
Abstract
Colorectal cancer (CRC) and hepatocellular carcinoma (HCC) are the second and third most common causes of death by cancer, respectively. The etiologies of the two cancers are either infectious insult or due to chronic use of alcohol, smoking, diet, obesity and diabetes. Pathological changes in the composition of the gut microbiota that lead to intestinal inflammation are a common factor for both HCC and CRC. However, the gut microbiota of the cancer patient evolves with disease pathogenesis in unique ways that are affected by etiologies and environmental factors. In this review, we examine the changes that occur in the composition of the gut microbiota across the stages of the HCC and CRC. Based on the idea that the gut microbiota are an additional “lifeline” and contribute to the tumor microenvironment, we can observe from previously published literature how the microbiota can cause a shift in the balance from normal → inflammation → diminished inflammation from early to later disease stages. This pattern leads to the hypothesis that tumor survival depends on a less pro-inflammatory tumor microenvironment. The differences observed in the gut microbiota composition between different disease etiologies as well as between HCC and CRC suggest that the tumor microenvironment is unique for each case.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of death by cancer with a high mortality rate (Patel et al., 2012). Approximately 50% of HCC are induced by hepatitis B (HBV) infection and 20% by hepatitis C (HCV) infection. Viral infection acts as a first hit to produce liver inflammation that can develop into cirrhosis over time, and approximately 70%–90% of HCC cases occur in conjunction with cirrhosis (Guo et al., 2018). CRC is the second most common cause of death from cancer and has high incidence in Western countries. The incidence is currently increasing in Asian countries, and environmental factors such as diet have a large impact (Park et al., 2018). Excessive consumption of red meat and high fat influences the composition of the gut microbiota which in turn, can produce metabolites that contribute to intestinal inflammation resulting in the initial carcinogenic milieu for CRC (Feng et al., 2015). Pathological changes in the gut microbiota, referred to as “gut dysbiosis”, that lead to inflammation in the intestine is a common feature of both CRC and HCC. However, primary CRC and HCC develop as distinctive tumors in the intestine and liver, respectively. The connection between gut microbiota composition and both CRC and HCC has been well studied in animal models (Xie et al., 2016b; Wong et al., 2017). There are three categories of gut dysbiosis: 1) loss of beneficial, commensal bacteria, 2) enhanced abundance of pathobionts, and 3) loss of overall microbial diversity. These categories often occur concurrently (Petersen and Round, 2014).
HCC and CRC Development
Tumors are composed of a variety of cells such as fibroblasts, leukocytes, endothelial cells as well as cancer cells which together comprise the tumor micro-environment (TME). Within this TME, different cell types can signal to one another and recent studies have suggested that it is the cancer cells that are the “organizers” of the TME and are the principle source of the cellular signaling responsible for the induction and final formation of the TME (Li and Stanger, 2019). In the liver, cells such as hepatic stellate cells (HSC), Kupfer cells (liver macrophages, KC), pit cells, dendritic cells, natural killer T-cells (NKT) along with hepatic sinusoid endothelial cells (LSEC) form the surrounding environment or stroma for hepatocytes/HCC tumor cells (Ohtani and Kawada, 2019).
The path to HCC involves several stages which are the results of chronic hepatocyte death, inflammation and repair of liver tissue (Wu et al., 2014b). In non-viral HCC, the gut microbiota can gain access to the liver as result of a chronic liver disease (CLD) associated dysfunction of the intestinal barrier that is the result of high-fat diet (HFD), alcohol, and/or increased amounts of secondary bile acids (BAs) such as deoxycholic acid (DCA) (Ohtani and Kawada, 2019). The “leaky” intestinal membrane allows for translocation of bacteria-derived LPS (gram-negative bacteria) and lipoteichoic acid (LTA, derived from gram-positive bacteria) which bind to and activate the toll-like receptor-4 (TLR-4). Subsequently, the activation of TLR-4 by LPS and/or LTA initiates the nuclear factor-kappa-B (NF-κB) inflammatory signaling pathway and ultimately leads to production of the inflammatory cytokines IL-1β and IL-18 (Loo et al., 2017). HCC with cirrhosis or non-alcoholic steatohepatitis (NASH) has often been associated with continuous LPS/LTA-TLR-4 signaling activation. Increased levels of gut microbiota derived DCA has been associated with HCC with NASH (Stenman et al., 2013; Loo et al., 2017). The inflammatory pathway JNK/p38 → NF-κB → IL-18/1β → ↑HSC activation, ROS →↑fibrosis, can be activated not only by the lipotoxicity derived from excess hepatic lipid storage but also from DNA damage caused by DCA in NASH (Bernstein et al., 2005; Ferreira et al., 2014; Chow et al., 2017; Ohtani and Kawada, 2019). NASH is also closely related to mitochondrial dysfunction as lipid accumulation in hepatocytes causes increased activation of peroxisome proliferator activated receptors (PPARs) leading to elevated ROS, increased oxidative stress, cell death and activation of fibrogenic HSCs (George et al., 2003). Mitochondrial derived ROS can also induce production of the inflammatory cytokines, TGF-β, TNF-α and IL-6 (Chen et al., 2019). Another gut microbiota derived metabolite associated with NASH and liver inflammation is trimethylamine (TMA) which is derived from choline metabolism and can be converted in the liver to a highly toxic and pro-inflammatory compound, trimethylamine oxide (TMAO) which not only causes hepatocyte damage but also results in choline deficiency and progression of hepatic steatosis with increased HCC risk (Chu et al., 2019).
Virus-associated HCC begins with an acute immunogenic insult due to infection with either hepatitis B or C virus (HBV or HCV). First, there is necrosis of hepatocytes due to the viral infection which activates the HSCs through the paracrine action of cytokines released by the necrotic hepatocytes. Second, HSCs are further stimulated by infiltrating activated KC cells and leukocytes to evolve into fibrogenic myofibroblasts which in turn release cytokines to stimulate over-production of ECM components including collagens I, III, laminin and fibronectin that result in an imbalance of ECM production and degradation which is termed fibrosis (Wu et al., 2014b; Ohtani and Kawada, 2019). HSCs also contribute to the development of the TME. An activated HSC can undergo senescence and become a senescence-associated secretory phenotype (SASP) that secretes tumor progression promoting cytokines (IL-6, IL-8), chemokines and proteases (matrix metalloproteinases, MMPs) which act to remodel the ECM and enhance inflammation (Rodier and Campisi, 2011; Rao and Jackson, 2016).
KCs are resident liver macrophages that perform phagocytosis to remove microbial debris in the form of damage-associated molecular patterns (DAMPs) primarily derived from damaged hepatocytes, microbial associated patterns (MAMPs) such as lipopolysaccharides (LPS), and bacteria from the liver blood flow. When activated by inflammatory cytokines, they secrete further inflammatory cytokines and chemokines to influence activation of HSCs and modulate the immune response by facilitating infiltrations of other types of leukocytes (Krenkel and Tacke, 2017; Ohtani and Kawada, 2019). Liver macrophages can also be restorative though their immune modulating effect on T-, B-, T- and NK-T cells which act to inactivate HSCs and allow resolution of the inflammation. When an imbalance exists between fibrosis and resolution of the inflammation driving it, increased fibrosis and liver cirrhosis results which often leads to HCC (Krenkel and Tacke, 2017). Figure 1 summarizes the path from inflammation to CLD to HCC.

Tumor formation in HCC. CLD → HCC starts with injury to the liver usually by viral infection with either HBV or HCV virus, or exposure to toxins such as TMAO and DCA (secondary BAs). The initial injury causes hepatocyte death with subsequent activation of KCs and HSCs that release pro-inflammatory substances and initiate a vicious cycle of liver damage and repair. The gut microbiota also produces substances such as LTA and LPS which are capable of activating TLR4 inflammatory pathways in the liver. HSC and KC activation lead to increased production of growth factors, PDGF, VEGF and FGF2 which contribute to the TME by enhancing hepatocyte proliferation and neovascularization. Increased production of TGFβ and MMPs acts together to increase ECM production while increased COX2 and PGE2 levels modulate the immune system in such a way as to create an immunosuppressive environment to protect the HCC tumor. The major inflammatory pathways that contribute to increased fibrosis/cirrhosis are written out in the top right of the figure.
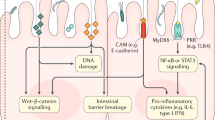
CRC, like HCC, develops slowly over more than 10 years in an environment of chronic inflammation and repair. Like HCC, the etiology of CRC varies and this invokes different aberrations from normal intestinal mucosa. The two major types of CRC, sporadic (SCRC) and colitis-associated (CCRC) differ in their histologic presentation and the timing/sequence of cellular mutations (Ullman and Itzkowitz, 2011; Dekker et al., 2019). Figure 2 summarizes the differences/similarities between sporadic and colitis-associated CRC types. Colitis, like CLD is often triggered by environmental insults such as bacterial or viral infections which in turn, activate an immune response that leads to the initial inflammation setting up a cycle of prolonged, repeated ulceration followed by re-epithelialization with increasingly abnormal clones of aneuploid cells (Xavier and Podolsky, 2007). In the intestine, tumor-associated myeloid cells produce IL-23 which affect Th17 cell polarization and the subsequent production of the cytokines IL-17A, IL-21, TNF-α and IL-6 which in turn, provide the pro-tumorigenic inflammatory response in CRC (De Simone et al., 2013; Long et al., 2017). IL-6 activates the signal transducer and activator of transcription-3 (STAT3) signaling pathway which in turn, acts to promote tumor growth (De Simone et al., 2015) and anti-tumoral immunity (Wu et al., 2014a). IL-6 also activates the NF-κB signaling pathway which helps to perpetuate inflammation and promote tumorigenesis (Koliaraki et al., 2015). Both phosphorylated STAT3 (activated) (Lin et al., 2011) and NF-κB (Myant et al., 2013) levels have been reported to be high in tumor-initiating cells, a subpopulation within the bulk tumor with “stem-like” characteristics (Schwitalla et al., 2013; Long et al., 2017).

Tumor formation in sporadic and colitis-associated CRCs. Sporadic CRC begins with a mutation in the tumor suppressor gene, adenomatous polyposis coli (APC). The resulting dysplastic adenomas typically develop in a chronically inflamed mucosa which promotes chromosomal and microsatellite instability, hypermethylation of gene promoter regions and oxidative stress. The adenomas progress sequentially from early (APC mutation) → intermediate→ late adenoma→ carcinoma (loss of p53 tumor suppressor gene). Colitis-associated CRC (CCRC) develops dysplastic lesions that are often flat rather than distinct adenoma polyps and the progression to CRC starts with colitis with no dysplasia (loss of p53) → indefinite dysplasia→ low-grade dysplasia → high-grade dysplasia (loss of APC) → carcinoma.
The intestine is densely populated with microorganisms and several species have been associated with CRC. In general, early stages of CRC have been characterized by a decrease in commensal bacteria such as Lactobacillus, Bifidobacterium and Clostridium which are known to produce anti-inflammatory SCFAs and were negatively correlated with increased markers for damaged intestinal epithelium such as diamine oxide (DAO), D-lactate and LPS. Fusobacterium nucleatum and Escherichia coli (pks+) were found to be positively correlated with DAO. LPS and D-lactate (Liu et al., 2020).
Fusobacterium nucleatum has been shown to interact directly with CRC tumor cells where its adhesin, FadA binds to E-cadherin on the CRC cell surface and activates Wnt/β-catenin signaling (Rubinstein et al., 2013). F. nucleatum has been shown to bind to the T-cell inhibitory immune receptor of natural killer cells (NK) through another adhesin, Fap2 (Gur et al., 2015). Fap2 has been reported to facilitate increased binding of F. nucleatum to CRC cells via its ability to bind to a highly expressed CRC cell surface marker, the disaccharide sugar motif [galactose N-acetyl-D-galactosamine (Gal-GalNAc)] (Coppenhagen-Glazer et al., 2015). F. nucleatum has also been shown to activate the cellular survival mechanism, autophagy, through TLR4 receptors located on CRC cell surfaces in response to oxaliplatin chemotherapy (Yu et al., 2017). The enterotoxic bacteria, Bacteroides fragilis (ETBF), causing GI inflammation has been reported to inhabit biofilms coating human CRCs and precancerous colonic adenomas (Boleij et al., 2015). Similarly, Escherichia coli expressing the genomic island polyketide synthase (pks+), have been detected in human CRC tissues. The genotoxin produced by pks+ E. coli, colibactin, alkylates the DNA of colonic epithelial cells and is thus a source of potential mutations leading to CRC (Arthur et al., 2012). It must be emphasized that many other bacteria as yet to be well characterized may also play a role in CRC carcinogenesis and progression.
An important topic of continuing research is an understanding of what compositional changes in the microbiota, if any, occur as pathogenesis to the final tumor stage of both CRC and HCC occurs. In this review we will examine several recent studies that highlighted the differences in gut microbiota composition between different disease etiologies and stages for CRC and HCC. The papers discussed in this review were selected using the following criteria: 1) published within the last five years, 2) focused on comparison of at least two stages of disease, 3) for HCC, focused on a single etiology for liver disease, 4) were human studies only, and 5) taxonomy sequencing was done at the genus or species level. Few studies met all of these criteria and the most current of these, with the largest sample sizes, were chosen in this review. The tables in this review listed changes in microbiota genera and also whether the changes would either promote or inhibit inflammation (anti- or pro-). For some cases the change can be either anti- or pro-inflammation depending on the bacterial species or presence of other immune-modulating bacteria. Details are given in the text below the table along with references for each genus listed with pro- or anti-inflammation properties.
A Comparison of Differential Bacterial Compositions Associated with HCV vs. Healthy Controls
Our discussion of viral induced HCC begins with the examination of the impact on the gut microbiota by the initial “hit” of virus infection. Several studies have suggested that not only the stage of liver disease but also the stage of HCV infection were responsible for changes in the gut microbiota composition (Aly et al., 2016; Heidrich et al., 2018; Inoue et al., 2018). HCV infection progresses through 4 chronological stages: 1) persistently normal serum alanine aminotransferase (PNALT), 2) chronic hepatitis (CH), 3) liver cirrhosis (LC) and 4) HCC (Inoue et al., 2018).
A recent study (Table 1) of the fecal microbiome for six chronic, stage 4 HCV patients with no other underlying disease, and eight healthy controls (HC) from the same geographical region was performed and their results showed that the microbiota richness and diversity of HCs was higher than that of HCV patients (Aly et al., 2016). Lower diversity has been shown by others to indicate chronic inflammation (Lozupone et al., 2012; Chen et al., 2015; Giloteaux et al., 2016).
Most of the bacteria enhanced in HCs are plant polysaccharides and fiber fermenters, such as Ruminococcus, Clostridium, Lachnospira, and Bacteroides, and may participate in cross-feeding each other, as well as other commensal bacteria (La Reau and Suen, 2018). Lachnospira, Butyricimonas and Clostridium especially those in cluster XIVa and IV are acetic acid and butyric acid-producing bacteria, are anti-inflammatory and promote healthy colonocytes (Sakamoto et al., 2009; Lopetuso et al., 2013; Hibberd et al., 2017). Bifidobacterium and Bacteroides have bile salt hydrolase allowing them to deconjugute BAs and exert metabolic benefits via BA signaling (Wexler, 2007; O’Callaghan and van Sinderen, 2016). Commensal Parabacteroides has recently been found to have metabolic benefits via secondary BA signaling as well as succinate-activated intestinal gluconeogenesis (Wang et al., 2019). Among those bacteria enhanced in HCV-HCC, Prevotella and Phascolarctobacteria can be a source of LPS as they are gram negative organisms and thus can be pro-inflammatory (Wu et al., 2017; Schwenger et al., 2018). Some reports also showed that these two bacteria could be anti-inflammatory, depending on the microenvironment and their species or strains. Similarly, Veillonella can also be either pro-inflammatory or anti-inflammatory. When co-inhabitating the gut with a specific strain of Streptococcus, co-stimulation of Veillonella with S. salvarius strain 1 led to decreased amounts of inflammatory cytokine production whereas with strain 2, there was a notable increase relative to either microbe alone (van den Bogert et al., 2014).
It appears from this small study that HCV-HCC generates quite a few anti-inflammatory microbes and several that are immune-modulating. This is not surprising because one could hypothesize that as a tumor matures, it needs to acquire homeostasis with its environment in order to survive and thus an anti-inflammatory, immunosuppressive TME is more desirable.
Another cross-sectional study (Table 2) aimed to delineate the effect of hepatitis C viral infection from liver cirrhosis on the gut microbiota. The intestinal microbiota of 95 patients chronically infected with HCV (n = 57 without cirrhosis (NCIR); n = 38 with cirrhosis (CIR)) and 50 HC were examined. Microbial diversity was significantly decreased from HC to NCIR to CIR (Heidrich et al., 2018). The alpha diversity was found to be according to the rank order, CIR < NCIR < HC.
An important finding here was that infection with the virus had a unique impact on the composition of the gut microbiota independent of CLD. In pattern 3A, two bacteria are not detectable until the disease is present and one bacterium exhibits a slight increase with CLD present that does not increase with CLD progression to CIR. Pattern 3B is comprised of 4 bacteria that decrease quite a bit in abundance when CLD reaches the fibrosis state. As to fibrosis, Pattern 1A shows increased Streptococcus and Veillonella which increase even more as CLD progresses to CIR and can be pro-inflammatory or anti-inflammatory depending on the strain of Streptococcus. Lactobacillus are normally considered beneficial to host health but they produce bile salt hydrolase which can lead to the production of more primary unconjugated BAs that are better activators of FXR and can thus decrease BA production as well as increased secondary BAs such as DCA that may be hepatotoxic (Ridlon et al., 2006; Bernstein et al., 2011). Pattern 1B contains mostly Clostridium species and their decrease with disease progression may signify loss of commensal beneficial bacteria. Pattern 2A has two beneficial bacterial genera, Akkermansia and Bifidobacterium that decrease in abundance in the NCIR but then increase in the CIR state. The die-off in NCIR followed by a resurgence in CIR may be due to a mutation or opportunistic overgrowth of a particular species in either of these two genera and thus taxonomic classification will have be done at the species level in order to fully understand the data.
To summarize this section, we can see that the infection with HCV by itself alters the gut microbiota allowing pro-inflammatory Citrobacter to appear and causing loss of Clostridia spp., thus setting the stage for bacterial translocation to occur to the liver. In the fibrotic, NCIR stage there is shift to even more genera of Clostridium being decreased accompanied by increased amounts of pro-inflammatory genera which further increases the inflammatory environment in both gut and liver. The trend towards a more pro-inflammatory gut continues for CIR.
The last study (Table 3) to be discussed for HCV pursued the idea of looking at different stages of hepatitis C infection and the effect on gut microbiota composition (Inoue et al., 2018). This study involved 23 HC (healthy control), 18 PNALT (persistently normal alanine aminotransferase), 84 CH (chronic hepatitis), 40 LC (liver cirrhosis) and 24 HCC patients. Microbial diversity was decreased in HCC patients and was associated with the clinical severity of the disease; HCC< LC < CH < PNALT << HC.
When looking at the changes caused by viral infection (PNALT and CH), one can understand how an unfavorable gut microenvironment is developed. Loss of the genera Clostridium along with decreased Ruminococcus and Lachnospiraceae, all dominant commensal bacteria that are short chain fatty acid (SCFA) producing signifies reduced control of inflammation as SCFAs, especially, butyrate regulate the differentiation of Treg cells (Furusawa et al., 2013). SCFAs are also nutrients for colonic epithelial cells and modulate colonic pH and their decrease results in an increased fecal pH and increased levels of ammonia (Wong et al., 2006). The most noteworthy change in this study was the large overgrowth in Streptococcus, a urease-producing bacteria, which has been associated with minimal hepatic encephalopathy (MHE), a condition often found in LC patients (Zhang et al., 2013). Streptococcus has also been reported to downregulate the innate immune system and as it is most dominant microbe change observed in HCC, may contribute to tumor survival and modeling of the TME (Cosseau et al., 2008). Its increased abundance was also observed in the second study discussed in this section (Heidrich et al., 2018). Another interesting change is the increase and then disappearance of Fusicatenibacter, butyrate producing bacteria which are part of Clostridium cluster IV (Rapozo et al., 2017). In this study, the inflammation status of the organisms present remains even across the stages of disease based on their fold changes.
A Comparison of Differential Bacterial Compositions Associated with HBV vs. Healthy Controls
We then examined the impact of HBV infection on the gut microbiota. The first study (Table 4), regarding HBV, focused on early CHB before the onset of liver damage and metabolic disorders in order to determine whether gut dysbiosis occurred prior to and if it contributed to the pathogenesis of liver disease (Wang et al., 2017).
The alterations due to those bacteria which are reduced in CHB patients result in reduced productions of SCFAs and antibacterial peptides while those bacteria which are enhanced in CHB were closely connected to the host’s physical indices and the accumulation of serum metabolites. These included cholesterol, L-aspartic acid, L-tyrosine, L-phenylalanine, octanoic acid and 1-napthol. The accumulation of aromatic amino acids such as L-phenylalanine and L-tyrosine, may affect cerebral functions and play a causal role in hepatic encephalopathy (Dejong et al., 2007). Notice also the “pairing” of Veillonella and Streptococcus as seen for HCV-HCC. It should be noted that there is increased abundance of Lachnospiraceae, a member of the order Clostridia. Certain strains have been associated with the development of diabetes (Kameyama and Itoh, 2014). This highlights the importance of deeper taxonomic classification. Actinomyces inhabits the gut and becomes an opportunistic pathogen, especially in immune-compromised patients, causing inflammation (Kameyama and Itoh, 2014). Overall, infection with hepatitis B causes a lot of inflammation and this is reflected in the gut microbiota composition.
In a second study (Table 5), changes in gut microbiota composition for HC (n = 15), CHB (n = 21), LC (n = 25) and HCC (n = 21) groupings were determined (Zeng et al., 2020).
The findings that are different here from previous studies are: 1) decrease in Bifidobacterium for all stages but less of a decrease seen for HCC, 2) the occurrence of another pro-inflammatory bacteria, Dorea which increases with stage of CLD (Leclercq et al., 2014), 3) the appearance of increased Coprococcus, a pro-inflammatory bacteria associated with western HFD which increases in CHB and LC but decreases in abundance in HCC (Pedersen et al., 2019), and 4) the decrease in Roseburia, an organism that cross-feeds other beneficial bacteria via it ability to degrade dietary β-mannans, with increasing disease stage (La Rosa et al., 2019).
Changes in Gut Microbiota Composition Associated with HBV and Non-hepatitis Virus Related HCC
As mentioned above, more than 70% of HCC originates from an initial “hit” from hepatitis virus (Guo et al., 2018). A recent study (Table 6) examined the changes in composition of the gut microbiota associated with hepatitis virus induced HCC vs. non-virus induced HCC (Liu et al., 2019). Both etiological routes to HCC, virus vs. non-virus, ultimately led to liver inflammation and cirrhosis but the changes in gut microbiota were found to be different.
Several remarkable differences between bacterial relative abundances related to viral-induced HCC relative to non-viral induced HCC are shown in Table 6. The pro-inflammatory, endotoxin-producing Escherichia-Shigella, Enterococcus and Proteus are increased in non-viral HCC but reduced in HBV-HCC. There are more members of the phylum Firmicutes that are decreased in the non-viral HCC group which contributes to a more pro-inflammatory state. Overall, the non-viral-HCC appears to have a more pro-inflammatory profile relative to HBV-HCC indicating perhaps a slightly different TME for non-viral HCC. This may be because the non-viral-HCC begins on an already chronically inflamed infrastructure whereas viral-HCC starts with a dramatic immunogenic response.
In the last study (Table 7) to be discussed regarding HCC in mouse models, we are going to look at the association of BA composition with various stages of non-viral liver disease (Xie et al., 2016a). Examining the shifts in BA metabolism is another approach for assessing the impact the gut microbiota on the modeling of the TME. This is the only mouse centered study that was chosen because it was one of a kind in both its approach to study liver disease stages as well as having gut microbiota data that was sequenced to the genus level. In this study, mice were induced to develop HCC using a combination of HFD and a tumor inducer, streptozotocin. Samples were taken at each stage, steatosis, fibrosis, cirrhosis and HCC and bile acids (BAs) were analyzed at each stage both in liver and feces (Xie et al., 2016a).
There are a few new genera not encountered in the other studies such as Desulfovibrio, Parasutterella, Barnsiella and Odoribacter. One can see from these data that organisms that are negatively associated with the disease stages tend to decrease BAs in the liver which cause inflammation while those organisms that are positively associated with the disease stages tend to increase toxic BAs in the liver. There are some discrepancies but this may be due to the necessity for pursuing taxonomy to specific strains or species to identify strains that are more pro-inflammatory. The use of BA profiles can further our understanding of the inflammatory status of the TME in a non-invasive way.
Thus far we have focused mainly on changes in the gut microbiota composition correlated with disease progression and have observed a shift in microbiota composition. It starts with an almost abrupt decrease in anti-inflammatory commensal bacteria followed by increased pro-inflammatory species as one progresses from early virus infection to fibrosis. The progression to cirrhosis seems to be made mainly by fold-changes and the appearance of a few new species in order to further elevate inflammation. As one proceeds to HCC, however, there is shift in the balance between inflammatory and anti-inflammatory genera of bacteria and more anti-inflammatory or immune-modulating species appear to create a new homeostatic condition to preserve the life of the tumor.
Changes in the Gut Microbiota Associated with CRC
CRC-related microbiota could be classified to three distinct patterns (Mizutani et al., 2020). First, the abundances of gut microbes such as pro-inflammatory bacteria were elevated from stage 0 to more advanced stages, such as Fusobacterium nucleatum, Peptostreptococcus anaerobius, Peptostreptococcus stomatis, and Parvimonas micra., P. anaerobius, P. stomatis and P. micra were predominantly enriched in stage I/II and stage III/IV, and their abundances decreased after tumor resection, which implies that these species might not cause carcinogenesis but were adapted to the cancerous environment (Yachida et al., 2019). Second, the abundances of some microbes, such as Atopobium parvulum and Actinomyces odontolyticus, increased in multiple polypoid adenomas and/or in stage 0 but were not increased in more advanced stages. Third, those anti-inflammatory microbes or some probiotics, such as butyrate producers (i.e., Lachnospira multipara and Eubacterium eligens) and Bifidobacterium longum were depleted with the progression of CRC. Like HCC, there are changes in the composition of the microbiota as the disease progresses through different stages (Liu et al., 2020).
In the first study for discussion of CRC (Table 8), a metagenomics shotgun sequencing of 156 fecal samples was done (Feng et al., 2015). The samples were from 55 HCs, 42 advanced adenoma and 41 carcinoma patients.
Some microbes are associated with CLD and HCC as well as new organisms more specific for CRC. Thetaiotaomicron, Difficile, Oscillibacter, Ovatus, Methanobrevibacter, and Porphyromonas have been reported as opportunistic pathogens, and are implicated in IBD, IBS, ulcerative colitis, and CRC (Saitoh et al., 2002; Kim et al., 2012; Dai et al., 2018; Porter et al., 2018; Sandhu and McBride, 2018; Wu et al., 2019). Some other opportunistic pathogens, such as Parvimonas and Dialister are implicated with oral infections and liver abscesses (Chaucer et al., 2018; Soeiro et al., 2019). Coprococcus, Holdemania, Xylanisolvens, and Acidominococcus have been associated with increased consumption of dietary fiber and are considered as commensal bacteria (Voreades et al., 2014; Despres et al., 2016; Barrett et al., 2018). Marvinbryantia is a member of Clostridia and ferments cellulose and methylcellulose. It also boosts the production of succinate (Rey et al., 2010).
What we can conclude from this initial study is that the contribution of the gut microbiota for CRC appears to be relatively pro-inflammatory relative to the adenoma stage although there are anti-inflammatory microbes present. One can also see that there is a different group of gut microbiota from what was seen in HCC that has shifted and thus the TME for CRC, if it receives contributions from the gut microbiota for its modeling, is different from that of HCC based on the idea that the gut microbiota fuels the tumor as well as the host and are a lifeline for the cancer to continue to grow.
A second study (Table 9) was done across all three stages of the disease, polyps, advanced adenoma, and carcinoma (Zhang et al., 2018). The cohort studied included 130 CRC, 88 advanced adenoma, 62 patients with polyps and 130 HCs. What was unique is that all the taxonomy was reported at the genus/species level.
The results in Table 9 demonstrate just how different conclusions can be made if taxonomy is taken to the species level. There is a clear increase in inflammation across the stages of the disease with little abatement at the tumor stage as was seen in all the other studies that were discussed.
The final summary table (Table 10) is a comparison of gut microbiota shifts between CRC and HCC. The three types of HCC, HCV, HBV and non-viral gut microbiota shifts are listed separately. It is immediately apparent that there are many more bacteria genera reported for CRC than for HCC. The table also indicates that there are definite differences in gut microbiota shifts for all three types of HCC and that there are also common organisms reported for both CRC and HCC. Some increasing and decreasing trends are also sometime opposite for the different cancer types. The emerging picture is that CRC is distinct from HCC in their respective, gut microbiota driven progression and maintenance. It cannot, however be excluded that there are some functional redundancies amongst the different microbiota associated with the two cancers.
Conclusion
In this review we sought to discover if the tumor microenvironment was similar for the various types of HCC and to compare HCC vs. CRC. If we use the idea that the microbiota co-metabolizes with the tumor in the same way as it provides for the host, then we can think of it as a lifeline for the tumor. Targeting the lifeline of a tumor is not a new concept as one of the first attempts to do this was with anti-angiogenic drugs with the notion of “starving the tumor” by denying it a blood supply. Perhaps we can manipulate the microbiota to achieve a similar effect. If the gut microbiota do contribute to the modelling of the TME via providing nutrients or immune modulation then from the data presented here, HCC and CRC have a very different TME. In addition, it can be seen from the Table 10 that HCC spawned from three different etiologies may also have distinct differences in their TME. Although we just chose just a few isolated studies to discuss, certain consistencies did reveal themselves. For instance, the overgrowth of Streptococcus that was more dominant for HCV-HCC relative to HBV-HCC. There was also a slightly more pro-inflammatory profile for non-viral HCC when compared to viral HCC and perhaps this is because non-viral HCC begins on a background of an already “homeostatic” inflamed intestine due to the shift in the balance which happened to cause IBD or metabolic syndrome. It must be emphasized that more studies addressing evolutionary changes in the gut microbiota correlated to disease etiology and/or stage need to be done. It should also be cautioned that some results may differ, especially between different labs as a result of different sampling techniques, depth of sequencing and patient populations. However, the ability to manipulate the gut microbiota to kill tumors is certainly something worth reaching for.
The limitation of the review is that fecal microbiota composition is not equivalent to the concept of “TME”. The microbiome profile of tumor tissue would be more appropriate than fecal microbiome. In a study conducted by Sung and Yu groups, gut mucosal microbiome of adenoma and adenoma-adjacent mucosae, carcinoma and carcinoma-adjacent mucosae have been analyzed. The results suggest that a taxonomically defined microbial consortium, such as Bacteroides fragilis, Gemella, Parvimonas, Peptostreptococcus, and Granulicatella are implicated in the development of CRC (Nakatsu et al., 2015). Another study on CRC also found that the colorectal mucosal microbiota were changed with the progression of CRC, with an increasing trend in the abundances of Bacteroidetes, Firmicutes and Fusobacteria, and a decreasing trend in the abundance of Proteobacteria from stage I to IV (Pan et al., 2020). As to liver diseases, the alcohol-induced liver disease mouse models showed that bacterial translocation to the liver might be associated with microbiota changes in the distal gastrointestinal tract (Bluemel et al., 2019).
Change history
18 December 2020
The original version is updated due to inclusion of envelop symbol to the corresponding author.
References
Aly AM, Adel A, El-Gendy AO, Essam TM, Aziz RK (2016) Gut microbiome alterations in patients with stage 4 hepatitis C. Gut Pathog 8:42
Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB et al (2012) Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338:120–123
Barrett HL, Gomez-Arango LF, Wilkinson SA, McIntyre HD, Callaway LK, Morrison M, Dekker Nitert M (2018) A vegetarian diet is a major determinant of gut microbiota composition in early pregnancy. Nutrients 10:890
Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H (2005) Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res 589:47–65
Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, Bernstein H (2011) Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol 85:863–871
Bluemel S, Wang L, Kuelbs C, Moncera K, Torralba M, Singh H, Fouts DE, Schnabl B (2019) Intestinal and hepatic microbiota changes associated with chronic ethanol administration in mice. Gut Microbes 14:1–11
Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC et al (2015) The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis 60:208–215
Chaucer B, Smith N, Beatty D, Yadav M (2018) Multiple hepatic abscess from parvimonas micra: an emerging gastrointestinal microbe. ACG Case Rep J 5:e70
Chen Y, Guo J, Qian G, Fang D, Shi D, Guo L, Li L (2015) Gut dysbiosis in acute-on-chronic liver failure and its predictive value for mortality. J Gastroenterol Hepatol 30:1429–1437
Chen K, Ma J, Jia X, Ai W, Ma Z, Pan Q (2019) Advancing the understanding of NAFLD to hepatocellular carcinoma development: from experimental models to humans. Biochim Biophys Acta Rev Cancer 1871:117–125
Chow MD, Lee YH, Guo GL (2017) The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol Aspects Med 56:34–44
Chu H, Duan Y, Yang L, Schnabl B (2019) Small metabolites, possible big changes: a microbiota-centered view of non-alcoholic fatty liver disease. Gut 68:359–370
Coppenhagen-Glazer S, Sol A, Abed J, Naor R, Zhang X, Han YW, Bachrach G (2015) Fap2 of Fusobacterium nucleatum is a galactose-inhibitable adhesin involved in coaggregation, cell adhesion, and preterm birth. Infect Immun 83:1104–1113
Cosseau C, Devine DA, Dullaghan E, Gardy JL, Chikatamarla A, Gellatly S, Yu LL, Pistolic J, Falsafi R, Tagg J et al (2008) The commensal Streptococcus salivarius K12 downregulates the innate immune responses of human epithelial cells and promotes host-microbe homeostasis. Infect Immun 76:4163–4175
Dai Z, Coker OO, Nakatsu G, Wu WKK, Zhao L, Chen Z, Chan FKL, Kristiansen K, Sung JJY, Wong SH et al (2018) Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome 6:70
De Simone V, Pallone F, Monteleone G, Stolfi C (2013) Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2:e26617
De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D, Sica GS, Sileri P, MacDonald TT, Pallone F et al (2015) Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene 34:3493–3503
Dejong CH, van de Poll MC, Soeters PB, Jalan R, Olde Damink SW (2007) Aromatic amino acid metabolism during liver failure. J Nutr 137:1579S–1585S
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB (2019) Colorectal cancer. Lancet 394:1467–1480
Despres J, Forano E, Lepercq P, Comtet-Marre S, Jubelin G, Chambon C, Yeoman CJ, Berg Miller ME, Fields CJ, Martens E et al (2016) Xylan degradation by the human gut Bacteroides xylanisolvens XB1A(T) involves two distinct gene clusters that are linked at the transcriptional level. BMC Genomics 17:326
Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z et al (2015) Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun 6:6528
Ferreira DM, Afonso MB, Rodrigues PM, Simao AL, Pereira DM, Borralho PM, Rodrigues CM, Castro RE (2014) c-Jun N-terminal kinase 1/c-Jun activation of the p53/microRNA 34a/sirtuin 1 pathway contributes to apoptosis induced by deoxycholic acid in rat liver. Mol Cell Biol 34:1100–1120
Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T et al (2013) Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504:446–450
George J, Pera N, Phung N, Leclercq I, Yun Hou J, Farrell G (2003) Lipid peroxidation, stellate cell activation and hepatic fibrogenesis in a rat model of chronic steatohepatitis. J Hepatol 39:756–764
Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR (2016) Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome 4:30
Greathouse KL, Harris CC, Bultman SJ (2015) Dysfunctional families: Clostridium scindens and secondary bile acids inhibit the growth of Clostridium difficile. Cell Metab 21:9–10
Guo W, Tan HY, Wang N, Wang X, Feng Y (2018) Deciphering hepatocellular carcinoma through metabolomics: from biomarker discovery to therapy evaluation. Cancer Manag Res 10:715–734
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, Enk J, Bar-On Y, Stanietsky-Kaynan N, Coppenhagen-Glazer S et al (2015) Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42:344–355
Heidrich B, Vital M, Plumeier I, Doscher N, Kahl S, Kirschner J, Ziegert S, Solbach P, Lenzen H, Potthoff A et al (2018) Intestinal microbiota in patients with chronic hepatitis C with and without cirrhosis compared with healthy controls. Liver Int 38:50–58
Hibberd AA, Lyra A, Ouwehand AC, Rolny P, Lindegren H, Cedgard L, Wettergren Y (2017) Intestinal microbiota is altered in patients with colon cancer and modified by probiotic intervention. BMJ Open Gastroenterol 4:e000145
Inoue T, Nakayama J, Moriya K, Kawaratani H, Momoda R, Ito K, Iio E, Nojiri S, Fujiwara K, Yoneda M et al (2018) Gut dysbiosis associated with hepatitis C virus infection. Clin Infect Dis 67:869–877
Kameyama K, Itoh K (2014) Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice. Microbes Environ 29:427–430
Kim G, Deepinder F, Morales W, Hwang L, Weitsman S, Chang C, Gunsalus R, Pimentel M (2012) Methanobrevibacter smithii is the predominant methanogen in patients with constipation-predominant IBS and methane on breath. Dig Dis Sci 57:3213–3218
Koliaraki V, Pasparakis M, Kollias G (2015) IKKbeta in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J Exp Med 212:2235–2251
Krenkel O, Tacke F (2017) Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 17:306–321
La Reau AJ, Suen G (2018) The Ruminococci: key symbionts of the gut ecosystem. J Microbiol 56:199–208
La Rosa SL, Leth ML, Michalak L, Hansen ME, Pudlo NA, Glowacki R, Pereira G, Workman CT, Arntzen MO, Pope PB et al (2019) The human gut Firmicute Roseburia intestinalis is a primary degrader of dietary beta-mannans. Nat Commun 10:905
Leclercq S, Matamoros S, Cani PD, Neyrinck AM, Jamar F, Starkel P, Windey K, Tremaroli V, Backhed F, Verbeke K et al (2014) Intestinal permeability, gut-bacterial dysbiosis, and behavioral markers of alcohol-dependence severity. Proc Natl Acad Sci USA 111:E4485–4493
Li J, Stanger BZ (2019) The tumor as organizer model. Science 363:1038–1039
Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, Lin J (2011) STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer Res 71:7226–7237
Liu Q, Li F, Zhuang Y, Xu J, Wang J, Mao X, Zhang Y, Liu X (2019) Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog 11:1
Liu X, Cheng Y, Shao L, Ling Z (2020) Alterations of the predominant fecal microbiota and disruption of the gut mucosal barrier in patients with early-stage colorectal cancer. Biomed Res Int 2020:2948282
Long AG, Lundsmith ET, Hamilton KE (2017) Inflammation and colorectal cancer. Curr Colorectal Cancer Rep 13:341–351
Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y et al (2017) Gut microbiota promotes obesity-associated liver cancer through PGE2-mediated suppression of antitumor immunity. Cancer Discov 7:522–538
Lopetuso LR, Scaldaferri F, Petito V, Gasbarrini A (2013) Commensal Clostridia: leading players in the maintenance of gut homeostasis. Gut Pathog 5:23
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489:220–230
Mizutani S, Yamada T, Yachida S (2020) Significance of the gut microbiome in multistep colorectal carcinogenesis. Cancer Sci 111:766–773
Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D et al (2013) ROS production and NF-kappaB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 12:761–773
Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WK, Ng SC, Tsoi H, Dong Y, Zhang N et al (2015) Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun 6:8727
O’Callaghan A, van Sinderen D (2016) Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol 7:925
Ohtani N, Kawada N (2019) Role of the gut-liver axis in liver inflammation, fibrosis, and cancer: a special focus on the gut microbiota relationship. Hepatol Commun 3:456–470
Pan HW, Du LT, Li W, Yang YM, Zhang Y, Wang CX (2020) Biodiversity and richness shifts of mucosa-associated gut microbiota with progression of colorectal cancer. Res Microbiol 1:12. https://doi.org/10.1016/j.resmic.2020.01.001
Park CH, Eun CS, Han DS (2018) Intestinal microbiota, chronic inflammation, and colorectal cancer. Intest Res 16:338–345
Patel M, Shariff MI, Ladep NG, Thillainayagam AV, Thomas HC, Khan SA, Taylor-Robinson SD (2012) Hepatocellular carcinoma: diagnostics and screening. J Eval Clin Pract 18:335–342
Pedersen KB, Pulliam CF, Patel A, Del Piero F, Watanabe TTN, Wankhade UD, Shankar K, Hicks C, Ronis MJ (2019) Liver tumorigenesis is promoted by a high saturated fat diet specifically in male mice and is associated with hepatic expression of the proto-oncogene Agap2 and enrichment of the intestinal microbiome with Coprococcus. Carcinogenesis 40:349–359
Petersen C, Round JL (2014) Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol 16:1024–1033
Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH (2010) Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105:2420–2428
Porter NT, Luis AS, Martens EC (2018) Bacteroides thetaiotaomicron. Trends Microbiol 26:966–967
Rao SG, Jackson JG (2016) SASP: tumor suppressor or promoter? Yes! Trends Cancer 2:676–687
Rapozo DC, Bernardazzi C, de Souza HS (2017) Diet and microbiota in inflammatory bowel disease: the gut in disharmony. World J Gastroenterol 23:2124–2140
Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, Gordon JI (2010) Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem 285:22082–22090
Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259
Rodier F, Campisi J (2011) Four faces of cellular senescence. J Cell Biol 192:547–556
Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW (2013) Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14:195–206
Saitoh S, Noda S, Aiba Y, Takagi A, Sakamoto M, Benno Y, Koga Y (2002) Bacteroides ovatus as the predominant commensal intestinal microbe causing a systemic antibody response in inflammatory bowel disease. Clin Diagn Lab Immunol 9:54–59
Sakamoto M, Takagaki A, Matsumoto K, Kato Y, Goto K, Benno Y (2009) Butyricimonas synergistica gen. nov., sp. nov. and Butyricimonas virosa sp. nov., butyric acid-producing bacteria in the family ‘Porphyromonadaceae’ isolated from rat faeces. Int J Syst Evol Microbiol 59:1748–1753
Sandhu BK, McBride SM (2018) Clostridioides difficile. Trends Microbiol 26:1049–1050
Schwenger KJP, Chen L, Chelliah A, Da Silva HE, Teterina A, Comelli EM, Taibi A, Arendt BM, Fischer S, Allard JP (2018) Markers of activated inflammatory cells are associated with disease severity and intestinal microbiota in adults with nonalcoholic fatty liver disease. Int J Mol Med 42:2229–2237
Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G et al (2013) Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 152:25–38
Soeiro C, Quilici IR, Legoff A, Oussalah MB, Morin M, Alauzet C, Charmillon A (2019) Hepatic abscess due to Dialister pneumosintes—a case report. Anaerobe 59:35–37
Stenman LK, Holma R, Eggert A, Korpela R (2013) A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 304:G227–234
Takeshita K, Mizuno S, Mikami Y, Sujino T, Saigusa K, Matsuoka K, Naganuma M, Sato T, Takada T, Tsuji H et al (2016) A single species of Clostridium Subcluster XIVa decreased in ulcerative colitis patients. Inflamm Bowel Dis 22:2802–2810
Thota VR, Dacha S, Natarajan A, Nerad J (2011) Eggerthella lenta bacteremia in a Crohn’s disease patient after ileocecal resection. Fut Microbiol 6:595–597
Ullman TA, Itzkowitz SH (2011) Intestinal inflammation and cancer. Gastroenterology 140:1807–1816
Upadhyaya B, McCormack L, Fardin-Kia AR, Juenemann R, Nichenametla S, Clapper J, Specker B, Dey M (2016) Impact of dietary resistant starch type 4 on human gut microbiota and immunometabolic functions. Sci Rep 6:28797
van den Bogert B, Meijerink M, Zoetendal EG, Wells JM, Kleerebezem M (2014) Immunomodulatory properties of Streptococcus and Veillonella isolates from the human small intestine microbiota. PLoS ONE 9:e114277
Voreades N, Kozil A, Weir TL (2014) Diet and the development of the human intestinal microbiome. Front Microbiol 5:494
Wang J, Wang Y, Zhang X, Liu J, Zhang Q, Zhao Y, Peng J, Feng Q, Dai J, Sun S et al (2017) Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic hepatitis B patients. Front Microbiol 8:2222
Wang K, Liao M, Zhou N, Bao L, Ma K, Zheng Z, Wang Y, Liu C, Wang W, Wang J et al (2019) Parabacteroides distasonis alleviates obesity and metabolic dysfunctions via production of succinate and secondary bile acids. Cell Rep 26(222–235):e225
Wexler HM (2007) Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20:593–621
Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ (2006) Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40:235–243
Wong SH, Zhao L, Zhang X, Nakatsu G, Han J, Xu W, Xiao X, Kwong TNY, Tsoi H, Wu WKK et al (2017) Gavage of fecal samples from patients with colorectal cancer promotes intestinal carcinogenesis in germ-free and conventional mice. Gastroenterology 153(1621–1633):e1626
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, Wang Z, Wang C, Zhang Z, Xia W et al (2014a) gammadeltaT17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity 40:785–800
Wu YJ, Xu MY, Lu LG (2014b) Clinical advances in fibrosis progression of chronic hepatitis B and C. J Clin Transl Hepatol 2:222–227
Wu F, Guo X, Zhang J, Zhang M, Ou Z, Peng Y (2017) Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp Ther Med 14:3122–3126
Wu M, Li P, An Y, Ren J, Yan D, Cui J, Li D, Li M, Wang M, Zhong G (2019) Phloretin ameliorates dextran sulfate sodium-induced ulcerative colitis in mice by regulating the gut microbiota. Pharmacol Res 150:104489
Xavier RJ, Podolsky DK (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434
Xie G, Wang X, Huang F, Zhao A, Chen W, Yan J, Zhang Y, Lei S, Ge K, Zheng X et al (2016a) Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int J Cancer 139:1764–1775
Xie G, Wang X, Liu P, Wei R, Chen W, Rajani C, Hernandez BY, Alegado R, Dong B, Li D et al (2016b) Distinctly altered gut microbiota in the progression of liver disease. Oncotarget 7:19355–19366
Xie YH, Gao QY, Cai GX, Sun XM, Sun XM, Zou TH, Chen HM, Yu SY, Qiu YW, Gu WQ et al (2017) Fecal Clostridium symbiosum for noninvasive detection of early and advanced colorectal cancer: test and validation studies. EBioMedicine 25:32–40
Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M et al (2019) Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med 25:968–976
Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N et al (2017) Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170(548–563):e516
Zeng Y, Chen S, Fu Y, Wu W, Chen T, Chen J, Yang B, Ou Q (2020) Gut microbiota dysbiosis in patients with hepatitis B virus-induced chronic liver disease covering chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. J Viral Hepat 27:143–155
Zhang Z, Zhai H, Geng J, Yu R, Ren H, Fan H, Shi P (2013) Large-scale survey of gut microbiota associated with MHE Via 16S rRNA-based pyrosequencing. Am J Gastroenterol 108:1601–1611
Zhang Y, Yu X, Yu E, Wang N, Cai Q, Shuai Q, Yan F, Jiang L, Wang H, Liu J et al (2018) Changes in gut microbiota and plasma inflammatory factors across the stages of colorectal tumorigenesis: a case-control study. BMC Microbiol 18:92
Abbreviations
BA, bile acid; CLD, chronic liver disease; DCA, deoxycholic acid; COX2, cyclooxygenase-2; ECM, extracellular matrix; FGF2, fibroblast growth factor-2; HSC, hepatic stellate cell; HBV/HCV, hepatitis B/C; HCC, hepatocellular carcinoma; ASC, inflammasome adaptor protein apoptosis-associated speck-like protein containing CARD; IL-6/1β/18, interleukin-6/1β/18; KC, Kupfer cell; LPS, lipopolysaccharides; LTA, lipoteichoic acid; MMP, matrix metalloproteinase; NASH, non-alcoholic steatohepatitis; NF-κb, nuclear factor-kappa-B; PDGF, platelet-derived growth factor; PGE2, prostaglandin E2; ROS, reactive oxygen species; TLR-4, toll-like receptor-4; TMA, trimethylamine; TMAO, trimethylamine oxide; TGFβ, tumor growth factor-β; TME, tumor microenvironment; TNFα, tumor necrosis factor-α; TNFR, tumor necrosis factor receptor; VEGF, vascular endothelium growth factor
Compliance with Ethics Guidelines
Wei Jia, Cynthia Rajani, Hongxi Xu and Xiaojiao Zheng declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jia, W., Rajani, C., Xu, H. et al. Gut microbiota alterations are distinct for primary colorectal cancer and hepatocellular carcinoma. Protein Cell 12, 374–393 (2021). https://doi.org/10.1007/s13238-020-00748-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-020-00748-0