3 and then removed via replication-coupled passive dilution (Gu et al., 2011; Inoue and Zhang, 2011). However, another study on mouse zygotes shows that the early loss of paternal 5mC is unaffected when 5hmC formation is abrogated by small-molecule inhibition of TET activity (Amouroux et al., 2016). TET3 seems to be unrequired for loss of 5mC in the early zygote. Instead, TET3 targets new 5mC generated by DNMT3A and DNMT1, which indicates that accumulation of paternal 5hmC is resulted from de novo DNA methylation rather than the TET3-driven hydroxylation of paternal 5mC (Amouroux et al., 2016). This provides another possible mechanism determining paternal DNA methylation dynamics. The maternal genome is more resistant to this initial wave of demethylation, di-methylated histone H3 lysine 9 (H3K9me2) is reported to promote CG methylation maintenance in maternal genome by recruiting the maternal factor PGC7 (also known as STELLA, DPPA3 (Nakamura et al., 2007; Nakamura et al., 2012; Han et al., 2018; Zeng et al., 2019). However, the appropriateness of this conclusion is queried by a recent study which shows a global increase in 5mC level induced by uncontrolled de novo methylation by DNMT1 in STELLA-deficient zygotes (Li et al., 2018b). Besides, another study shows H3K9me2 enrichment is reduced by oocyte specific deletion of G9a (an H3K9me2 methyltransferase), but the CG methylation is minimally affected (Au Yeung et al., 2019). Thus, the role of H3K9me2 and Stella in DNA methylation protection of the maternal genome are worthy of further verification. In addition, the maternal genome is more prone to passive demethylation during sequential cleavages, which is DNA replication dependent, giving rise to epigenetic asymmetry in the early embryo (Stewart et al., 2016; Guo et al., 2017; Xu and Xie, 2018).
Within the developing embryos, the initial global hypomethylation level maintains naïve pluripotency and guarantees accurate future differentiation regulation (Nichols and Smith, 2009; Theunissen et al., 2016; Peng et al., 2019). DNA methylation is re-established in lineage-specific regions beginning in the blastocyst stage (Zhang et al., 2018). The exit from pluripotency and entry into lineage-specific differentiation have been proven to be associated with genome-wide de novo DNA methylation, during which the co-expression of the DNMT3 and TET enzymes promotes coherent genome-wide oscillations of CpG-density-dependent DNA methylation (Smith et al., 2017; Rulands et al., 2018). These findings provide insights into the emergence of epigenetic heterogeneity during early embryo development, indicating that dynamic changes in DNA methylation might influence early cell fate decisions (Rulands et al., 2018).
Aberrant reprogramming of the DNA methylome may lead to developmental defects and embryonic arrest. In somatic cell nuclear transfer (SCNT) embryos, the efficiency of embryonic development is much lower than that in normally fertilized embryos, owing to the multiple epigenetic barriers that impede SCNT-mediated reprogramming, including abnormally higher levels of DNA methylation in cloned embryos (Dean et al., 2001; Yang et al., 2007; Peat and Reik, 2012; Teperek and Miyamoto, 2013). By comparing the DNA methylome of SCNT embryos and fertilized embryos, differentially methylated regions (DMRs) are identified (Gao et al., 2018b). Unexpected re-methylation is found in cloned embryos, which possesses higher methylation level than the former stage, termed as re-methylated DMRs (rDMRs). rDMRs are enriched at promoters, SINE and long terminal repeat (LTR). Re-methylation-affected downregulated genes at 2-cell stage of SCNT embryos are highly enriched of totipotent- and developmental-related genes (Gao et al., 2018b). This finding is compatible with the perspective that an aberrant DNA methylome at the ZGA stage may lead to developmental defects and female infertility (Wang and Dey, 2006). Moreover, the totipotency marker Mouse endogenous retrovirus type L (MERVL), one of the endogenous retroviruses (ERVs) specifically expressed at the 2-cell stage (Svoboda et al., 2004; Ribet et al., 2008; Eckersley-Maslin et al., 2016; Huang et al., 2017), exhibits a dramatically higher remnant methylation state in SCNT embryos than in fertilized embryos, and its transcription activity was evidently repressed (Gao et al., 2018b). These observations collectively suggest that the impact of methylation memory from gametes or donor cells should be considered when investigating the epigenetic reprogramming of offspring.
Generally similar reprogramming patterns but with different details in human pre-implantation embryos to those in mouse
Abundant studies on the reprogramming of the DNA methylome have paved the way for deciphering the mechanism of DNA methylation reprogramming in early human embryos (Fulka et al., 2004; Lister et al., 2009; Molaro et al., 2011; Smith and Meissner, 2013; Guo et al., 2014b). However, compared to the commonly utilized mammalian animal models, human embryos show a much more complex genetic background, which may present barriers to their analysis. Remarkable work has been accomplished on the genome-wide profiling of DNA methylation in human pre-implantation embryos, yet multiple issues remain to be addressed. In contrast to previous observations in mice, which show genome-wide demethylation occurs mainly at the 1-cell stage, the initial rapid DNA demethylation occurs from fertilization to 2-cell stage in human embryos and keep stable until the morula stage, then followed by a second reduction from morula stage to blastocyst stage (Guo et al., 2014b). A set of transient, maternal DMRs are found both in mouse and human early embryos, but more DMRs are resolving to hypermethylation in human and most of these short-lived DMRs are not equivalently regulated in mouse. Also, the repetitive element regulation is more diverse in human than that in mouse (Smith et al., 2014). Notably, the human paternal genome undergoes demethylation at a much faster rate than the maternal genome, which is similar to the situation in early mouse embryos (Fulka et al., 2004; Okae et al., 2014a; Smith et al., 2014; Zhu et al., 2018). The paternal genome shows rather lower DNA methylation level than the maternal one by the end of the zygotic stage (Fulka et al., 2004; Okae et al., 2014a; Smith et al., 2014). Moreover, H3K4me3-marked active genes in human ESCs are essentially devoid of DNA methylation in both mature gametes and human pre-implantation embryos (Guo et al., 2014b). Taking advantage of single-cell post-bisulfite adapter tagging (PBAT) DNA methylome sequencing analysis at the single-cell level and single-base resolution, issues such as the inclusion of aneuploid embryos in samples and the heterogeneity of DNA methylation among individuals can be addressed (Zhu et al., 2018). The global DNA methylation pattern displays a dynamic balance between dramatic demethylation and focused intensive de novo methylation. The major demethylation occurs in a stepwise and wave-like manner, while focused de novo methylation prevails at the 8-cell stage. Furthermore, the maternal genome is maintained in a more hypermethylated state at a wide variety of genomic loci during pre-implantation. In summary, these findings paved the way for revealing the mechanisms of the regulatory network of DNA methylation during early human embryogenesis. It remains to be determined whether the distinct features of asymmetric DNA methylomes may affect the transcriptome or play a role in cell fate decisions.
HISTONE MODIFICATIONS
Histone modifications are key regulatory events throughout pre-implantation embryogenesis that influence the interactions of transcriptional regulators with chromatin (Stewart et al., 2015; Skvortsova et al., 2018; Wu et al., 2018; Xu and Xie, 2018). However, previous studies on histone modification reprogramming during mammalian early embryo development are restricted to observations based on immunofluorescence staining, which exhibits a relatively low resolution for the analysis of underlying mechanisms.
Taking advantage of the newly launched low-input ChIP-seq and CUT&RUN methods, a high-resolution map for investigating the reprogramming of histone modifications during mammalian pre-implantation embryos was finally profiled. Here, we review recent progress in understanding the molecular mechanisms of histone modification reprogramming in early embryogenesis generated by these newly developed methods.
H3K4me3
A unique pattern in mouse oocytes and major resetting of H3K4me3 after fertilization
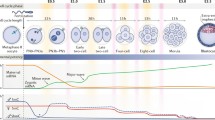
Histone modifications play critical roles in the spatiotemporal regulation of gene expression in mammals (Bannister and Kouzarides, 2011; Sadakierska-Chudy and Filip, 2015; Huang et al., 2019; Xia et al., 2019). Recently, low-input ChIP-seq methods were developed and utilized to profile the genome-wide landscape of histone modifications during ZGA and the first cell fate decision in mouse early embryos (Dahl et al., 2016; Liu et al., 2016b; Zhang et al., 2016) (Fig. 1). After fertilization, H3K4me3 (a mark of active promoters) in the paternal genome is rapidly depleted, but this mark is re-established during major ZGA. By contrast, the noncanonical form of H3K4me3 (ncH3K4me3), covering broad domains in both promoters and distal regions, is found in the maternal genome (making up ~ 22% of the genome) (Dahl et al., 2016; Zhang et al., 2016). ncH3K4me3 is readily established in mature oocytes and is not replaced by canonical H3K4me3 until the major ZGA stage. The establishment and removal of distal ncH3K4me3 is a unique characteristic of oocyte and early embryo genomes. However, the mechanism and function of ncH3K4me3 dynamics still need to be further investigated. DNA methylation in the maternal genome is anti-correlated with ncH3K4me3, as ∼ 18% versus ∼ 57% CpG methylation is observed in oocytes with or without ncH3K4me3 (Wang et al., 2014; Dahl et al., 2016). Active removal of broad H3K4me3 domains by the lysine demethylases KDM5A and KDM5B is essential for normal ZGA and early embryogenesis(Dahl et al., 2016), while Kdm5b overexpression results in transcriptome reactivation in mature oocytes (Zhang et al., 2016), suggesting that ncH3K4me3 may be responsible for the genome-wide silencing state. Transposable elements (TEs), including B1/B2/B4 and ERVL, show high overlap with ncH3K4me3 in distal regions, indicating that ncH3K4me3 in distal regions can be correlated with the activities of repeats (Zhang et al., 2016). ncH3K4me3 is reprogrammed upon ZGA at the late 2-cell stage, which requires both zygotic transcription and active demethylation, rather than passive dilution (Zhang et al., 2016). Maternal factors within the oocyte cytoplasm must direct the rapid change of the H3K4me3 landscape. Identifying key maternal factors may provide more insight into the mechanism of H3K4me3 remodeling. On the other hand, H3.3 is cooperated to the genome after fertilization and is essential for embryo development (Lin et al., 2013; Lin et al., 2014; Wen et al., 2014). The turnover of H3.3 is associated with histone modification changes such as H3K27me3 and H3K36me3 (Kraushaar et al., 2013; Lin et al., 2013), but the correlation between H3.3 and ncH3K4me3 is unclear and whether the replacement of H3.3 is responsible for the removal of ncH3K4me3 deserves further investigation.

Epigenome reprogramming of histone modifications and chromatin accessibility during early mouse embryo development. H3K4me3: After fertilization, H3K4me3 in the paternal genome is rapidly depleted but re-established during major ZGA. By contrast, noncanonical H3K4me3 with broad domains in both promoters and distal regions is found in MII oocytes and is replaced by canonical H3K4me3 at the late 2-cell stage (ZGA). Broad ncH3K4me3 domains are correlated with partial DNA methylation domains (PMDs). Broad promoter H3K4me3 domains are much more abundant in early embryos than in MII oocytes or derived cell lines and are associated with high levels of gene expression. During ZGA, chromatin accessibility is observed at both the TSS and TES sites of active genes. Transposable elements (TEs) are also accessible and are enriched in distal H3K4me3. Developmental genes are primed to be active until the blastocyst stage, marked by bivalent H3K4me3/H3K27me3. H3K27me3: During mouse early embryo development, H3K27me3 in promoter regions exhibits a widespread loss at the 2-cell stage, a caused by the global erasure of H3K27me3 in the paternal genome and the selective depletion of promoter H3K27me3 in the maternal genome. H3K27me3 and DNA methylation are negatively correlated with H3K4me3. H3K27me3 also appears in non-promoter regions in a highly pervasive and promiscuous manner. H3K9me3: H3K9me3 peaks mainly fall in LTRs in early embryos. The number of H3K9me3-marked LTRs gradually increases and remains high during pre-implantation development. Most of the parental H3K9me3 regions are established de novo upon fertilization. Promoter H3K9me3 marks are erased upon fertilization and are reestablished postimplantation. Most LTR-enriched H3K9me3 domains are progressively established after the 4-cell stage and are responsible for LTR silencing. During the early cleavage stage, H3K9me3 domains overlap with H3K27me3-marked facultative heterochromatin. Maternal-specific H3K9me3 regions are much more abundant than paternal-specific regions during early embryogenesis, but this divergence gradually diminishes. Transposable elements: Mouse endogenous retrovirus type L (MERVL), a member of the ERV3 family member, is expressed in both 2-cell-like ESCs and cleavage-stage embryos, where it drives the expression of many transcripts specific to ZGA and totipotency. Upon fertilization, LINE1 is actively transcribed, with the increase in LINE1 RNA reaching its highest level at the 2-cell stage. LINE1 is essential for Dux silencing, the synthesis of rRNA, exit from the 2-cell stage and chromatin remodeling over accessible regions during pre-implantation embryo development. Chromatin accessibility: Open chromatin exists around both the promoters and transcription end sites (TES) of actively transcribed genes at the 2-cell stage. The transient and active transcription of transposable elements is probably associated with increased chromatin accessibility at the 2-cell stage of early embryos
The canonical form of H3K4me3 in the promoter region is established at the 2-cell stage. Interestingly, the width of H3K4me3 peaks is highly dynamic during mouse pre-implantation embryonic development and is positively correlated with gene expression level (Liu et al., 2016b). The dynamicity of the extent of H3K4me3 marks may provide a novel mechanism of epigenetic regulation in early cleavage embryos when most repressive makers are removed from the promoter region (Liu et al., 2016b). Broad H3K4me3 domains (> 5 kb) in the promoters are much more abundant in early embryos than in MII oocytes or derived cell lines. Lack of KDM5B can extend the width of promoter H3K4me3 domains and disrupt precise lineage differentiation, which indicates that broad promoter H3K4me3 marks may help maintain the transcription of key cell-type-specific factors in a steady state (Liu et al., 2016b). The mechanism of H3K4me3 reprogramming remains unknown, but a recent study showed that the establishment of broad H3K4me3 domains may be catalyzed by KMT2B, a deficiency of which results in slower ovulation and embryonic arrest (Andreu-Vieyra et al., 2010). However, which transcription factors are involved in this precisely controlled process requires further investigation.
Canonical pattern of H3K4me3 in GV oocytes and species-specific dynamics of H3K4me3 in humans
The CUT&RUN (cleavage under targets and release using nuclease) method (Skene and Henikoff, 2017) reduces the requirement for materials for the genome-wide analysis of histone modifications. Thus, the reprogramming of key histone modifications in human pre-implantation embryo development is finally accessible to profiling (Fig. 2). Unexpectedly, H3K4me3 was found to show sharp peaks at promoters in human GV and MI oocytes (Xia et al., 2019), unlike the abundant broad ncH3K4me3 domains distributed in distal partially methylated regions in mouse mature oocytes. These differences indicate a different epigenetic mechanism of genome silencing in human oocytes that is regulated by ncH3K4me3 in mice. Notably, weaker (compared to promoter H3K4me3) but widespread distal H3K4me3 marks have been discovered in pre-ZGA embryos, which indicates the de novo deposition of H3K4me3 and characterizes a primed epigenetic state for subsequent transcriptional regulation (Xia et al., 2019). The function of these distal H3K4me3 marks remains unknown, and whether their function is similar to that of ncH3K4me3 in mice before ZGA is worthy of elucidation. The profiling of such major reprogramming of both promoter and distal H3K4me3 marks will pave the way for future investigations of parental-to-zygotic transition in human pre-implantation embryos.

Dynamic histone modifications and chromatin accessibility during human pre-implantation embryogenesis. H3K4me3: H3K4me3 shows sharp peaks at promoters in human GV oocytes. During early human embryogenesis, wider promoter H3K4me3 marks are easily observable at the 4-cell stage (pre-ZGA), and 53% of these marks remain at the 8-cell stage (peri-ZGA) and are preferentially activated. The remainder of these sites (~ 47%), where H3K4me3 is lost, are in the promoters of genes related to development and differentiation, which remain inactive during ZGA. Weaker (compared to promoter H3K4me3) but widespread distal H3K4me3 marks are observed in pre-ZGA embryos, which indicates the de novo deposition of H3K4me3, and are decreased at the 8-cell stage. Distal H3K4me3 is deposited in CpG-rich and hypomethylated regions. Most of the distal H3K4me3 marks overlap with cis-regulatory elements and are highly chromatin accessible at the 4-cell stage. H3K27me3: H3K27me3 in human GV oocytes is deposited in the promoters of developmental genes and partially methylated domains. Human embryos at the ZGA stage (8-cell stage) show almost no H3K27me3 signals, indicating the global erasure of H3K27me3 in the maternal genome. The inaccessible cis-regulatory elements located in distal chromatin regions near developmental genes are correlated with H3K27me3 in the human ICM. Chromatin accessibility: Widespread accessible chromatin regions also highly overlap with cis-regulatory elements and transposable elements in human early embryos. High chromatin accessibility exists in distal regions enriched in transcription factor binding sites, overlapping with hypomethylated DNA regions in oocytes. Distal accessible chromatin at the 4-cell stage is also enriched for distal H3K4me3. These distal regions become inaccessible after the 8-cell stage
H3K27me3
Large-scale loss of promoter H3K27me3 marks after fertilization and pervasive ncH3K27me3 marks
H3K27me3 is a polycomb-based chromatin signature associated with gene repression (Bernstein et al., 2006; Simon and Kingston, 2009; Di Croce and Helin, 2013). H3K27me3 has been proved to be intergenerationally inherited from the maternal genome during early embryogenesis, regulating the activation of enhancers and lineage-specific genes (Inoue et al., 2017a; Zenk et al., 2017). During mouse early embryo development, H3K27me3 in promoter regions of both maternal and paternal alleles exhibits a widespread loss as early as in PN5 zygotes, followed by rapid dynamics during the transition from the morula to inner cell mass (ICM) and trophectoderm (TE) stages (Santos et al., 2005; Liu et al., 2016b; Zheng et al., 2016; Inoue et al., 2017a) (Fig. 1). This rapid loss is accomplished by the global erasure of H3K27me3 in the paternal genome and the selective depletion of promoter H3K27me3 in the maternal genome. In addition, dual-omics analysis indicate a negative correlation between H3K27me3 and DNA methylation in MII oocytes (Zheng et al., 2016) and some genomic regions in post implantation embryos (Yang et al., 2018b), while the anti-correlation is indistinct in cultured cells like ESCs and neural stem cells (NSCs) (Bartke et al., 2010; Wu et al., 2010; Brinkman et al., 2012), as well as mouse pre-implantation embryos, in which H3K27me3 is negatively correlated with H3K4me3 (Liu et al., 2016b; Zheng et al., 2016). On the other hand, H3K27me3 also appears in non-promoter regions, where it is highly pervasive and dramatically promiscuous. This supports the hypothesis that polycomb complexes constantly scan the whole genome for H3K27me3 deposition to compensate for the transcription repression function, which can also be realized by DNA methylation and H3K9me3. However, the functions and molecular mechanisms of such non-canonical H3K27me3 patterns remain to be verified.
In SCNT embryos, strong H3K27me3 signals are observed in all pseudopronuclei at the 1-cell stage (Zhang et al., 2009; Xie et al., 2016), which are highly distinct from the asymmetric H3K27me3 marks in parental genomes in normally fertilized embryos. In addition, ZGA failure in SCNT embryos at the 2-cell stage seems to be compatible with the defect of H3K27me3 erasure inherited from donor somatic cells, suggesting that H3K27me3 represents an epigenetic barrier to epigenome reprogramming (Zhang et al., 2009; Matoba et al., 2018). But simply eliminating H3K27me3 signals does little to promote the SCNT efficiency. A recent study shows that the overexpression of Kdm6a (an H3K27me3 demethylase) facilitates the expression of ZGA-related genes in SCNT embryos but results in no improvement of the birth rate or ntESC establishment efficiency. Conversely, knockdown of Kdm6b (another H3K27me3 demethylase) promotes both ZGA and SCNT efficiency (Yang et al., 2018a). H3K27me3-dependent imprinting genes are aberrant in SCNT embryos (Okae et al., 2014b; Inoue et al., 2017a) and may be amended by knockdown of Kdm6b. Kdm6b reduction also disrupts the ectopic expression of Xist, in which H3K27me3 serves as an imprinting mark and is correlated with abnormality of X chromosome inactivation in SCNT embryos (Inoue et al., 2010; Matoba et al., 2011; Inoue et al., 2017b; Matoba et al., 2018; Yang et al., 2018a).
Maternal H3K27me3-mediated imprinting has been discovered in early mouse embryos (Inoue et al., 2017a), as it can mediate silencing of DNA hypomethylated promoters, achieved by polycomb recruitment (Deaton and Bird, 2011). The H3K27me3 imprinting is probably established during oogenesis and maintained in pre-implantation embryos, which will be diluted in ICM and most of the H3K27me3-mediate imprinting is lost in the epiblast (EPI) of E6.5 embryos (Inoue et al., 2017a). Recent studies reveal that oocyte-derived H3K27me3-mediated imprinting will switch to being DNA methylation-dependent in extra-embryonic cells after implantation, in which both maternal EED and zygotic DNMT3A/B may take part (Smith et al., 2017; Inoue et al., 2018; Chen et al., 2019b), reflecting the complementary roles of H3K27me3 and DNA methylation in controlling imprinting. Recently, the complete loss of H3K27me3-dependent imprinting patterns in maternal allele-specific genes has been found in SCNT blastocysts, probably due to the absence of H3K27me3 marks at these loci in donor somatic cells (Okae et al., 2014b; Matoba et al., 2018). These epigenetic abnormalities of pre-implantation SCNT embryos are highly correlated with subsequent placenta overgrowth and embryonic lethality (Miri et al., 2013; Okae et al., 2014b; Inoue et al., 2017b). Intriguingly, the upregulation of clustered miRNAs of Sfmbt2, one of the H3K27me3-dependent imprinted genes (Matoba et al., 2018; Chen et al., 2020), has recently been identified as the major cause of placental hyperplasia in SCNT mice (Inoue et al., 2020). These observations highlight the crucial function of H3K27me3-imprinted genes in both pre-implantation and postimplantation embryos, although the detailed molecular mechanisms and consequences of intergenerational epigenetic inheritance remain elusive.
Global erasure of H3K27me3 at human ZGA
H3K27me3 in human GV oocytes is deposited in promoters of developmental genes and partially methylated domains, differing from the pattern in mouse oocytes (Xia et al., 2019). During human pre-implantation embryo development, the resetting of H3K27me3 is also different from that in mice. Human embryos at ZGA (8-cell stage) show almost no H3K27me3 signal, indicating the global erasure of H3K27me3 on both parental genomes (Xia et al., 2019) (Fig. 2). The absence of the core components of polycomb repression complex 2 (PRC2) in human embryos may be correlated with the loss of H3K27me3 (Saha et al., 2013). On the other hand, the global loss of H3K27me3 in human embryos predicts the absence of imprinting regulation, such as X chromosome inactivation (XCI), which is one of the critical events during early mouse embryogenesis (Petropoulos et al., 2016; Inoue et al., 2017b). Moreover, the H3K27me3-mediated imprinted genes identified in mouse early embryos, which have human orthologs, seems to experience de novo deposition of H3K27me3 (Xia et al., 2019). Further study is required to verify whether H3K27me3-controlled imprinting exist in human early embryos. Intriguingly, an analysis revealed asymmetric H3K27me3 patterning between ICM- and TE-specific genes in human pre-implantation embryos. Considering that ICM samples are a mixture of EPI and PE (primitive endoderm) cells, further studies are urgently needed to determine whether preferential H3K27me3 deposition also exists between EPI and PE cells (Xia et al., 2019).
Bivalent H3K4me3 and H3K27me3
Bivalent domains are considered to provide developmental regulators for later transcriptional activation upon differentiation (Vastenhouw and Schier, 2012). Although an H3K4me3/H3K27me3 bivalent state has been observed in lineage control genes in ESCs (Bernstein et al., 2006), zebrafish blastomeres (Vastenhouw et al., 2010), and primordial germ cells (PGCs) (Sachs et al., 2013), whether the associated regulatory characteristics differ remains unclear. It has been shown that the number of bivalent peaks is much lower in mouse pre-implantation embryos than in ESCs (Liu et al., 2016b), which indicates that bivalency may be more important in a stable cell line than in a transient stage. Bivalency is absent in developmental genes and is not established until the blastocyst stage, when lineage differentiation begins. Notably, most bivalent ICM and TE genes can be inherited by ESCs and TSCs and exhibit lower levels of expression (Liu et al., 2016b; Zheng et al., 2016). It is worth mentioning that EZH2 and SUZ12, the core components of PRC2, which are responsible for H3K27me3 deposition, target most of the inherited bivalent genes (Margueron and Reinberg, 2011; Liu et al., 2016b). This finding may help us understand the regulatory pattern of PRC2-mediated H3K27me3 upon the exit from totipotency to differentiation in early embryo development.
There is an absence of H3K4me3 and H3K27me3 bivalency in developmental genes upon implantation (Zheng et al., 2016). Intriguingly, in the EPI on embryonic day 6.5 (E6.5), stronger bivalency, defined as “super bivalency”, is found in the promoters of developmental genes, which is also evident in the E7.5 ectoderm but is much weaker in the E6.5 visceral endoderm (VE), mESCs and somatic lineages (Xiang et al., 2020). This raises the question of the function of this transient super bivalent state. Evidence shows that “super bivalency” is correlated with lineage-specific gene activation at later stages, such as cortex or heart differentiation. In addition, these super bivalent genes in the E6.5 EPI exhibit a unique higher-order chromatin organization (Xiang et al., 2020). These findings raise the possibility that strong bivalency in the primed EPI can help key developmental genes to maintain a unique spatial distribution and remain in a poised state for activation. KMT2B (MLL2) is known to be a methyltransferase for ncH3K4me3 in mouse oocytes (Hanna et al., 2018) and is responsible for the deposition of H3K4me3 in bivalent regions. In Kmt2b knockout embryos, H3K4me3 is globally downregulated at bivalent promoters, which is associated with aberrant developmental gene activation. Unexpectedly, bivalency is partially restored in some fraction of developmental gene promoters in the E8.5 head, indicating that Kmt2b plays a critical role in the super bivalency of the E6.5 EPI, while compensation mechanisms are involved in later embryonic development (Xiang et al., 2020). Furthermore, DNA hypomethylation is also suggested to participate in bivalency maintenance, in that Tet1/2 double knockout (DKO) leads to increased DNA methylation levels, followed by a significant H3K4me3 decrease in bivalent developmental genes (Xiang et al., 2020). Taken together, these results reveal a unique chromatin state transition that specifically appears during development from pre-implantation embryos to cell fate-determined lineages.
H3K9me3
H3K9me3 is a barrier to cell fate transition
H3K9me3-dependent heterochromatin is regarded as a barrier to cell fate changes, as it occludes the DNA from transcription factor binding (Tachibana et al., 2002; Burton and Torres-Padilla, 2010; Soufi et al., 2012; Becker et al., 2016). The principles and mechanisms of H3K9me3-dependent heterochromatin formation and function have recently been well reviewed elsewhere (Allshire and Madhani, 2018).
Early study has discovered gradual but incomplete demethylation patterns of H3K9me2 and H3K9me3 in SCNT embryos, in contrast to the asymmetric ones present in the parental genomes of fertilized embryos (Wang et al., 2007). The aberrant H3K9me3 reprogramming is deemed to directly causes ZGA failure, especially in SCNT embryos (Schultz, 2002; Matoba et al., 2014). At the 2-cell stage, reprogramming-resistant regions (RRRs) marked by H3K9me3 are defined, which are regions inherited from donor cells that fail to be successfully reprogrammed upon embryonic development (Matoba et al., 2014). Kdm4d (an H3K9 demethylase) overexpression in embryos and Suv39h1/h2 (H3K9me3 methyltransferases) knockdown in donor cells can rescue the transcription of ZGA-related genes impeded by H3K9me3, which dramatically improves the developmental rate of blastocysts. Notably, subsequent experiments using an embryo biopsy system and single-cell transcriptome sequencing identified Kdm4b, another H3K9me3 demethylase, as a key factor in the 2-cell arrest of SCNT embryos (Matoba et al., 2014; Liu et al., 2016a). Both studies emphasize the critical role of H3K9me3 reprogramming during early embryogenesis. The analysis of H3K9me3 in donor cells and 2-cell embryos showed a reduction in resistant H3K9me3 signals in Kdm4d-overexpressing SCNT embryos (Matoba et al., 2014). Moreover, overexpression of KDM4A also drastically increased the blastocyst rate of human SCNT and improves the derivation of human ntESC, with the similar mechanism as in mice (Chung et al., 2015). Our recent study also shows that H3K9me3 in donor cells also prevent the removal of topologically associated domains (TADs) during SCNT (Chen et al., 2020). These findings suggest that some H3K9me3, mainly associated with heterochromatin, is resistant to the cell conversion methods and presents as a barrier to reprogramming to pluripotency that impairs both the efficiency of reprogramming and the quality of embryogenesis.
On the other hand, H3K9me3 modification regulates the expression of repeats elements and some protein coding genes in mouse pre-implantation embryos (Hatanaka et al., 2015; Wang et al., 2018a). Kdm4b overexpression can improve cloning efficiency, but injection of high levels of Kdm4b mRNA have been proved to interrupt TE differentiation and an optimized Kdm4b mRNA injection dose can further increase the implantation and birth rates of cloned mice (Liu et al., 2016a). These indicate the level of H3K9me3 modification need to be well balanced during reprogramming and embryo development.
H3K9me3 regulates the proper expression of LTRs in early mouse embryos
Upon fertilization, a large fraction of repeat-rich sequences, including LTR retrotransposons, undergo dramatic demethylation and become accessible for highly active transcription, which has been shown to be critical for ZGA (Peaston et al., 2004; Wang and Dey, 2006; Zhang et al., 2019). LTRs must be properly regulated because they pose a risk to genome integrity through their potential for illicit recombination and self-duplication. Since major DNA demethylation occurs upon fertilization, the regulation of the transcription of LTRs requires a switch from DNA methylation to other types of epigenetic modifications, including repressive histone modifications, H3K9me3 and H3K27me3 (Wang et al., 2014; Becker et al., 2016). Previous research has shown that CAF-1 is responsible for the deposition of repressive histone marks, including H4K20me3 and H3K9me3, in retrotransposon regions and results in the silencing of retrotransposons in the mouse morula (Hatanaka et al., 2015; Ishiuchi et al., 2015), which provides a possible mechanism of retrotransposon regulation in early embryos. Recently, a high-resolution map for investigating the reprogramming of H3K9me3-dependent heterochromatin in mouse pre-implantation embryos was profiled (Wang et al., 2018a). As expected, it was found that the H3K9me3 peaks fall mainly within LTRs in early embryos and the number of H3K9me3-marked LTRs gradually increases, which remains high during pre-implantation development (Wang et al., 2018a). In contrast, promoter H3K9me3 marks are erased upon fertilization and will be established postimplantation, indicating the involvement of different regulatory mechanisms from those of H3K9me3 in LTRs (Wang et al., 2018a) (Fig. 1). The distribution characteristics of H3K9me3 echo its function in LTR regulation. Further analysis showed that increased H3K9me3 levels are correlated with the silencing of LTRs in the corresponding regions. The proper decoration of H3K9me3 by the CHAF1A complex in LTRs may be critical for blastocyst formation, cell fate decisions and the totipotency-pluripotency transition (Wang et al., 2018a). These results shed light on the molecular mechanisms of LTR silencing by H3K9me3-dependent heterochromatin. Further studies are required to reveal the molecular mechanisms of H3K9me3-dependent chromatin organization for transposable element (TE) regulation in mammalian embryos.
At the postimplantation stage, H3K9me3 marks are re-established in promoter regions. The expression level of lineage-specific genes is repressed owing to the formation of H3K9me3-decorated chromatin in their promoter regions (Wang et al., 2018a). Motif enrichment analysis indicated that Pou5f1, Sox12, Sox11, Lhx1, Zfp105 and Foxa2 are potential transcription factors contributing to the formation of epiblast-specific H3K9me3, while Zbed6, Elf4, Glis2, Creb3l2 and Ascl2 are involved in extraembryonic-specific H3K9me3 formation. Whether these transcription factors all play roles in chromatin organization during embryogenesis remains to be verified. Excitingly, endoderm-specific triple-knockout mutant (TKO) mice for all three histone H3K9 methyltransferases (SETDB1 and SUV39H1/H2) show a three-fold reduction in body weight and are much smaller in size compared to the control mice (Nicetto et al., 2019). In addition, the livers of the TKO mice show remarkable derepression of nonhepatic genes and fail to express mature hepatocyte genes (Nicetto et al., 2019). This highlights the significance of the precise regulation of H3K9me3 deposition at lineage-specific genes during mammalian embryo development.
OTHER HISTONE MODIFICATIONS
H3K36me3
H3K36me3, catalyzed by SETD2, is associated with transcriptionally active chromatin (Edmunds et al., 2008). SETD2 can mediate RNA polymerase II interaction and couple H3K36me3 with transcript elongation (Kizer et al., 2005). Unlike H3K4me3, H3K36me3 positively correlates with DNA methylation at gene bodies by recruiting DNMT3A/B (Baubec et al., 2015), which is conserved in most mammalian cells (Hawkins et al., 2010). SETD2 depleted oocytes exhibit huge loss of H3K36me3 and leads to H3K4me3 and H3K27me3 invasions into the former H3K36me3 regions (Xu et al., 2019). In addition, oocytes lacking SETD2 results in an aberrant DNA methylome which includes loss of maternal imprints and aberrant H3K4me3 deposition instead of DNA methylation, especially at ICRs (Xu et al., 2019). Furthermore, SETD2 scarcity results in oocyte maturation defects and embryonic lethality (Xu et al., 2019). These observations emphasize the essential role of H3K36me3 in establishing and safeguarding the maternal DNA methylome during mouse oogenesis and early embryo development.
H3R26me2
Mouse ESCs and blastomeres in early embryos with increased H3R26me2, an activating mark, show higher expression of a subset of pluripotency genes, is highly correlated with cell fate decision and pluripotency (Torres-Padilla et al., 2007; Wu et al., 2009; Goolam et al., 2016; White et al., 2016). Overexpression of coactivator-associated-protein-arginine-methyltransferase 1 (CARM1) in mouse ESCs and embryos elevates expression of key pluripotent genes, like Oct4/Pou5f1 (Wu et al., 2009; Goolam et al., 2016), Nanog (Torres-Padilla et al., 2007; Wu et al., 2009) and Sox2 (Goolam et al., 2016; White et al., 2016), of which promoters display detectable levels of H3R17/26 methylation. Differentially expressed H3R26me2 between 4-cell blastomeres are reported to be mediated by the heterogeneous activity of CARM1, with CARM1-activated blastomeres are prone to develop into ICM rather than TE (Torres-Padilla et al., 2007; Parfitt and Zernicka-Goetz, 2010; Shi et al., 2015). Intriguingly, the earliest cell fate decision in mouse early embryos has recently been advanced to emerge as early as late 2-cell stage, when a long noncoding RNA, LincGET, is transiently and asymmetrically expressed in the nucleus from 2-cell to 4-cell stage (Wang et al., 2018b). Notably, LincGET physically binds to CARM1 (Wang et al., 2018b), promoting CARM1 to accumulate in nuclear granules that requires paraspeckle component NEAT1 and its partner P54NRB (Hupalowska et al., 2018). This further gives rise to the increasing of H3R26me2 level, activating ICM-specific gene expression, upregulating transposons, and increasing global chromatin accessibility (Wang et al., 2018b). The mechanisms of H3R26me2 deposition and its potential effect on nuclear organization and lineage allocation during early embryo development require further investigations.
TRANSPOSABLE ELEMENTS
Transposable elements are repetitive DNA sequences that account for approximately half of the mammalian genome and have been considered deleterious to cells and a cause of cancer or apoptosis (Burns, 2017; Malki et al., 2019). They are silenced to prevent cells from experiencing promiscuous gene activation and potential mutations (Babaian and Mager, 2016; Burns, 2017) resulting from their transposable nature. Most of these elements are maintained in a silenced state by DNA hypermethylation or repressive histone modifications inducing H3K9me2/3 marks (Karimi et al., 2011; Leung et al., 2014). Transposable elements are highly expressed during mouse embryonic genome activation at the 2-cell stage (Evsikov et al., 2004). The proper regulation of retrotransposon expression is critical for the sequential reprogramming of the embryonic genome (Peaston et al., 2004).
MERVL
ERVs represent almost 10% of the mouse and human genome (Stocking and Kozak, 2008). MERVL is a member of the ERV3 family member that is expressed in both 2-cell-like ESCs and cleavage-stage embryos, where it drives the expression of many transcripts specific to ZGA and totipotency (Kigami et al., 2003; Svoboda et al., 2004; Macfarlan et al., 2012; Wu et al., 2016). The transcription of MERVL is under stringent surveillance to ensure stage-specific regulation during pre-implantation embryo development.
DUX plays critical roles in converting ESCs into a 2C-like state (Hendrickson et al., 2017) by controlling MERVL through the Dux-miR-344-Zmym2/Lsd1 axis (Yang et al., 2020). A previous study suggested that Dux is a ZGA inducer, as it is transiently expressed at the early 2-cell stage and robustly activates ZGA-related genes (De Iaco et al., 2017; Whiddon et al., 2017). Developmental pluripotency-associated 2 (DPPA2) and DPPA4 have been proved to drive the expression of Dux by directly bind to its promoter and gene body in 2C-like ESCs (Eckersley-Maslin et al., 2019). However, ~ 20% of embryos with zygotic depletion of DUX is able to reach a later embryonic stage without defective ZGA. Recently, Zhang and our group demonstrated that Dux is indeed important but is not a prerequisite for in vivo early embryo development (Chen and Zhang, 2019; Guo et al., 2019). Indeed, Dux deletion delays ZGA and decreases the developmental potential of embryos, but Dux-KO mice can survive to adulthood. Furthermore, the prolonged expression of Dux leads to developmental arrest and embryo death, which emphasizes the importance of proper silencing and degradation processes (Guo et al., 2019). Despite the absence of Dux, MERVL is highly expressed at the mid- and late 2-cell stages, which indicates the existence of compensation mechanisms that are probably regulated by other transcription factors or chromatin remodelers (Guo et al., 2019).
Distinct from the transient but bursting existence of Dux at the early 2-cell stage (initiated as early as the zygotic stage (Macfarlan et al., 2012; Deng et al., 2014; Abe et al., 2018)), Zscan4c, a transcription factor with zinc finger domains, is expressed at the 2-cell/4-cell stages, which is compatible with the existence of MERVL transcripts. A recent study in mESCs indicated that Zscan4c acts as an activator of MERVL and genes playing roles in 2-cell/4-cell embryo (Zhang et al., 2019). Zscan4c activates MERVL via direct binding to MT2 (the LTR of MERVL) loci and activates 2-cell/4-cell embryo genes by regulating the enhancer activity of MT2, associated with the increased deposition of H3K4me1, H3K27ac, and H3K14ac (Zhang et al., 2019). In contrast to Dux, Zscan4c is suggested to reinforce the activation of the 2C-like transcriptional network, rather than drive it (Eckersley-Maslin et al., 2019).
LINE1
The long interspersed element 1 (LINE1) retroelements are the most abundant class of retroelements in mammals (Richardson et al., 2015). LINE1 is highly expressed during mouse pre-implantation embryo development (Fadloun et al., 2013), which implies its critical roles in gene regulatory networks (Bourque, 2009). Upon fertilization, LINE1 is actively transcribed and peaks at the 2-cell stage. By contrast, the retrotransposition rate of LINE1 is rather low, giving rise to the hypothesis that LINE1 functions in a retrotransposition-independent manner, which has recently been verified (Jachowicz et al., 2017). Chromatin-associated LINE1 is found in mESCs and interacts with a nucleolin-KAP1/TRIM28 complex (Jachowicz et al., 2017; Percharde et al., 2018). Notably, nucleolin is essential for rRNA synthesis and processing (Ginisty et al., 1998) and has been identified as a repressor of Dux and the 2C program in ESCs (Gabellini et al., 2002). PRC1.6 and corepressor tripartite motif-containing protein 28 (TRIM28/KAP1) have been shown to directly bind and repress Dux in mESCs (Cossec et al., 2018; Percharde et al., 2018). The reduced abundance of LINE1 RNA affects the interaction between Dux and nucleolin in peri-nucleolar heterochromatin, which probably facilitates entry into the 2C state (Percharde et al., 2018). Whether this is consistent in early embryos deserves further in vivo exploration. Notably, LINE1 is essential for maintaining an open chromatin state in early embryogenesis (Jachowicz et al., 2017). Prematurely decreased chromatin accessibility occurs if LINE1 is repressed. In contrast, the prevention of chromatin condensation accompanies artificially prolonged LINE1 transcription (Percharde et al., 2018). Taken together, these findings indicate that LINE1 is essential for Dux silencing, the synthesis of rRNA, exit from the 2-cell stage and chromatin remodeling over accessible regions during pre-implantation embryo development. The proper regulation and degradation of LINE1 transcripts are critical for normal oocyte maturation and embryogenesis. Our group recently demonstrated a new posttranscriptional regulation mechanism of LINE1 mediated by ZCCHC8, a central factor in the nuclear exosome targeting (NEXT) complex(Wu et al., 2019). Zcchc8-depleted ESCs exhibit defects in proliferation, pluripotency maintenance, and differentiation. Intriguingly, the maternal loss of ZCCHC8 in mouse oocytes and early embryos indicates sustained abundant LINE1 RNA, accompanied by higher chromatin accessibility (Wu et al., 2019). ZCCHC8 has been reported to show a functional correlation with RNA export and translation (Lubas et al., 2011; Roundtree et al., 2017; Kasowitz et al., 2018; Mure et al., 2018); thus, the further investigation of RNA modifications may shed light on the underlying regulatory mechanisms.
CHROMATIN REMODELING
Chromatin accessibility
During mammalian pre-implantation development, major chromatin reorganization is critical for epigenetic reprogramming to convert terminally differentiated gametes into a totipotent state (Burton and Torres-Padilla, 2014). When global transcription takes place, open chromatin is newly established, while epigenetic modifications can be partially inherited from maternal genomes (Tsompana and Buck, 2014; Wu et al., 2016). The molecular mechanism of how cell memory of open chromatin can be erased and re-established during early embryo development remains unclear. Recent work based on low-input DNase I sequencing (liDNase-seq) suggests that DNase I-hypersensitive sites (DHSs) are progressively established and show a major increase in 8-cell embryos (Jin et al., 2015; Lu et al., 2016). Another study using improved ATAC-seq (an assay for transposase-accessible chromatin using sequencing) revealed the landscape of chromatin accessibility dynamics in early mouse embryo development (Buenrostro et al., 2015; Cusanovich et al., 2015; Wu et al., 2016) (Fig. 1). In contrast to the asymmetric reprogramming patterns of DNA methylation and histone modifications in the parental genomes after fertilization, chromatin accessibility seems to be more synchronized, except for a few instances of allele-specific open chromatin and transcription (Tsompana and Buck, 2014; Wu et al., 2016). Notably, open chromatin exists around both the promoters and transcription end sites of actively transcribed genes at the 2-cell stage, which is distinguished from the map of cis-regulatory sequences in other mouse tissues and cell types (Shen et al., 2012). Additionally, the transient and active transcription of transposable elements is probably associated with increased chromatin accessibility at the 2-cell stage of early embryos (Peaston et al., 2004). In addition, by combining the analysis of transcriptome and chromatin accessibility, NR5A2, which promotes the expression of Pou5f1 and Nanog, has been shown to participate in the regulatory network involved in the lineage specification of ICM and TE, and this regulation may occur as early as the 8-cell stage (Lu et al., 2016; Wu et al., 2016). These findings provide hints about the regulatory circuity during pre-implantation embryogenesis, while multiomics analyses are necessary to complement the panorama of epigenome remodeling.
During SCNT embryo development, chromatin accessibility undergoes major reprogramming and is mostly completed before the first embryonic cleavage (Djekidel et al., 2018). Additionally, this reprogramming appears to be DNA replication independent, indicating the importance of maternal factors involved in chromatin remodeling (Djekidel et al., 2018). The search for the maternal factors responsible for the chromatin state transition is an important research topic that deserves increasing attention.
In human embryogenesis, widely spread accessible chromatin regions are readily detected in 2-cell embryos, before ZGA occurs around the 4-cell to 8-cell stage (Lee et al., 2014; Wu et al., 2018). These regions show a preference for CpG-rich promoters and are correlated with gene activation. High chromatin accessibility also exists in distal regions, which are enriched in transcription factor binding sites and overlap with hypomethylated DNA regions in oocytes. Distal accessible chromatin at the 4-cell stage is also enriched in distal H3K4me3 (Xia et al., 2019) (Fig. 2). Putative transcription factors such as CTCF, KLF, SOX2, POU5F1, GATA and TEAD are well conserved in both mice and humans according to transcriptome or chromatin accessibility analysis (Xia et al., 2019). However, the low-input ATAC-seq method cannot avoid aneuploid human embryos, and the heterogeneity of individuals is much more extensive because of their complex genetic backgrounds. It is worth mentioning that the single-cell chromatin overall omic-scale landscape sequencing (scCOOL-seq) technique, which enables the simultaneous analysis of the chromatin state, nucleosome positioning, DNA methylation, and copy number variation in the same individual cell, has been developed (Li et al., 2018a). The results obtained using this approach indicated that the most dramatic chromosome remodeling in human embryos occurs between the 4- and 8-cell stages (Li et al., 2018a). Studies of DNA methylation and chromatin accessibility in individual cells from the same embryo demonstrate cell-to-cell variance and the unsynchronized reprogramming of the DNA methylome and chromatin organization in different genomic regions. Collectively, these data pave the way for deciphering the mechanism of epigenomic reprogramming in pre-implantation human embryos and indicate potential applications in clinical diagnosis.
3D chromatin
The nucleus of eukaryotic interphase cells contains chromatin packaged in a hierarchical structure, which is essential for gene regulatory networks (Fullwood et al., 2009; Atlasi and Stunnenberg, 2017). The 3D chromatin architecture plays critical roles in wide-ranging biological events, including RNA transcription, DNA replication, cell division and meiosis (Gorkin et al., 2014; Bonev and Cavalli, 2016; Beagrie et al., 2017; Hug and Vaquerizas, 2018). The role of the 3D genome organization in development and cell differentiation has been reviewed in detail elsewhere (Zheng and Xie, 2019). Nevertheless, the dynamics of the 3D chromatin structure during mammalian embryogenesis remain elusive, owing to the limited technology available for investigating the 3D genome organization.
Substantially distinct 3D structures in gametes and gradually established chromatin organization during embryogenesis
Recent studies utilizing the low-input Hi-C (genome-wide chromosome conformation capture) method revealed the dynamics of 3D genome architecture reprogramming in mouse early embryo development (Du et al., 2017; Ke et al., 2017) (Fig. 3). Notably, MII oocytes lacks TADs and compartments given its mitotic nature, while earlier stages of oocytes show Polycomb-associating domains (PADs), which is marked by H3K27me3 and briefly reappear on the maternal genome upon fertilization (Du et al., 2020). By contrast, sperm present both topologically associating domains (TADs) and A/B compartments (Du et al., 2017). The sperm genome also shows frequent extra-long-range interactions (> 4 Mb) and inter-chromosomal interactions(Battulin et al., 2015). Following fertilization, the higher-order structures of the chromatin of both parents are obscure at the zygotic and ZGA stages but are spatially separated from each other and show distinct compartmentalization. Such allele separation and compartmentalization are maintained until the 8-cell stage and coincide with accumulations of H3K27me3 ( Du et al., 2017; Borsos et al., 2019; Collombet et al., 2020). The gradual establishment of the parental chromatin organization occurs throughout the development of pre-implantation embryos, with slow consolidation of TADs and the A/B compartments. Notably, the re-establishment of chromatin compartments in the maternal genome appears to be much weaker than that in the paternal genome, which is compatible with the distinct 3D nature of the chromatin of oocytes and sperm to some extent (Du et al., 2017). In addition, the lamina-associated domains (LADs) (van Steensel and Belmont, 2017) of both parental genomes experience de novo but distinct establishment (Borsos et al., 2019), which precedes consolidation of TADs.

The higher-order chromatin organization in the gametes and pre-implantation embryos of mouse and human. Mouse: 3D chromatin: MII oocytes lacks TADs and compartments given its mitotic. PADs and their compartmental interactions appear to emerge only on the maternal allele in early 2-cell embryos, and begin to fade away in the 8-cell embryos. By contrast, sperm present frequent extra-long-range interactions (> 4 Mb) and inter-chromosomal interactions. Upon fertilization, the higher-order structures of both parental chromatins are obscure at the zygotic and ZGA stages but are spatially separated from each other with distinct compartmentalization. The gradual establishment of the parental chromatin organization occurs throughout the development of pre-implantation embryos, with slow consolidation of TADs and the A/B compartments. Such allele separation and compartmentalization are maintained until the 8-cell stage. Human: Human sperm lack TADs and expression of the chromatin regulator CTCF. Consistent with findings in mice, TADs and A/B compartmentalization are gradually established during human embryogenesis. CTCF is highly expressed at the ZGA stage of human embryos, which coincides with the time at which TADs are observed
Dramatic higher-order chromatin structure reprogramming indicates the importance of epigenetic reorganization during mammalian early development. However, little is known about the molecular basis of the reprogramming of the 3D chromatin structure. The establishment of TADs depends on DNA replication rather than ZGA, which reveals the function of cell cycle transition in genome structure dynamics (Ke et al., 2017). Furthermore, the reprogramming of other epigenetic modifications is dramatically interrelated with the reorganization of the 3D genome during mouse early embryogenesis. Large-scale DNA demethylation, for instance, appears to preferentially occur in compartment A rather than compartment B, resulting in an increase in the accessibility of chromatin to DNA demethylases (Du et al., 2017; Ke et al., 2017).
Moreover, a recent study by our group revealed spatiotemporally dynamic chromatin reorganization in SCNT embryos (Chen et al., 2020). Both aberrant TADs and compartment A/B organization are observed throughout early SCNT embryo development. The overexpression of Kdm4b partially ameliorates abnormal 3D chromatin structures, suggesting that H3K9me3 modification in donor cells is a barrier to chromatin structure reprogramming. This indicates a correlation between the organization of the 3D genome architecture and histone modifications.
No TADs or CTCF-dependent 3D chromatin structure in human sperm
The dynamics of the 3D chromatin architecture during human embryogenesis have recently been revealed. Unlike mouse sperm, human sperm lack TADs and the expression of the chromatin regulator CTCF (Chen et al., 2019a) (Fig. 3). Consistent with findings in mice, TADs and A/B compartmentalization are gradually established during human embryogenesis. CTCF is highly expressed at the ZGA stage of human embryos, which coincides with the time at which TADs are observed, indicating the critical role of CTCF in the establishment of the higher-order chromatin structure during early human embryo development. Intriguingly, the establishment of TADs in human embryos is ZGA dependent, which leads to the hypothesis that the correlation between TAD formation, DNA replication and gene activation differs among species or cell types (Ke et al., 2017; Chen et al., 2019a).
PERSPECTIVES
During the development of pre-implantation embryos, ZGA occurs predominantly at the two-cell stage in mice and the eight-cell stage in humans, which involves global DNA demethylation, chromatin remodeling, spatial reorganization of the genome and substantial transcriptional changes. Recent discoveries have shed light on the potential regulatory mechanisms that drive subsequent biological events upon fertilization, but many such mechanisms remain to be investigated. Future work is needed to further elucidate the molecular details that underlie the interaction between epigenetic remodeling and the totipotency transition as well as cell fate decisions in early mammalian embryo development.
Epigenetic reprogramming during early embryonic development is an exquisitely controlled process, involving both global re-establishment of most epigenetic marks and locus-specific regulation. In recent years, due partly to single-cell and low-cell-number epigenomic studies, our understanding of the epigenetic reprogramming landscape of pre-implantation development has improved considerably. However, how reprogramming is regulated at different genome loci remains unknown. Different transcription factors must play important roles in determining the locus-specific epigenetic transition pattern. The identification of these factors and the underlying mechanisms will improve our understanding of cell fate transitions and mammalian early development. Cell fate transitions are coordinated by the synergistic action of multiple types of epigenetic remodeling. Multiomics analysis is needed to clarify the fundamental principles underlying totipotency acquisition and cell fate decisions.
The mechanism of epigenetic remodeling seems to vary in different species; for example, TAD establishment depends on DNA replication in mouse embryos, while that in human embryos requires ZGA. The exploration of the epigenetic regulation mechanism of human early embryo development will continue to be an important area of research, the results of which will be beneficial for diagnosing and treating human infertility. It is worth mentioning that human embryos show high individual heterogeneity, including a high proportion of aneuploid embryos, which may to some extent conceal the detailed characteristics of the histone modifications and 3D chromatin structures that exist during human embryogenesis. Notably, remarkable progress has recently been made in mapping the molecular architecture of lineage specification during gastrulation and early organogenesis in mouse embryos (Peng et al., 2019; Pijuan-Sala et al., 2019; Weinreb et al., 2020). Inspiringly, it is now possible to study the development of postimplantation primate embryos through in vitro culture (Ma et al., 2019; Niu et al., 2019). This breakthrough may also provide insight into studies on pre-implantation development.
In summary, recent advances in sequencing technologies have given rise to research probing the regulatory network based on epigenome reprogramming during mammalian pre-implantation embryonic development, and further studies are urgently needed to reveal the underlying molecular mechanisms.
References
Abe KI, Funaya S, Tsukioka D, Kawamura M, Suzuki Y, Suzuki MG, Schultz RM, Aoki F (2018) Minor zygotic gene activation is essential for mouse pre-implantation development. Proc Natl Acad Sci USA 115:E6780–E6788
Allshire RC, Madhani HD (2018) Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19:229–244
Amdani SN, Yeste M, Jones C, Coward K (2015) Sperm factors and oocyte activation: current controversies and considerations. Biol Reprod 93:50
Amouroux R, Nashun B, Shirane K, Nakagawa S, Hill PW, D'Souza Z, Nakayama M, Matsuda M, Turp A, Ndjetehe E et al (2016) De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat Cell Biol 18:225–233
Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM (2010) MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. https://doi.org/10.1371/journal.pbio.1000453
Atlasi Y, Stunnenberg HG (2017) The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet 18:643–658
Au Yeung WK, Brind'Amour J, Hatano Y, Yamagata K, Feil R, Lorincz MC, Tachibana M, Shinkai Y, Sasaki H (2019) Histone H3K9 methyltransferase G9a in oocytes is essential for preimplantation development but dispensable for CG methylation protection. Cell Rep 27(282–293):e284
Babaian A, Mager DL (2016) Endogenous retroviral promoter exaptation in human cancer. Mob DNA 7:24
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21:381–395
Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T (2010) Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143:470–484
Battulin N, Fishman VS, Mazur AM, Pomaznoy M, Khabarova AA, Afonnikov DA, Prokhortchouk EB, Serov OL (2015) Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol 16:77
Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, Schubeler D (2015) Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 520:243–247
Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR et al (2017) Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543:519–524
Becker JS, Nicetto D, Zaret KS (2016) H3K9me3-dependent heterochromatin: barrier to cell fate changes. Trends Genet 32:29–41
Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K et al (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326
Bonev B, Cavalli G (2016) Organization and function of the 3D genome. Nat Rev Genet 17:772
Bonte D, Reddy Guggilla R, Stamatiadis P, De Sutter P, Heindryckx B (2018) Chapter 14—unraveling the causes of failed fertilization after intracytoplasmic sperm injection due to oocyte activation deficiency. In: Horcajadas JA, Gosálvez J (eds) Reproductomics. Academic Press, London, pp 243–277
Borsos M, Perricone SM, Schauer T, Pontabry J, de Luca KL, de Vries SS, Ruiz-Morales ER, Torres-Padilla ME, Kind J (2019) Genome–lamina interactions are established de novo in the early mouse embryo. Nature 569:729–733
Bourque G (2009) Transposable elements in gene regulation and in the evolution of vertebrate genomes. Curr Opin Genet Dev 19:607–612
Brinkman AB, Gu H, Bartels SJ, Zhang Y, Matarese F, Simmer F, Marks H, Bock C, Gnirke A, Meissner A et al (2012) Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22:1128–1138
Buenrostro JD, Wu B, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY, Greenleaf WJ (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523:486–490
Burns KH (2017) Transposable elements in cancer. Nat Rev Cancer 17:415–424
Burton A, Torres-Padilla ME (2010) Epigenetic reprogramming and development: a unique heterochromatin organization in the pre-implantation mouse embryo. Brief Funct Genomics 9:444–454
Burton A, Torres-Padilla ME (2014) Chromatin dynamics in the regulation of cell fate allocation during early embryogenesis. Nat Rev Mol Cell Biol 15:723–734
Canovas S, Ross PJ (2016) Epigenetics in pre-implantation mammalian development. Theriogenology 86:69–79
Chen Z, Zhang Y (2019) Loss of DUX causes minor defects in zygotic genome activation and is compatible with mouse development. Nat Genet 51:947–951
Chen X, Ke Y, Wu K, Zhao H, Sun Y, Gao L, Liu Z, Zhang J, Tao W, Hou Z et al (2019a) Key role for CTCF in establishing chromatin structure in human embryos. Nature 576:306–310
Chen Z, Yin Q, Inoue A, Zhang C, Zhang Y (2019b) Allelic H3K27me3 to allelic DNA methylation switch maintains noncanonical imprinting in extraembryonic cells. Sci Adv 5:eaay7246
Chen M, Zhu Q, Li C, Kou X, Zhao Y, Li Y, Xu R, Yang L, Yang L, Gu L et al (2020) Chromatin architecture reorganization in murine somatic cell nuclear transfer embryos. Nat Commun 11:1813
Chung YG, Matoba S, Liu Y, Eum JH, Lu F, Jiang W, Lee JE, Sepilian V, Cha KY, Lee DR et al (2015) Histone demethylase expression enhances human somatic cell nuclear transfer efficiency and promotes derivation of pluripotent stem cells. Cell Stem Cell 17:758–766
Collombet S, Ranisavljevic N, Nagano T, Varnai C, Shisode T, Leung W, Piolot T, Galupa R, Borensztein M, Servant N et al (2020) Parental-to-embryo switch of chromosome organization in early embryogenesis. Nature 580:142–146
Cossec JC, Theurillat I, Chica C, Bua Aguin S, Gaume X, Andrieux A, Iturbide A, Jouvion G, Li H, Bossis G et al (2018) SUMO safeguards somatic and pluripotent cell identities by enforcing distinct chromatin states. Cell Stem Cell 23(742–757):e748
Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C, Shendure J (2015) Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348:910–914
Dahl JA, Jung I, Aanes H, Greggains GD, Manaf A, Lerdrup M, Li G, Kuan S, Li B, Lee AY et al (2016) Broad histone H3K4me3 domains in mouse oocytes modulate maternal-to-zygotic transition. Nature 537:548–552
Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, Wolf E, Reik W (2001) Conservation of methylation reprogramming in mammalian development: aberrant reprogramming in cloned embryos. Proc Natl Acad Sci USA 98:13734–13738
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25:1010–1022
De Iaco A, Planet E, Coluccio A, Verp S, Duc J, Trono D (2017) DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat Genet 49:941–945
Deng Q, Ramskold D, Reinius B, Sandberg R (2014) Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343:193–196
Di Croce L, Helin K (2013) Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol 20:1147–1155
Djekidel MN, Inoue A, Matoba S, Suzuki T, Zhang CX, Lu FL, Jiang L, Zhang Y (2018) Reprogramming of chromatin accessibility in somatic cell nuclear transfer is DNA replication independent. Cell Reports 23:1939–1947
Du Z, Zheng H, Huang B, Ma R, Wu J, Zhang X, He J, Xiang Y, Wang Q, Li Y et al (2017) Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547:232–235
Du Z, Zheng H, Kawamura YK, Zhang K, Gassler J, Powell S, Xu Q, Lin Z, Xu K, Zhou Q et al (2020) Polycomb group proteins regulate chromatin architecture in mouse oocytes and early embryos. Mol Cell 77(825–839):e827
Eckersley-Maslin MA, Svensson V, Krueger C, Stubbs TM, Giehr P, Krueger F, Miragaia RJ, Kyriakopoulos C, Berrens RV, Milagre I et al (2016) MERVL/Zscan4 network activation results in transient genome-wide DNA demethylation of mESCs. Cell Rep 17:179–192
Eckersley-Maslin MA, Alda-Catalinas C, Reik W (2018) Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat Rev Mol Cell Biol 19:436–450
Eckersley-Maslin M, Alda-Catalinas C, Blotenburg M, Kreibich E, Krueger C, Reik W (2019) Dppa2 and Dppa4 directly regulate the Dux-driven zygotic transcriptional program. Genes Dev 33:194–208
Edmunds JW, Mahadevan LC, Clayton AL (2008) Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 27:406–420
Evsikov AV, de Vries WN, Peaston AE, Radford EE, Fancher KS, Chen FH, Blake JA, Bult CJ, Latham KE, Solter D et al (2004) Systems biology of the 2-cell mouse embryo. Cytogenet Genome Res 105:240–250
Fadloun A, Le Gras S, Jost B, Ziegler-Birling C, Takahashi H, Gorab E, Carninci P, Torres-Padilla ME (2013) Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat Struct Mol Biol 20:332–338
Fulka H, Mrazek M, Tepla O, Fulka J Jr (2004) DNA methylation pattern in human zygotes and developing embryos. Reproduction 128:703–708
Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A, Mei PH et al (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462:58–64
Gabellini D, Green MR, Tupler R (2002) Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 110:339–348
Gao L, Wu K, Liu Z, Yao X, Yuan S, Tao W, Yi L, Yu G, Hou Z, Fan D et al (2018a) Chromatin accessibility landscape in human early embryos and its association with evolution. Cell 173(248–259):e215
Gao R, Wang C, Gao Y, Xiu W, Chen J, Kou X, Zhao Y, Liao Y, Bai D, Qiao Z et al (2018b) Inhibition of aberrant DNA re-methylation improves post-implantation development of somatic cell nuclear transfer embryos. Cell Stem Cell 23(426–435):e425
Ginisty H, Amalric F, Bouvet P (1998) Nucleolin functions in the first step of ribosomal RNA processing. EMBO J 17:1476–1486
Goolam M, Scialdone A, Graham SJL, Macaulay IC, Jedrusik A, Hupalowska A, Voet T, Marioni JC, Zernicka-Goetz M (2016) Heterogeneity in Oct4 and Sox2 targets biases cell fate in 4-cell mouse embryos. Cell 165:61–74
Gorkin DU, Leung D, Ren B (2014) The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14:762–775
Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG et al (2011) The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477:606–610
Guo F, Li X, Liang D, Li T, Zhu P, Guo H, Wu X, Wen L, Gu TP, Hu B et al (2014a) Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell 15:447–459
Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, Yan J, Ren X, Lin S, Li J et al (2014b) The DNA methylation landscape of human early embryos. Nature 511:606–610
Guo F, Li L, Li J, Wu X, Hu B, Zhu P, Wen L, Tang F (2017) Single-cell multi-omics sequencing of mouse early embryos and embryonic stem cells. Cell Res 27:967–988
Guo M, Zhang Y, Zhou J, Bi Y, Xu J, Xu C, Kou X, Zhao Y, Li Y, Tu Z et al (2019) Precise temporal regulation of Dux is important for embryo development. Cell Res 29:956–959
Han L, Ren C, Li L, Li X, Ge J, Wang H, Miao YL, Guo X, Moley KH, Shu W et al (2018) Embryonic defects induced by maternal obesity in mice derive from Stella insufficiency in oocytes. Nat Genet 50:432–442
Hanna CW, Taudt A, Huang J, Gahurova L, Kranz A, Andrews S, Dean W, Stewart AF, Colome-Tatche M, Kelsey G (2018) MLL2 conveys transcription-independent H3K4 trimethylation in oocytes. Nat Struct Mol Biol 25:73–82
Hatanaka Y, Inoue K, Oikawa M, Kamimura S, Ogonuki N, Kodama EN, Ohkawa Y, Tsukada Y, Ogura A (2015) Histone chaperone CAF-1 mediates repressive histone modifications to protect pre-implantation mouse embryos from endogenous retrotransposons. Proc Natl Acad Sci USA 112:14641–14646
Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S et al (2010) Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell 6:479–491
Hendrickson PG, Dorais JA, Grow EJ, Whiddon JL, Lim JW, Wike CL, Weaver BD, Pflueger C, Emery BR, Wilcox AL et al (2017) Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat Genet 49:925–934
Huang Y, Kim JK, Do DV, Lee C, Penfold CA, Zylicz JJ, Marioni JC, Hackett JA, Surani MA (2017) Stella modulates transcriptional and endogenous retrovirus programs during maternal-to-zygotic transition. Elife. https://doi.org/10.7554/eLife.22345
Huang X, Gao X, Li W, Jiang S, Li R, Hong H, Zhao C, Zhou P, Chen H, Bo X et al (2019) Stable H3K4me3 is associated with transcription initiation during early embryo development. Bioinformatics 35:3931–3936
Hug CB, Vaquerizas JM (2018) The birth of the 3D genome during early embryonic development. Trends Genet 34:903–914
Hupalowska A, Jedrusik A, Zhu M, Bedford MT, Glover DM, Zernicka-Goetz M (2018) CARM1 and paraspeckles regulate pre-implantation mouse embryo development. Cell 175(1902–1916):e1913
Inoue A, Zhang Y (2011) Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334:194–194
Inoue A, Zhang Y (2014) Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat Struct Mol Biol 21:609–616
Inoue K, Kohda T, Sugimoto M, Sado T, Ogonuki N, Matoba S, Shiura H, Ikeda R, Mochida K, Fujii T et al (2010) Impeding Xist expression from the active X chromosome improves mouse somatic cell nuclear transfer. Science 330:496–499
Inoue A, Jiang L, Lu F, Suzuki T, Zhang Y (2017a) Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547:419–424
Inoue A, Jiang L, Lu F, Zhang Y (2017b) Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31:1927–1932
Inoue A, Chen Z, Yin Q, Zhang Y (2018) Maternal Eed knockout causes loss of H3K27me3 imprinting and random X inactivation in the extraembryonic cells. Genes Dev 32:1525–1536
Inoue K, Ogonuki N, Kamimura S, Inoue H, Matoba S, Hirose M, Honda A, Miura K, Hada M, Hasegawa A et al (2020) Loss of H3K27me3 imprinting in the Sfmbt2 miRNA cluster causes enlargement of cloned mouse placentas. Nat Commun 11:2150
Ishiuchi T, Enriquez-Gasca R, Mizutani E, Boskovic A, Ziegler-Birling C, Rodriguez-Terrones D, Wakayama T, Vaquerizas JM, Torres-Padilla ME (2015) Early embryonic-like cells are induced by downregulating replication-dependent chromatin assembly. Nat Struct Mol Biol 22:662–671
Iurlaro M, von Meyenn F, Reik W (2017) DNA methylation homeostasis in human and mouse development. Curr Opin Genet Dev 43:101–109
Jachowicz JW, Bing X, Pontabry J, Boskovic A, Rando OJ, Torres-Padilla ME (2017) LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat Genet 49:1502–1510
Jin WF, Tang QS, Wan MM, Cui KR, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R et al (2015) Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528:142
Jukam D, Shariati SAM, Skotheim JM (2017) Zygotic genome activation in vertebrates. Dev Cell 42:316–332
Karimi MM, Goyal P, Maksakova IA, Bilenky M, Leung D, Tang JX, Shinkai Y, Mager DL, Jones S, Hirst M et al (2011) DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 8:676–687
Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, Schultz RM, Wang PJ (2018) Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet 14:e1007412
Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L et al (2017) 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170(367–381):e320
Kigami D, Minami N, Takayama H, Imai H (2003) MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol Reprod 68:651–654
Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD (2005) A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol Cell Biol 25:3305–3316
Kohli RM, Zhang Y (2013) TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502:472–479
Kragesteen BK, Spielmann M, Paliou C, Heinrich V, Schopflin R, Esposito A, Annunziatella C, Bianco S, Chiariello AM, Jerkovic I et al (2018) Dynamic 3D chromatin architecture contributes to enhancer specificity and limb morphogenesis. Nat Genet 50:1463–1473
Kraushaar DC, Jin W, Maunakea A, Abraham B, Ha M, Zhao K (2013) Genome-wide incorporation dynamics reveal distinct categories of turnover for the histone variant H3.3. Genome Biol 14:R121
Lee MT, Bonneau AR, Giraldez AJ (2014) Zygotic genome activation during the maternal-to-zygotic transition. Annu Rev Cell Dev Biol 30:581–613
Legault LM, Bertrand-Lehouillier V, McGraw S (2018) Pre-implantation alcohol exposure and developmental programming of FASD: an epigenetic perspective. Biochem Cell Biol 96:117–130
Leung D, Du T, Wagner U, Xie W, Lee AY, Goyal P, Li Y, Szulwach KE, Jin P, Lorincz MC et al (2014) Regulation of DNA methylation turnover at LTR retrotransposons and imprinted loci by the histone methyltransferase Setdb1. Proc Natl Acad Sci USA 111:6690–6695
Li E, Zhang Y (2014) DNA methylation in mammals. Cold Spring Harb Perspect Biol 6:a019133
Li L, Guo F, Gao Y, Ren Y, Yuan P, Yan L, Li R, Lian Y, Li J, Hu B et al (2018a) Single-cell multi-omics sequencing of human early embryos. Nat Cell Biol 20:847–858
Li Y, Zhang Z, Chen J, Liu W, Lai W, Liu B, Li X, Liu L, Xu S, Dong Q et al (2018b) Stella safeguards the oocyte methylome by preventing de novo methylation mediated by DNMT1. Nature 564:136–140
Lin CJ, Conti M, Ramalho-Santos M (2013) Histone variant H3.3 maintains a decondensed chromatin state essential for mouse pre-implantation development. Development 140:3624–3634
Lin CJ, Koh FM, Wong P, Conti M, Ramalho-Santos M (2014) Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Dev Cell 30:268–279
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM et al (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462:315–322
Liu W, Liu X, Wang C, Gao Y, Gao R, Kou X, Zhao Y, Li J, Wu Y, Xiu W et al (2016a) Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov 2:16010
Liu X, Wang C, Liu W, Li J, Li C, Kou X, Chen J, Zhao Y, Gao H, Wang H et al (2016b) Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 537:558–562
Loppin B, Bonnefoy E, Anselme C, Laurencon A, Karr TL, Couble P (2005) The histone H3.3 chaperone HIRA is essential for chromatin assembly in the male pronucleus. Nature 437:1386–1390
Lu F, Liu Y, Inoue A, Suzuki T, Zhao K, Zhang Y (2016) Establishing chromatin regulatory landscape during mouse preimplantation development. Cell 165:1375–1388
Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, Lykke-Andersen S, Andersen JS, Dziembowski A, Jensen TH (2011) Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43:624–637
Ma H, Zhai J, Wan H, Jiang X, Wang X, Wang L, Xiang Y, He X, Zhao ZA, Zhao B et al (2019) In vitro culture of cynomolgus monkey embryos beyond early gastrulation. Science. https://doi.org/10.1126/science.aax7890
Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL (2012) Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487:57–63
Malki S, van der Heijden GW, O'Donnell KA, Martin SL, Bortvin A (2019) A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev Cell 51:658
Margueron R, Reinberg D (2011) The Polycomb complex PRC2 and its mark in life. Nature 469:343–349
Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, Nakamura T, Abe K, Nakano T, Ishino F et al (2011) RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc Natl Acad Sci U S A 108:20621–20626
Matoba S, Liu Y, Lu F, Iwabuchi KA, Shen L, Inoue A, Zhang Y (2014) Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 159:884–895
Matoba S, Wang H, Jiang L, Lu F, Iwabuchi KA, Wu X, Inoue K, Yang L, Press W, Lee JT et al (2018) Loss of H3K27me3 imprinting in somatic cell nuclear transfer embryos disrupts post-implantation development. Cell Stem Cell 23(343–354):e345
Messerschmidt DM, Knowles BB, Solter D (2014) DNA methylation dynamics during epigenetic reprogramming in the germline and pre-implantation embryos. Genes Dev 28:812–828
Mihajlovic AI, Bruce AW (2017) The first cell-fate decision of mouse pre-implantation embryo development: integrating cell position and polarity. Open Biol. https://doi.org/10.1098/rsob.170210
Minami N, Suzuki T, Tsukamoto S (2007) Zygotic gene activation and maternal factors in mammals. J Reprod Dev 53:707–715
Miri K, Latham K, Panning B, Zhong Z, Andersen A, Varmuza S (2013) The imprinted polycomb group gene Sfmbt2 is required for trophoblast maintenance and placenta development. Development 140:4480–4489
Molaro A, Hodges E, Fang F, Song Q, McCombie WR, Hannon GJ, Smith AD (2011) Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 146:1028–1040
Mure F, Corbin A, Benbahouche NEH, Bertrand E, Manet E, Gruffat H (2018) The splicing factor SRSF3 is functionally connected to the nuclear RNA exosome for intronless mRNA decay. Sci Rep 8:12901
Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M et al (2007) PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol 9:64–71
Nakamura T, Liu YJ, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T (2012) PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 486:415–419
Nicetto D, Donahue G, Jain T, Peng T, Sidoli S, Sheng L, Montavon T, Becker JS, Grindheim JM, Blahnik K et al (2019) H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 363:294–297
Nichols J, Smith A (2009) Naive and primed pluripotent states. Cell Stem Cell 4:487–492
Niu Y, Sun N, Li C, Lei Y, Huang Z, Wu J, Si C, Dai X, Liu C, Wei J et al (2019) Dissecting primate early post-implantation development using long-term in vitro embryo culture. Science. https://doi.org/10.1126/science.aaw5754
Okae H, Chiba H, Hiura H, Hamada H, Sato A, Utsunomiya T, Kikuchi H, Yoshida H, Tanaka A, Suyama M et al (2014a) Genome-wide analysis of DNA methylation dynamics during early human development. PLoS Genet 10:e1004868
Okae H, Matoba S, Nagashima T, Mizutani E, Inoue K, Ogonuki N, Chiba H, Funayama R, Tanaka S, Yaegashi N et al (2014b) RNA sequencing-based identification of aberrant imprinting in cloned mice. Hum Mol Genet 23:992–1001
Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99:247–257
Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J (2000) Active demethylation of the paternal genome in the mouse zygote. Curr Biol 10:475–478
Parfitt DE, Zernicka-Goetz M (2010) Epigenetic modification affecting expression of cell polarity and cell fate genes to regulate lineage specification in the early mouse embryo. Mol Biol Cell 21:2649–2660
Peaston AE, Evsikov AV, Graber JH, de Vries WN, Holbrook AE, Solter D, Knowles BB (2004) Retrotransposons regulate host genes in mouse oocytes and pre-implantation embryos. Dev Cell 7:597–606
Peat JR, Reik W (2012) Incomplete methylation reprogramming in SCNT embryos. Nat Genet 44:965–966
Peng G, Suo S, Cui G, Yu F, Wang R, Chen J, Chen S, Liu Z, Chen G, Qian Y et al (2019) Molecular architecture of lineage allocation and tissue organization in early mouse embryo. Nature 572:528–532
Percharde M, Lin CJ, Yin Y, Guan J, Peixoto GA, Bulut-Karslioglu A, Biechele S, Huang B, Shen X, Ramalho-Santos M (2018) A LINE1-nucleolin partnership regulates early development and ESC identity. Cell 174(391–405):e319
Petropoulos S, Edsgard D, Reinius B, Deng Q, Panula SP, Codeluppi S, Reyes AP, Linnarsson S, Sandberg R, Lanner F (2016) Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 167:285
Pijuan-Sala B, Griffiths JA, Guibentif C, Hiscock TW, Jawaid W, Calero-Nieto FJ, Mulas C, Ibarra-Soria X, Tyser RCV, Ho DLL et al (2019) A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 566:490–495
Ribet D, Louvet-Vallee S, Harper F, de Parseval N, Dewannieux M, Heidmann O, Pierron G, Maro B, Heidmann T (2008) Murine endogenous retrovirus MuERV-L is the progenitor of the "orphan" epsilon viruslike particles of the early mouse embryo. J Virol 82:1622–1625
Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV (2015) The influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr. https://doi.org/10.1128/microbiolspec.MDNA3-0061-2014
Risal S, Pei Y, Lu H, Manti M, Fornes R, Pui HP, Zhao Z, Massart J, Ohlsson C, Lindgren E et al (2019) Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat Med 25:1894–1904
Rivera RM, Ross JW (2013) Epigenetics in fertilization and pre-implantation embryo development. Prog Biophys Mol Biol 113:423–432
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P et al (2017) YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. https://doi.org/10.7554/eLife.31311
Rulands S, Lee HJ, Clark SJ, Angermueller C, Smallwood SA, Krueger F, Mohammed H, Dean W, Nichols J, Rugg-Gunn P et al (2018) Genome-scale oscillations in DNA methylation during exit from pluripotency. Cell Syst 7(63–76):e12
Sachs M, Onodera C, Blaschke K, Ebata KT, Song JS, Ramalho-Santos M (2013) Bivalent chromatin marks developmental regulatory genes in the mouse embryonic germline in vivo. Cell Rep 3:1777–1784
Sadakierska-Chudy A, Filip M (2015) A Comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox Res 27:172–197
Saha B, Home P, Ray S, Larson M, Paul A, Rajendran G, Behr B, Paul S (2013) EED and KDM6B coordinate the first mammalian cell lineage commitment to ensure embryo implantation. Mol Cell Biol 33:2691–2705
SanMiguel JM, Bartolomei MS (2018) DNA methylation dynamics of genomic imprinting in mouse development. Biol Reprod 99:252–262
Santos F, Peters AH, Otte AP, Reik W, Dean W (2005) Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev Biol 280:225–236
Schultz RM (2002) The molecular foundations of the maternal to zygotic transition in the pre-implantation embryo. Hum Reprod Update 8:323–331
Schulz KN, Harrison MM (2019) Mechanisms regulating zygotic genome activation. Nat Rev Genet 20:221–234
Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV et al (2012) A map of the cis-regulatory sequences in the mouse genome. Nature 488:116–120
Shen L, Inoue A, He J, Liu Y, Lu F, Zhang Y (2014) Tet3 and DNA replication mediate demethylation of both the maternal and paternal genomes in mouse zygotes. Cell Stem Cell 15:459–471
Shi J, Chen Q, Li X, Zheng X, Zhang Y, Qiao J, Tang F, Tao Y, Zhou Q, Duan E (2015) Dynamic transcriptional symmetry-breaking in pre-implantation mammalian embryo development revealed by single-cell RNA-seq. Development 142:3468–3477
Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708
Skene PJ, Henikoff S (2013) Histone variants in pluripotency and disease. Development 140:2513–2524
Skene PJ, Henikoff S (2017) An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. https://doi.org/10.7554/eLife.21856
Skvortsova K, Iovino N, Bogdanovic O (2018) Functions and mechanisms of epigenetic inheritance in animals. Nat Rev Mol Cell Biol 19:774–790
Smith ZD, Meissner A (2013) DNA methylation: roles in mammalian development. Nat Rev Genet 14:204–220
Smith ZD, Chan MM, Mikkelsen TS, Gu H, Gnirke A, Regev A, Meissner A (2012) A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature 484:339–344
Smith ZD, Chan MM, Humm KC, Karnik R, Mekhoubad S, Regev A, Eggan K, Meissner A (2014) DNA methylation dynamics of the human pre-implantation embryo. Nature 511:611–615
Smith ZD, Shi J, Gu H, Donaghey J, Clement K, Cacchiarelli D, Gnirke A, Michor F, Meissner A (2017) Epigenetic restriction of extraembryonic lineages mirrors the somatic transition to cancer. Nature 549:543–547
Soufi A, Donahue G, Zaret KS (2012) Facilitators and impediments of the pluripotency reprogramming factors' initial engagement with the genome. Cell 151:994–1004
Stewart KR, Veselovska L, Kim J, Huang J, Saadeh H, Tomizawa S, Smallwood SA, Chen T, Kelsey G (2015) Dynamic changes in histone modifications precede de novo DNA methylation in oocytes. Genes Dev 29:2449–2462
Stewart KR, Veselovska L, Kelsey G (2016) Establishment and functions of DNA methylation in the germline. Epigenomics 8:1399–1413
Stocking C, Kozak CA (2008) Murine endogenous retroviruses. Cell Mol Life Sci 65:3383–3398
Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM (2004) RNAi and expression of retrotransposons MuERV-L and IAP in pre-implantation mouse embryos. Dev Biol 269:276–285
Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H et al (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16:1779–1791
Tadros W, Lipshitz HD (2009) The maternal-to-zygotic transition: a play in two acts. Development 136:3033–3042
Teperek M, Miyamoto K (2013) Nuclear reprogramming of sperm and somatic nuclei in eggs and oocytes. Reprod Med Biol 12:133–149
Theunissen TW, Friedli M, He Y, Planet E, O'Neil RC, Markoulaki S, Pontis J, Wang H, Iouranova A, Imbeault M et al (2016) Molecular criteria for defining the naive human pluripotent state. Cell Stem Cell 19:502–515
Torres-Padilla ME, Parfitt DE, Kouzarides T, Zernicka-Goetz M (2007) Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 445:214–218
Tsompana M, Buck MJ (2014) Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7:33
van Steensel B, Belmont AS (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169:780–791
Vastenhouw NL, Schier AF (2012) Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol 24:374–386
Vastenhouw NL, Zhang Y, Woods IG, Imam F, Regev A, Liu XS, Rinn J, Schier AF (2010) Chromatin signature of embryonic pluripotency is established during genome activation. Nature 464:922–926
Verma N, Pan H, Dore LC, Shukla A, Li QV, Pelham-Webb B, Teijeiro V, Gonzalez F, Krivtsov A, Chang CJ et al (2018) TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat Genet 50:83–95
Wang H, Dey SK (2006) Roadmap to embryo implantation: clues from mouse models. Nat Rev Genet 7:185–199
Wang F, Kou Z, Zhang Y, Gao S (2007) Dynamic reprogramming of histone acetylation and methylation in the first cell cycle of cloned mouse embryos. Biol Reprod 77:1007–1016
Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W et al (2014) Programming and inheritance of parental DNA methylomes in mammals. Cell 157:979–991
Wang C, Liu X, Gao Y, Yang L, Li C, Liu W, Chen C, Kou X, Zhao Y, Chen J et al (2018a) Reprogramming of H3K9me3-dependent heterochromatin during mammalian embryo development. Nat Cell Biol 20:620–631
Wang J, Wang L, Feng G, Wang Y, Li Y, Li X, Liu C, Jiao G, Huang C, Shi J et al (2018b) Asymmetric expression of LincGET biases cell fate in two-cell mouse embryos. Cell 175(1887–1901):e1818
Weinreb C, Rodriguez-Fraticelli A, Camargo FD, Klein AM (2020) Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. https://doi.org/10.1126/science.aaw3381
Wen D, Banaszynski LA, Liu Y, Geng F, Noh KM, Xiang J, Elemento O, Rosenwaks Z, Allis CD, Rafii S (2014) Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proc Natl Acad Sci USA 111:7325–7330
Whiddon JL, Langford AT, Wong CJ, Zhong JW, Tapscott SJ (2017) Conservation and innovation in the DUX4-family gene network. Nat Genet 49:935–940
White MD, Angiolini JF, Alvarez YD, Kaur G, Zhao ZW, Mocskos E, Bruno L, Bissiere S, Levi V, Plachta N (2016) Long-lived binding of Sox2 to DNA predicts cell fate in the four-cell mouse embryo. Cell 165:75–87
Wu H, Zhang Y (2014) Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell 156:45–68
Wu X, Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet 18:517–534
Wu Q, Bruce AW, Jedrusik A, Ellis PD, Andrews RM, Langford CF, Glover DM, Zernicka-Goetz M (2009) CARM1 is required in embryonic stem cells to maintain pluripotency and resist differentiation. Stem Cells 27:2637–2645
Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, Sun YE (2010) Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329:444–448
Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W et al (2016) The landscape of accessible chromatin in mammalian pre-implantation embryos. Nature 534:652–657
Wu J, Xu J, Liu B, Yao G, Wang P, Lin Z, Huang B, Wang X, Li T, Shi S et al (2018) Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 557:256–260
Wu Y, Liu W, Chen J, Liu S, Wang M, Yang L, Chen C, Qi M, Xu Y, Qiao Z et al (2019) Nuclear exosome targeting complex core factor Zcchc8 regulates the degradation of LINE1 RNA in early embryos and embryonic stem cells. Cell Rep 29(2461–2472):e2466
Xia W, Xu J, Yu G, Yao G, Xu K, Ma X, Zhang N, Liu B, Li T, Lin Z et al (2019) Resetting histone modifications during human parental-to-zygotic transition. Science 365:353–360
Xiang Y, Zhang Y, Xu Q, Zhou C, Liu B, Du Z, Zhang K, Zhang B, Wang X, Gayen S et al (2020) Epigenomic analysis of gastrulation identifies a unique chromatin state for primed pluripotency. Nat Genet 52:95–105
Xie B, Zhang H, Wei R, Li Q, Weng X, Kong Q, Liu Z (2016) Histone H3 lysine 27 trimethylation acts as an epigenetic barrier in porcine nuclear reprogramming. Reproduction 151:9–16
Xu Q, Xie W (2018) Epigenome in early mammalian development: inheritance, reprogramming and establishment. Trends Cell Biol 28:237–253
Xu Q, Xiang Y, Wang Q, Wang L, Brind'Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W et al (2019) SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet 51:844–856
Yang X, Smith SL, Tian XC, Lewin HA, Renard JP, Wakayama T (2007) Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat Genet 39:295–302
Yang L, Song L, Liu X, Bai L, Li G (2018a) KDM6A and KDM6B play contrasting roles in nuclear transfer embryos revealed by MERVL reporter system. Embo Rep. https://doi.org/10.15252/embr.201846240
Yang X, Hu B, Hou Y, Qiao Y, Wang R, Chen Y, Qian Y, Feng S, Chen J, Liu C et al (2018b) Silencing of developmental genes by H3K27me3 and DNA methylation reflects the discrepant plasticity of embryonic and extraembryonic lineages. Cell Res 28:593–596
Yang F, Huang X, Zang R, Chen J, Fidalgo M, Sanchez-Priego C, Yang J, Caichen A, Ma F, Macfarlan T et al (2020) DUX-miR-344-ZMYM2-mediated activation of MERVL LTRs induces a totipotent 2C-like state. Cell Stem Cell 26(234–250):e237
Yao C, Zhang W, Shuai L (2019) The first cell fate decision in pre-implantation mouse embryos. Cell Regen (Lond) 8:51–57
Yeste M, Jones C, Amdani SN, Coward K (2017) Oocyte activation and fertilisation: crucial contributors from the sperm and oocyte. Results Probl Cell Differ 59:213–239
Yu B, Smith TH, Battle SL, Ferrell S, Hawkins RD (2019) Superovulation alters global DNA methylation in early mouse embryo development. Epigenetics 14:780–790
Zeng Y, Chen T (2019) DNA methylation reprogramming during mammalian development. Genes (Basel). https://doi.org/10.1126/science.aaw3381
Zeng TB, Han L, Pierce N, Pfeifer GP, Szabo PE (2019) EHMT2 and SETDB1 protect the maternal pronucleus from 5mC oxidation. Proc Natl Acad Sci U S A 116:10834–10841
Zenk F, Loeser E, Schiavo R, Kilpert F, Bogdanovic O, Iovino N (2017) Germ line-inherited H3K27me3 restricts enhancer function during maternal-to-zygotic transition. Science 357:212–216
Zhang M, Wang F, Kou Z, Zhang Y, Gao S (2009) Defective chromatin structure in somatic cell cloned mouse embryos. J Biol Chem 284:24981–24987
Zhang B, Zheng H, Huang B, Li W, Xiang Y, Peng X, Ming J, Wu X, Zhang Y, Xu Q et al (2016) Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537:553–557
Zhang Y, Xiang Y, Yin Q, Du Z, Peng X, Wang Q, Fidalgo M, Xia W, Li Y, Zhao ZA et al (2018) Dynamic epigenomic landscapes during early lineage specification in mouse embryos. Nat Genet 50:96–105
Zhang W, Chen F, Chen R, Xie D, Yang J, Zhao X, Guo R, Zhang Y, Shen Y, Goke J et al (2019) Zscan4c activates endogenous retrovirus MERVL and cleavage embryo genes. Nucleic Acids Res 47:8485–8501
Zheng H, Xie W (2019) The role of 3D genome organization in development and cell differentiation. Nat Rev Mol Cell Biol 20:535–550
Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X et al (2016) Resetting epigenetic memory by reprogramming of histone modifications in mammals. Mol Cell 63:1066–1079
Zhu P, Guo H, Ren Y, Hou Y, Dong J, Li R, Lian Y, Fan X, Hu B, Gao Y et al (2018) Single-cell DNA methylome sequencing of human pre-implantation embryos. Nat Genet 50:12–19
ACKNOWLEDGMENTS
This work was supported by the National Key R&D Program of China (2016YFA0100400 and 2018YFC1004000) and the National Natural Science Foundation of China (31721003, 31820103009, 31701262, 81630035).
ABBREVIATIONS
2C-like cells, 2-cell embryo-like embryonic stem cells; 3D, three dimensional; 5hmC, 5-hydroxymethylcytosine; 5mC, 5-methylcytosine; Ascl2, achaete-scute family bHLH transcription factor 2; ATAC-seq, an assay for transposase-accessible chromatin using sequencing; CAF-1, F1 capsule protein; CARM1, coactivator associated arginine methyltransferase 1; CHAF1A, chromatin assembly factor 1 subunit A; ChIP-seq, chromatin immunoprecipitation sequencing; CREB3L2, cAMP responsive element binding protein 3 like 2; CTCF, CCCTC-binding factor; CUT&RUN, cleavage under targets and release using nuclease; DHSs, DNase I-hypersensitive sites; DMRs, differentially methylated regions; DNA, deoxyribonucleic acid; DNMT1, DNA (cytosine-5)-methyltransferase 1; DNMT3A, DNA (cytosine-5)-methyltransferase 3A; DNMT3B, DNA (cytosine-5)-methyltransferase 3B; DPPA2, developmental pluripotency associated 2; DPPA3, developmental pluripotency associated 3; DPPA4, developmental pluripotency associated 4; DUX, double homeobox; E6.5, embryonic day 6.5; EED, embryonic ectoderm development; ELF4, E74 like ETS transcription factor 4; EPI, epiblast; ERVs, endogenous retroviruses; ESCs, embryonic stem cells; EZH2, enhancer of zeste 2 polycomb repressive complex 2 subunit; FOXA2, forkhead box A2; GATA, GATA binding protein; GLIS2, GLIS family zinc finger 2; GV, germinal vesicle; H3K14ac, the acetylation at the 14th lysine residue of the histone H3 protein; H3K27ac, the acetylation at the 27th lysine residue of the histone H3 protein; H3K27me3, the tri-methylation at the 27th lysine residue of the histone H3 protein; H3K36me3, the tri-methylation at the 36th lysine residue of the histone H3 protein; H3K4me1, the mono-methylation at the 4th lysine residue of the histone H3 protein; H3K4me3, the tri-methylation at the 4th lysine residue of the histone H3 protein; H3K9me2, the di-methylation at the 9th lysine residue of the histone H3 protein; H3K9me3, the tri-methylation at the 9th lysine residue of the histone H3 protein; H3R17, arginine 17 of histone H3; H3R26me2, the di-methylation at the 26th Arginine residue of the histone H3 protein; H4K20me3, the tri-methylation at the 20th lysine residue of the histone H4 protein; Hi-C, high-resolution chromosome conformation capture; ICM, inner cell mass; ICRs, imprinting control regions; KAP1, kinesin-ii-associated protein; KDM4A, lysine demethylase 4A; KDM4B, lysine demethylase 4B; KDM4D, lysine demethylase 4D; KDM5A, lysine demethylase 5A; KDM5B, lysine demethylase 5B; KDM6A, lysine demethylase 6A; KDM6B, lysine demethylase 6B; KLF, Kruppel-Like Factor; KMT2B, lysine methyltransferase 2B; LADs, lamina-associated domain; LHX1, LIM homeobox 1; liDNase-seq, low-input DNase I sequencing; LINE1, the long interspersed element 1; LTR, long terminal repeat; MERVL, mouse endogenous retrovirus type L; MII, metaphase II; MLL2, myeloid/lymphoid or mixed-lineage leukemia 2; MZT, maternal-to-zygotic transition; NANOG, Nanog homeobox; ncH3K4me3, noncanonical H3K4me3; NEAT1, nuclear paraspeckle assembly transcript 1; NEXT, nuclear exosome targeting; NR5A2, nuclear receptor subfamily 5 group A member 2; NSCs, neural stem cells; ntESC, nuclear transfer embryonic stem cell; OCT4, organic cation/carnitine transporter4; P54NRB, non-POU domain containing octamer binding; PADs, polycomb-associating domains; PBAT, post-bisulfite adapter tagging; PE, primitive endoderm; PGC7, developmental pluripotency-associated 3; PGCs, primordial germ cells; PN3, pronuclear stage 3; PN5, pronuclear stage 5; POU5F1, POU class 5 homeobox 1; PRC1.6, polycomb repressive complex 1.6; PRC2, polycomb repression complex 2; rDMRs, re-methylated DMRs; RNA, ribonucleic acid; rRNA, ribosomal RNA; RRRs, reprogramming-resistant regions; scCOOL-seq, single-cell chromatin overall omic-scale landscape sequencing; SCNT, somatic cell nuclear transfer; SETD2, SET domain containing 2; SETDB1, SET domain, bifurcated 1; SFMBT2, Scm-like with four mbt domains 2; SINE, short interspersed nuclear element; SOX11, SRY-box transcription factor 11; SOX12, SRY-box transcription factor 12; SOX2, SRY-box transcription factor 2; STELLA, developmental pluripotency associated 3; SUV39H1, suppressor of variegation 3-9 homolog 1; SUV39H2, suppressor of variegation 3-9 homolog 2; SUZ12, SUZ12 polycomb repressive complex 2 subunit; TADs, topologically associated domains; TE, trophectoderm; TEAD, TEA domain transcription factor; TEs, transposable elements; TET1, tet methylcytosine dioxygenase 1; TET2, tet methylcytosine dioxygenase 2; TET3, tet methylcytosine dioxygenase 3; TKO, triple-knockout; TRIM28, tripartite motif-containing protein 28; TSCs, trophoblast stem cells; VE, visceral endoderm; XCI, X chromosome inactivation; XIST, inactive X specific transcripts; ZBED6, zinc finger BED type containing 6; ZCCHC8, zinc finger CCHC-type containing 8; ZFP105, zinc finger protein 105; ZGA, zygotic gene activation; ZSCAN4C, zinc finger and SCAN domain containing 4C
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
COMPLIANCE WITH ETHICS GUIDELINES
Ruimin Xu, Chong Li, Xiaoyu Liu and Shaorong Gao declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Additional information
Ruimin Xu and Chong Li have contributed equally to this work.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, R., Li, C., Liu, X. et al. Insights into epigenetic patterns in mammalian early embryos. Protein Cell 12, 7–28 (2021). https://doi.org/10.1007/s13238-020-00757-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00757-z