
ABSTRACT
Neuropsychiatric disorders are complex disorders characterized by heterogeneous genetic variations, variable symptoms, and widespread changes in anatomical pathology. In the context of neuropsychiatric disorders, limited access to relevant tissue types presents challenges for understanding disease etiology and developing effective treatments. Induced pluripotent stem cells (iPSCs) reprogrammed from patient somatic cells offer an opportunity to recapitulate disease development in relevant cell types, and they provide novel approaches for understanding disease mechanisms and for development of effective treatments. Here we review recent progress and challenges in differentiation paradigms for generating disease-relevant cells and recent studies of neuropsychiatric disorders using human pluripotent stem cell (hPSC) models where cellular phenotypes linked to disease have been reported. The use of iPSC-based disease models holds great promise for understanding disease mechanisms and supporting discovery of effective treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
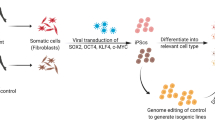
The recent advances in technologies that enable adult human somatic cells to be reprogrammed into iPSCs (Takahashi et al., 2007; Yu et al., 2007) hold enormous potential in the field of regenerative medicine (Shi et al., 2017). Despite the promise of iPSCs as an autologous cell source for cell transplantation therapy, concerns have been raised regarding the clinical applications of iPSCs (Gore et al., 2011; Zhao et al., 2011, 2015b; Ji et al., 2012; Kang et al., 2016). While much work remains to be done to improve the safety and reliability of the technology, iPSCs have provided a unique opportunity to study developmental neuropsychiatric diseases. In the past, this type of research has have been challenging due to the inaccessibility of relevant live tissues and cell types. The lack of reliable models has significantly hindered the progress in understanding the pathogenesis of the disease and development of novel treatments.
Animal models provide valuable insights into the mechanisms of specific functions of genes. However, modeling of human neuropsychiatric disorders in animals is extremely challenging given the complexity of the disorders, the subjective nature of many symptoms, and the lack of biomarkers and reliable tests (Nestler and Hyman, 2010). The differences in the brain structures of human and mouse have also made it difficult to study behaviors related to higher-function brain areas such as the dorsolateral prefrontal cortex, one of the most recently derived parts of the human brain that is often implicated in neuropsychiatric disorders (Tekin and Cummings, 2002). In addition, it is difficult to assess some complex phenotypes in mouse, such as psychosis.
Postmortem human brains serve as valuable sources for examining pathological changes in patients. However, these tissues only represent the disease endpoint, whereas the alterations that lead to the development of the diseases often occur early in development. Postmortem brain studies are thus limited in their ability to reveal dynamic cellular changes that are often important in the disease mechanisms.
iPSC technology has opened the field to new discoveries in neuropsychiatric diseases (see Table 1 for selected studies). iPSCs can be directly generated from somatic cells by the introduction of reprogramming factors (Takahashi et al., 2007). Moreover, iPSCs are capable of differentiating into adult cell types through directed differentiation with a combination of small molecules, growth factors, and morphogens, allowing the derivation of unlimited disease-relevant cells carrying the variations that caused or facilitated the development of the disease. These cells include human neurons, a previously inaccessible cell type.
Most neuropsychiatric diseases, including autism spectrum disorder (ASD), schizophrenia (SZ), bipolar disorder, and major depression, have a strong but complex genetic component. Multiple low penetrance genetic variants contribute to the etiology of those disorders (Schizophrenia Working Group of the Psychiatric Genomics, 2014; Bipolar et al., 2018; Wray et al., 2018; Grove et al., 2019). Because human iPSCs (hiPSCs) capture the genetic diversity of the patient, they are particularly useful for modeling how complex genetic variants lead to the pathogenesis of the disease.
NEURAL SPECIFICATION FROM HUMAN PLURIPOTENT STEM CELLS
The mammalian brain is composed of many cell populations that differ in their molecular, morphological, connectional, and functional properties. The recent advances in single cell technologies have enabled the identification of new neuronal subtypes in both mouse and human brains (Darmanis et al., 2015; Zeisel et al., 2015; Lake et al., 2016), offering new opportunities to study the diversity of the building blocks of the mammalian brain. However, how distinct subpopulations of cells respond to perturbation and how diseased brains differ in their molecular and functional signatures remain to be explored.
Substantial progress has been made in directing hPSCs differentiating into regional and functional specific neuronal subtypes. These differentiation protocols are often based upon key molecular signals in neural commitment and regionalization identified in animal models. Manipulations of the principles in early neural development have enabled the generation of highly enriched subpopulations of neurons and glial cells in vitro with functional aspects that resemble those in the brain (Mertens et al., 2016).
At the gastrulation stage, nervous system specification initiates from ectoderm cells, in a process triggered by molecules that inhibit bone morphogenetic protein (BMP) signaling and/or activate the fibroblast growth factor (FGF) pathways (Edlund and Jessell, 1999). The neural plate then folds to form the neural tube, from which the brain and spinal cord develop. Specification of subtypes of neural progenitor cells (NPCs) in the neural tube is achieved by the coordinated temporal and spatial gradient of morphogens and mitogens. For example, sonic hedgehog (SHH) is secreted from the notochord and floor plate of the ventral neural tube, whereas Wnt and BMP are secreted from the roof plate in the dorsal neural tube (Ciani and Salinas, 2005; Liu and Niswander, 2005; Fuccillo et al., 2006). This morphogen gradient specifies the neural subtypes along the dorsal-ventral (D-V) axis. The differentiation of the neural tube along the anterior-posterior (A-P) axis is coordinated by the gradient of timely and spatially regulated factors, including FGFs and retinoic acid (RA) (Maden, 2007; Mason, 2007; Guillemot and Zimmer, 2011).
Following this principle, hPSCs are first switched from the self-renewing condition to promote differentiation toward all three germ layers by the removal of self-renewal growth factors. Without extrinsic cues, the default differentiation is towards forebrain identity (Munoz-Sanjuan and Brivanlou, 2002). Coordinating morphogens along both the A-P axis and D-V axis is used for patterning of NPC subtypes (Tao and Zhang, 2016). Regionalized NPCs can be turned into specific neuronal subtypes through differentiation induction and neurotrophic support. For example, the progenitor cells with the dorsal forebrain identity become glutamatergic neurons, whereas the ventral progenitors become gamma-aminobutyric acid-ergic (GABAergic) or cholinergic neurons.
Specification of cortical excitatory neurons
Cortical excitatory neurons are born in the dorsal forebrain in vivo. The telencephalic/forebrain identity develops in the absence of any extrinsic cues and is enhanced by the inhibition of morphogens such as Wnts or BMPs (Wilson and Houart, 2004; Gaspard et al., 2008). The telecephalon then undergoes patterning along the D-V axis, from which cortical excitatory neural precursors are generated in the dorsal telecephalon (Sur and Rubenstein, 2005). The characteristics of corticogenesis are recapitulated in directed differentiation from hPSCs (Watanabe et al., 2005; Eiraku et al., 2008; Gaspard et al., 2008; Chambers et al., 2009; Shi et al., 2012). The layer-identity of a cortical neuron is linked to the timing of its generation: deep layer cortical neurons are generated first, followed by upper layer neurons. Analysis of a recent differentiation protocol has demonstrated that the in vitro generation of excitatory cortical neurons recapitulates the layer-specific sequential order (Espuny-Camacho et al., 2013). The proportion of each layer-specific neuronal subtype depends on differentiation conditions (Gaspard et al., 2008; Espuny-Camacho et al., 2013). Outer radial glial cells, in a region that has undergone significant evolutionary changes in cortical neurogenesis, have also been detected in cortical differentiation from hPSCs (Shi et al., 2012) but not from mouse PSCs (Lancaster et al., 2013). Alterations in corticogenesis have been implicated in many developmental neuropsychiatric disorders. Regional patterns of gene expression that typically differ in frontal and temporal cortex are significantly attenuated in the ASD brain based on transcriptomic analysis (Voineagu et al., 2011). Abnormal neurogenesis leading to functional defects in neuronal networks has also been reported using NPCs from iPSCs reprogrammed from ASD individuals. Interestingly, a drug currently in clinical trials could rescue defects in neuronal networks for ASD in vitro (Marchetto et al., 2017). In a recent study recapitulating cortical development from ASD patients-derived iPSCs, ASD-associated changes in the maturational sequence of early neuron development have been reported (Schafer et al., 2019). Temporal dysregulation of specific gene networks is tracked back to a pathologically primed stage in neural stem cells. Interestingly, circumventing the neural progenitor stage by direct conversion of ASD iPSCs into induced neurons abolished the ASD-associated phenotypes. These findings together support the idea that aberrant early cortical development contributes to the emergence of ASD.
Specification of hippocampal neurons
The hippocampus arises from the caudomedial edge of the dorsal telencephalic neuroepithelium adjacent to the cortical hem (Lee et al., 2000). To generate NPCs with hippocampal identity, embryoid bodies (EBs) (aggregates obtained from hPSC cultures) are treated with antagonists of the SHH pathway and a cocktail of factors mimicking the patterning of the forebrain, including Dickkopf-related protein 1 (DKK1), Noggin, and transforming growth factor-β (TGF-β) inhibitor SB431542, to instruct the dorsal telencephalic precursor identity (Yu et al., 2014). The hippocampal patterned NPCs express genes in early stages of hippocampal neurogenesis, such as SOX2, PAX6, EMX2 and FOXG1. The cortical hem provides a Wnt source to the progenitor cells (Lee et al., 2000). In the presence of high concentrations of Wnt3a, hippocampal dentate gyrus (DG)-like granule neurons that express prospero homeobox protein 1 (PROX1) can be generated from hippocampal NPCs (Yu et al., 2014). Hippocampal DG neurons generated from SZ patient iPSCs exhibited deficits in the generation of DG granule neurons, reduced neuronal activity, and reduced levels of spontaneous neurotransmitter release (Yu et al., 2014). Hippocampal DG neurons differentiated from bipolar patient iPSCs showed hyperexcitability in eletrophysiological recordings (Mertens et al., 2015; Stern et al., 2018). This hyperexcitability phenotype of young neurons in bipolar disorder can be selectively reversed by lithium treatment only in the neurons derived from patients who responded to lithium treatment (Mertens et al., 2015; Stern et al., 2018). The hippocampus proper is defined by the DG and cornu ammonis (CA). An efficient differentiation paradigm that generates multiple CA3 pyramidal neuron subtypes has also been developed using the same hippocampal patterned NPCs (Sarkar et al., 2018). During development, Wnt regulates cell proliferation and specification of the DG and CA fields of the hippocampus (Galceran et al., 2000; Lee et al., 2000). DG primordia locate next to the cortical hem whereas CA3 primodia locate more dorsally. To induce CA3 pyramidal neuron fate, hippocampal NPCs were exposed to lower amount of Wnt3a compared to the DG differentiation protocol (Sarkar et al., 2018). This differentiation paradigm produced several CA3 neuron subtypes, including a human specific SCGN-expressing CA3 neuron subtype.
Specification of inhibitory neurons
In vivo, GABAergic interneurons within the telencephalon populate from one of two embryonic subcortical progenitor zones, the medial and caudal ganglionic eminences (MGE and CGE) (Wonders and Anderson, 2006; Kelsom and Lu, 2013). The interneurons arising from each progenitor zone represent complementary interneuron subtypes (Kepecs and Fishell, 2014). Within the cortex, parvalbumin (PV) fast spiking interneurons and somatostatin interneurons arise from the MGE, whereas neurogliaform, bipolar, and VIP multipolar interneurons arise from the CGE (Lee et al., 2010; Miyoshi et al., 2010; Kepecs and Fishell, 2014). Several groups were able to generate MGE-like progenitors using small molecules that inhibit Wnt and SMAD signaling coupled with timely SHH exposure to induce ventral telencepahlic fate (Liu et al., 2013; Maroof et al., 2013; Nicholas et al., 2013). In culture, the MGE-like cells take a remarkably long time to develop into GABAergic interneuron subtypes with mature physiological properties, mimicking human neural development in vivo (Nicholas et al., 2013). A recent single cell study of the cortical inhibitory interneuron development and diversification has revealed that transcription factor MEF2C, a gene linked to neuropsychiatric and neurodevelopmental disorders, plays an important role in early precursors of PV-expressing interneurons (Mayer et al., 2018). Cortical interneurons generated from iPSCs derived from SZ subjects had dysregulated expression of protocadherin genes, which lie within documented SZ loci (Shao et al., 2019). SZ cortical interneurons showed defects in synaptic density and arborization that could be reversed by inhibitors of a downstream kinase in the protocadherin pathway. Similar phenotype is observed in mice lacking protocadherin-α. These findings reveal an intrinsic abnormality in SZ cortical interneurons, providing evidence that dysregulation of interneuron specification and development is linked to neuropsychiatric disorders.
Specification of microglia
Microglia is the resident macrophages of the central nervous system (CNS) and function in inflammation and the clearance of dead cells and debris. Microglia also play an important role in development and homeostasis, raising an interesting possibility that microglia dysfunction could contribute to neurological and psychiatric diseases. Neural networks are being constantly modified by the elimination and strengthening of the synapses. During late adolescence and early adulthood, synaptic pruning by microglia occurs to remove the excess synapses generated during development and make the brain connections more streamlined. Feinberg suggested that improper synaptic pruning during this period could lead to the development of SZ (Feinberg, 1982).
Microglia originate almost exclusively from erythromyeloid progenitors (EMP) generated during primitive hematopoiesis from yolk-sac cells (Ginhoux et al., 2010; Schulz et al., 2012; Kierdorf et al., 2013). EMPs further develop and migrate into the developing neural tube shortly before the closure of the blood-brain barrier. Microglia progenitors then mature in the CNS and facilitate CNS development. Recent murine studies identified key cytokines and transcription factors required for microglia development and maintenance in vivo, including IL-34, colony-stimulating factor1, and TGF-β (Greter et al., 2012; Wang et al., 2012; Butovsky et al., 2014; Yamasaki et al., 2014). A few studies have succeeded in generating microglia-like cells from hPSCs. These protocols all started with generating primitive hematopoietic cells and then differentiating those cells into microglia, recapitulating environmental cues present in the developing embryo (Muffat et al., 2016; Abud et al., 2017; Pandya et al., 2017). A human embryonic stem cell (hESC) model of Rett syndrome has indicated smaller primary microglial-like cells than their isogenic controls (Muffat et al., 2016). In an in vitro model that applied cellular reprogramming to study SZ-specific microglia-mediated synapse engulfment, excessive pruning was also detected in induced microglia-like cells generated from SZ patients (Sellgren et al., 2019).
Specification of astrocytes
Astrocytes are a major component of the human CNS and provide metabolic and trophic support to neurons. Astrocytes play an essential role in establishing and regulating neural circuits and have been implicated in many aspects of neurodevelopment and in neurodevelopmental diseases (Khakh and Sofroniew, 2015). During embryonic development, astrocytes are generated from radial glial cells at the subventricular zone (SVZ). Radial glial cells give birth to neurons in early embryonic development and to glial cells in the late phase and postnatal period. This neurogenesis-to-gliogenesis switch requires Notch signaling, which activates the JAK/STAT3 pathway and promotes gliogenesis (Morrison et al., 2000). The generation of astrocytes from hPSCs recapitulates the gliogenic switch during embryonic development. Glial progenitor differentiation from hPSCs requires neural induction, coupled with an extended time in culture for the gliogenic switch. To differentiate glial progenitors to astrocytes, BMP and ciliary neurotrophic factor are added to activate the JAK/STAT3 pathway (Krencik et al., 2011). Functional maturation of astrocytes can take up to six months (Krencik and Zhang, 2011; Krencik et al., 2011). An efficient protocol to derive functional astrocytes from hPSCs has been described (Santos et al., 2017), in which the glial progenitor cells can be propagated, expanded and frozen as intermediates. hPSCs are first converted into NPCs and then patterned to glial progenitors that could later give rise to functional astrocytes. Neural induction was achieved in EBs by SMAD inhibition. To induce JAK/STAT3 signaling, PDGF was added. The patterned glial progenitors expressed Nestin, A2B5, a cell surface ganglioside in astrocyte precursors, and nuclear factor IA, a transcription factor activated by Notch, which is required for gliogenesis. To induce differentiation and maturation of astrocytes, leukemia inhibitory factor was used in serum-free condition in neuronal medium. Astrocytic markers can be detected after four to six weeks of differentiation, such as CD44, S100 calcium-binding protein (S100β), glial fibrillary acidic protein (GFAP), and aldehyde dehydrogenase 1 family member L1, an early marker of astrocyte maturation. The hPSC-derived astrocytes are capable of glutamate uptake and display calcium transients. Recent studies of Alexander disease (AxD) have identified GFAP aggregates and Rosenthal fibers in AxD patient iPSC-derived astrocytes (Jones et al., 2018; Li et al., 2018). Moreover, these iPSC-based models also revealed that AxD astrocytes inhibited proliferation of human iPSC-derived oligodendrocyte progenitor cells (OPCs) in co-culture and reduced their myelination potential (Li et al., 2018), recapitulating the pathological hallmark of AxD.
Specification of oligodendrocytes
Recent evidence has suggested that there are abnormalities in oligodendrocyte function in psychiatric conditions (Hakak et al., 2001; Tkachev et al., 2003; Nave and Ehrenreich, 2014). Oligodendrocytes arise late during development, providing support and insulation to axons in the CNS. Oligodendrocytes originate from glial-restricted progenitor cells, known as OPCs, which are generated by subventricular cells in the developing brain and spinal cord (Emery, 2010). OPCs divide and migrate throughout the CNS (Emery, 2010). The process of oligodendrocyte differentiation from hPSCs essentially follows the well-characterized developmental origin of OPCs. Specification of OPCs starts with neural induction, and then followed by SHH activation. Oligodendrocytes generated from hPSCs are capable of producing myelin sheaths when co-cultured with neurons or transplanted into the mouse/rat brain or spinal cord (Keirstead et al., 2005; Nistor et al., 2005; Hu et al., 2009; Kerr et al., 2010; Sharp et al., 2010; Wang et al., 2013; Piao et al., 2015; Rodrigues et al., 2017). Direct conversion of OPCs from fibroblasts and induction of oligodendrocytes in human cortical spheroids have also been reported (Yang et al., 2013; Madhavan et al., 2018).
Functional circuits
Neurons are organized into circuits in neural networks to perform information processing and transmission. Dysregulated synaptic function has been hypothesized to underlie the altered neuronal function in complex neuropsychiatric disorders. It is therefore imperative to generate an in vitro model where synaptic function can be directly assessed. Neuronal maturation has been limited in vitro due to the difficulties of maintaining a long-term culture required for neurons to mature while still keeping them in a healthy condition. Substantial improvements in these areas have been made in the past decade. Synaptic markers such as synapsin and postsynaptic density protein 95 (PSD-95) can be detected in hPSCs-derived neural cultures. Moreover, spontaneous synaptic currents have been detected by electrophysiological recordings, indicating the formation of active neural networks. These advances have led to the discoveries of aberrant synaptic functions in iPSC models of SZ. In iPSCs derived from idiopathic SZ patients, diminished neuronal connectivity has been reported in conjunction with decreased neurite number (Brennand et al., 2011). A more recent study of iPSCs derived from SZ patients carrying a four base-pair deletion in Disrupted-in-schizophrenia 1 (DISC1), a gene that co-segregates with major psychiatric disorders, has reported aberrant synaptic formation and synaptic vesicle release deficits in patient-derived neurons (Wen et al., 2014).
Establishing a functional neural circuit in vitro using hPSC-derived neurons will help decipher the abnormalities at the circuit level in disease conditions. This is especially important for modeling neuropsychiatric disorders in which the imbalanced/abnormal neural networks have been implicated in the disease etiology. To recapitulate the disease status in vitro, it is also important to study the functional circuits in which the disease has been implicated. Attempts to model neuronal pathology of the cells with the functional targets have been made. Hippocampal circuitry has been studied using hPSC-derived DG and CA3 neurons (Sarkar et al., 2018). Microfluidic devices, involving a two-compartment system connected by narrow grooves, have been used to reconstruct DG-CA3 circuitry in which DG neurons are cultured in one compartment and CA3 neurons are cultured in the other compartment. In this system, axonal growth is allowed through the narrow grooves connecting the two compartments whereas cell migration is restricted. Rabies virus infection of the CA3 neurons permits the detection of presynaptic neurons that monosynaptically connect to the postsynaptic CA3 neurons. The presence of the DG-CA3 connection suggests that microfluidic device-based reconstruction of circuitry in vitro mimics the synaptic connections in vivo. Hippocampus-dependent functional impairment has been reported in SZ (Cirillo and Seidman, 2003; Freedman and Goldowitz, 2010; Tamminga et al., 2010; Rasetti et al., 2014). In postmortem studies, reduced glutamate transmissions in the mossy fiber pathway have also been indicated in SZ (Kolomeets et al., 2007; Tamminga et al., 2010; Li et al., 2015). Intriguingly, co-culture of the DG and CA3 neurons derived from SZ iPSCs has revealed decreased spontaneous activity (Sarkar et al., 2018). All together, these studies indeed suggest great promise in studying defects in transmission in neurons generated from patients with mental disorders.
A THREE-DIMENSIONAL (3D) BRAIN ORGANOID MODEL
Monolayer neuronal cultures often generate highly enriched uniform populations of targeted cell types (Pasca, 2018). The scalability and simplicity of two-dimensional (2D) cultures make the system more suitable for mechanistic studies and large-scale screening, e.g., CRISPR-Cas9 screening or high-throughput drug testing. However, the complex nature of brain tissues requires a better system to recapitulate the organogenesis, which involves local proliferation and patterning, cell fate determination, complex cell–cell interactions, and long-distance migration. Promisingly, 3D neural cultures, including brain organoids and neural spheroids, may serve this purpose.
Compared to 2D culture, in which cells are purified and isolated, 3D culture allows cells of different types to interact. However, understanding the cellular composition of brain organoids is challenging. To solve this issue, single-cell RNA sequencing allows systematic interrogation of many genes from large number of cells, thereby offering a good opportunity to isolate distinct cell clusters and study their molecular properties. A diversity of cell types, including NPCs, OPCs, glia, neurons, and retinal cells, has been identified in brain organoids (Quadrato et al., 2017). Within each cell type, subtypes can be further classified. For instance, 30 transcriptionally distinct subclusters can be identified in a neuronal cluster (Quadrato et al., 2017), and glial cells can be specified into at least three clusters (Sloan et al., 2017). Thus, endogenous cellular diversity can be recapitulated in organoids, and chances are that human-specific cell types that are transiently present in certain developmental stages might be discovered (Amiri et al., 2018).
The 3D organoid is far beyond a mixture of diverse cell types, but rather a self-assembly of cells under temporal and spatial regulation. In early organoid cultures, neural stem cells or radial glia reside next to the ventricle-like structure and give birth to neurons in the outer layers (Kadoshima et al., 2013; Lancaster et al., 2013; Pasca et al., 2015; Qian et al., 2016; Bershteyn et al., 2017). Astrocytes are rarely observed until six months or later (Lancaster et al., 2013; Pasca et al., 2015; Qian et al., 2016; Sloan et al., 2017). Oligodendrocytes can be induced by exogenous molecules, including PDGF-AA, IGF-1 (D50-60), T3, and PERK inhibitor, in the late stage of organoids culture (Madhavan et al., 2018). This timeline is consistent with early brain development, where neurogenesis starts prenatally and is followed by the gliogenesis postnatally (Miller and Gauthier, 2007). A recent study has confirmed the feasibility of using organoids to study molecular and cellular events in early cortical development (Amiri et al., 2018). Forebrain organoids were patterned and maintained in differentiation medium for 0, 11, or 30 days, and samples were collected for bulk or single-cell sequencing. At both transcriptomic and epigenomic level, those organoids recapitulate the temporal development of the human neocortex between 8 and 16 postconceptional week. Indeed, organoid system has been used in many studies to dissect temporal and spatial phenotypes in development. A decreased cell-cycle length was observed in early stage of organoids derived from ASD patients, but not in later stage (Mariani et al., 2015). In a separate study, an increased thickness of TBR1+ cortical regions was identified in ASD organoids (Schafer et al., 2019), which has been suggested to result from the increased NPC proliferation.
To better resemble the brain environment, CNS components that originate outside the ectoderm are generated and introduced to the brain organoids. In a study investigating neurite shaping and neuroinflammation, induced microglia from hPSCs have been introduced to organoid cultures (Abud et al., 2017). The neurovascular unit is crucial in CNS for supplying the brain with nutrients from blood stream and, although it is still missing in brain organoids, recent studies have successfully built functional blood vessels in vitro from hPSCs (Wimmer et al., 2019).
Early efforts to generate brain organoids from hPSCs largely depended on cell self-assembly with minimum external patterning (Lancaster et al., 2013). The “intrinsic protocol” offers the brain organoids more degrees of freedom to develop; however, it also makes the system less reliable in which the neural induction is less predictable, and a variety of brain region identities are randomly distributed in each individual organoid. Therefore, “directed approaches” have been developed to guide the organoids to acquire brain region-specific properties (Muguruma et al., 2015; Sakaguchi et al., 2015; Jo et al., 2016; Qian et al., 2016). In these protocols, hPSCs are first differentiated towards ectoderm and subsequently specified to the targeted brain regions by exogenous molecules (Di Lullo and Kriegstein, 2017). Dorsal forebrain is the default fate of the ectodermal tissues; therefore, minimal intervention is required to generate forebrain organoids. By manipulating pathways that are critical during development—for instance, Wnt, BMP, FGF, or SHH, CNS regions of ventral forebrain (Bagley et al., 2017; Birey et al., 2017), MGE (Xiang et al., 2017), midbrain (Jo et al., 2016; Qian et al., 2016; Xiang et al., 2017), hippocampus (Sakaguchi et al., 2015), hypothalamus (Qian et al., 2016), and cerebellum (Muguruma et al., 2015) can be induced in brain organoids (see Table 2 for the selected approaches). Intriguingly, many human brain-specific features are observed in brain region-specific organoids, which highlights the advantage of using organoid system to study human brain development and decode human-specific disorders. For instance, the human cerebral cortex, which is different from that in other species like mice, expands with folded surface (gyrencephalic) (Dehay et al., 2015). The complexification of the human cortex starts early in development and is associated with the emergence of the outer SVZ (oSVZ), particularly outer radial glia (oRG) (Dehay et al., 2015). In cortical organoids, the structure of the oSVZ has been observed (Qian et al., 2016; Bershteyn et al., 2017). Organoids derived from patients with nearly absent cortical folding, namely Miller-Dieker syndrome (MDS), were smaller in size and displayed deficits in precursor cleavage and early neuron migration (Iefremova et al., 2017). Consistently, in a separate study, mitotic delay in oRG was observed in MDS organoids (Bershteyn et al., 2017). On the other hand, the expansion and folding of the neuroepithelial structure are observed in PTEN mutant human cerebral organoids, which is linked to increased neural progenitor proliferation (Li et al., 2017).
Human brain development requires that cells migrate long distances from one brain region to another (Pasca, 2019), and cellular processes extend out to connect different brain regions. The fusion of two or more region-specific organoids may enable the study of inter-regional interactions and circuit assembly in human CNS. Glutamatergic pyramidal neurons and GABAergic interneurons are two major neuronal types that compose the neuronal circuits in the cerebral cortex. While glutamatergic neurons are generated in dorsal forebrain, GABAergic interneurons are primarily born in the ventral forebrain and migrate over long distances to integrate into cortical circuits. Disruption in interneuron migration is linked to neurodevelopmental disorders such as ASD and epilepsy (Birey et al., 2017). To study interneuron migration, region-specific organoids resembling MGE and dorsal forebrain cortical organoids are generated (Birey et al., 2017; Xiang et al., 2017) and fused together. The migration of interneurons has been identified specifically from ventral forebrain organoids to dorsal forebrain cortical organoids, whereas the reverse is not observed. However, abnormal interneuron migration is observed in organoids derived from patients with Timothy syndrome (Birey et al., 2017).
Thus far, organoid models have been largely used to study events in early brain development, including neuroprogenitor proliferation (Qian et al., 2016), interneuron migration (Birey et al., 2017; Xiang et al., 2017), and formation of cortical layers (Qian et al., 2016; Schafer et al., 2019). To build a model system that resembles the human brain in postnatal stage requires that brain organoids survive for extended periods of time and remain healthy. Although brain organoids can be kept in culture for up to two years (Pasca et al., 2015), the overall survival rate is low and the necrotic center becomes prominent in long-term culture, which might be a concern when organoids are used to recapitulate disease phenotype in postnatal brain. Lack of vascular circulation is believed to be one major factor that limits the life span of brain organoids. To address this issue, brain organoids have been generated in vitro and transplanted into adult mouse brain (Mansour et al., 2018). Intriguingly, the grafted organoids gained functional vascularization system soon after transplantation, showing progressive neuronal maturation and gliogenesis, and connecting to host brains by sending out extensive axons. Importantly, organoids can survive as long as 200+ days in mouse brain. While this strategy presents great promise in long-term maintenance of brain organoids, the developmental stage of those organoids remains to be determined. Another question is whether human brain tissue can respond to visual, auditory, or motor stimuli that are presented to the host mouse.
Brain organoids present powerful potential for studying human brain development. Despite certain limitations in reproducibility, scalability, and long-term survival, this model system offers a unique opportunity to tackle questions that are challenging to address in other systems such as 2D culture system and rodents.
CHALLENGES OF MODELING NEUROPSYCHIATRIC DISORDERS USING HIPSCS
Although the use of iPSCs to study neuropsychiatric disorders has provided unique insights to the field and has demonstrated great potentials to bridge the clinical features with cellular phenotypes that can be studied in the lab, there are a number of obstacles to be overcome. First of all, it has become increasingly clear that there is a great amount of variability in the reprogramming of hiPSCs from somatic cells and in the generation of brain cells from the hiPSCs. Variations may occur in any steps from reprogramming to differentiation. To reduce the technical variability and enable the identification of biological phenotypes, several approaches should be taken into consideration. Careful quality control is necessary at every step. For instance, only the iPSC clones that show full pluripotency should be selected for downstream experiments. Typically, a biological phenotype is ensured only if it’s observed in two or more iPSC clones from the same subject. Genetic background variations represent another challenge to iPSC disease modeling due to epistatic effects. Careful selection of control subjects with comparable age, gender, and genetic background is thus crucial to identifying disease-relevant phenotypes. Furthermore, although enormous attempts have been made to generate distinct neural cell types in vitro, it remains to be explored to what extent these iPSC-derived neural cells recapitulate the molecular and cellular signatures of their in vivo counterpart. In addition, many components of the building blocks of brains are still missing in an in vitro system, such as blood vessels, pericytes, and immune cells. These limitations should be considered and addressed when using hiPSC-derived cells to model human neuropsychiatric disorders.
CONCLUSIONS
In conclusion, hiPSC-based disease modeling technology has demonstrated the potential to advance many aspects of biomedical research. We present here recent progress in the directed differentiation of neuronal subtypes, glial cells, and 3D brain organoid models, and the use of these models to study neuropsychiatric disorders. While many challenges remain to be solved before translating hiPSC-based discoveries directly into clinics, the opportunity to investigate specific neural subtypes using patient-specific cells, to study disease etiology in a 3D cellular model recapitulating features of human brain, and to establish a scalable screening platform for drug discovery in vitro, all support the idea that hiPSC-based research could present a new way to discover disease-relevant functional changes, a better understanding of disease etiology, and a new paradigm for neuropsychiatric drug discovery.
References
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C et al (2017) iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94(278–293):e279
Amiri A, Coppola G, Scuderi S, Wu F, Roychowdhury T, Liu F, Pochareddy S, Shin Y, Safi A, Song L et al (2018) Transcriptome and epigenome landscape of human cortical development modeled in organoids. Science 362:eaat6720
Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA (2017) Fused cerebral organoids model interactions between brain regions. Nat Methods 14:743–751
Bavamian S, Mellios N, Lalonde J, Fass DM, Wang J, Sheridan SD, Madison JM, Zhou F, Rueckert EH, Barker D et al (2015) Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry 20:573–584
Bershteyn M, Nowakowski TJ, Pollen AA, Di Lullo E, Nene A, Wynshaw-Boris A, Kriegstein AR (2017) Human iPSC-derived cerebral organoids model cellular features of lissencephaly and reveal prolonged mitosis of outer radial glia. Cell Stem Cell 20(435–449):e434
Bipolar D, Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address, d.r.v.e., Bipolar, D., and Schizophrenia Working Group of the Psychiatric Genomics, C (2018) Genomic dissection of bipolar disorder and schizophrenia, including 28 SUBPHENOTYPES. Cell 173:1705e1716–1715e1716
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, Fan HC, Metzler KRC, Panagiotakos G, Thom N et al (2017) Assembly of functionally integrated human forebrain spheroids. Nature 545:54–59
Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, Beaumont KG, Kim HJ, Topol A, Ladran I et al (2015) Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry 20:361–368
Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D et al (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 473:221–225
Bundo M, Toyoshima M, Okada Y, Akamatsu W, Ueda J, Nemoto-Miyauchi T, Sunaga F, Toritsuka M, Ikawa D, Kakita A et al (2014) Increased l1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81:306–313
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE et al (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17:131–143
Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol 27:275–280
Ciani L, Salinas PC (2005) WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat Rev Neurosci 6:351–362
Cirillo MA, Seidman LJ (2003) Verbal declarative memory dysfunction in schizophrenia: from clinical assessment to genetics and brain mechanisms. Neuropsychol Rev 13:43–77
Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR (2015) A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 112:7285–7290
Dehay C, Kennedy H, Kosik KS (2015) The outer subventricular zone and primate-specific cortical complexification. Neuron 85:683–694
Deshpande A, Yadav S, Dao DQ, Wu ZY, Hokanson KC, Cahill MK, Wiita AP, Jan YN, Ullian EM, Weiss LA (2017) Cellular phenotypes in human iPSC-derived neurons from a genetic model of autism spectrum disorder. Cell Rep 21:2678–2687
Di Lullo E, Kriegstein AR (2017) The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci 18:573–584
Edlund T, Jessell TM (1999) Progression from extrinsic to intrinsic signalling in cell fate specification: a view from the nervous system. Cell 96:211–224
Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y (2008) Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3:519–532
Emery B (2010) Regulation of oligodendrocyte differentiation and myelination. Science 330:779–782
Espuny-Camacho I, Michelsen KA, Gall D, Linaro D, Hasche A, Bonnefont J, Bali C, Orduz D, Bilheu A, Herpoel A et al (2013) Pyramidal neurons derived from human pluripotent stem cells integrate efficiently into mouse brain circuits in vivo. Neuron 77:440–456
Feinberg I (1982) Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res 17:319–334
Freedman R, Goldowitz D (2010) Studies on the hippocampal formation: From basic development to clinical applications: Studies on schizophrenia. Prog Neurobiol 90:263–275
Fuccillo M, Joyner AL, Fishell G (2006) Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat Rev Neurosci 7:772–783
Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R (2000) Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development 127:469–482
Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, van den Ameele J, Espuny-Camacho I, Herpoel A, Passante L, Schiffmann SN et al (2008) An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455:351–357
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845
Gore A, Li Z, Fung HL, Young JE, Agarwal S, Antosiewicz-Bourget J, Canto I, Giorgetti A, Israel MA, Kiskinis E et al (2011) Somatic coding mutations in human induced pluripotent stem cells. Nature 471:63–67
Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kundig TM, Frei K, Ginhoux F, Merad M et al (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37:1050–1060
Griesi-Oliveira K, Acab A, Gupta AR, Sunaga DY, Chailangkarn T, Nicol X, Nunez Y, Walker MF, Murdoch JD, Sanders SJ et al (2015) Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol Psychiatry 20:1350–1365
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, Pallesen J, Agerbo E, Andreassen OA, Anney R et al (2019) Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 51:431–444
Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71:574–588
Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V, Fienberg AA (2001) Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A 98:4746–4751
Hashimoto-Torii K, Torii M, Fujimoto M, Nakai A, El Fatimy R, Mezger V, Ju MJ, Ishii S, Chao SH, Brennand KJ et al (2014) Roles of heat shock factor 1 in neuronal response to fetal environmental risks and its relevance to brain disorders. Neuron 82:560–572
Hook V, Brennand KJ, Kim Y, Toneff T, Funkelstein L, Lee KC, Ziegler M, Gage FH (2014) Human iPSC neurons display activity-dependent neurotransmitter secretion: aberrant catecholamine levels in schizophrenia neurons. Stem Cell Rep 3:531–538
Hu BY, Du ZW, Zhang SC (2009) Differentiation of human oligodendrocytes from pluripotent stem cells. Nat Protoc 4:1614–1622
Iefremova V, Manikakis G, Krefft O, Jabali A, Weynans K, Wilkens R, Marsoner F, Brandl B, Muller FJ, Koch P et al (2017) An organoid-based model of cortical development identifies non-cell-autonomous defects in Wnt signalling contributing to Miller-Dieker syndrome. Cell Rep 19:50–59
Ji J, Ng SH, Sharma V, Neculai D, Hussein S, Sam M, Trinh Q, Church GM, McPherson JD, Nagy A et al (2012) Elevated coding mutation rate during the reprogramming of human somatic cells into induced pluripotent stem cells. Stem Cells 30:435–440
Jo J, Xiao Y, Sun AX, Cukuroglu E, Tran HD, Goke J, Tan ZY, Saw TY, Tan CP, Lokman H et al (2016) Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19:248–257
Jones JR, Kong L, Hanna MGT, Hoffman B, Krencik R, Bradley R, Hagemann T, Choi J, Doers M, Dubovis M et al (2018) Mutations in GFAP disrupt the distribution and function of organelles in human astrocytes. Cell Rep 25:947–958
Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, Sasai Y (2013) Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A 110:20284–20289
Kang E, Wang X, Tippner-Hedges R, Ma H, Folmes CD, Gutierrez NM, Lee Y, Van Dyken C, Ahmed R, Li Y et al (2016) Age-related accumulation of somatic mitochondrial DNA mutations in adult-derived human iPSCs. Cell Stem Cell 18:625–636
Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O (2005) Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci 25:4694–4705
Kelsom C, Lu W (2013) Development and specification of GABAergic cortical interneurons. Cell Biosci 3:19
Kepecs A, Fishell G (2014) Interneuron cell types are fit to function. Nature 505:318–326
Kerr CL, Letzen BS, Hill CM, Agrawal G, Thakor NV, Sterneckert JL, Gearhart JD, All AH (2010) Efficient differentiation of human embryonic stem cells into oligodendrocyte progenitors for application in a rat contusion model of spinal cord injury. Int J Neurosci 120:305–313
Khakh BS, Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18:942–952
Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C et al (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci 16:273–280
Kolomeets NS, Orlovskaya DD, Uranova NA (2007) Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse 61:615–621
Krencik R, Weick JP, Liu Y, Zhang ZJ, Zhang SC (2011) Specification of transplantable astroglial subtypes from human pluripotent stem cells. Nat Biotechnol 29:528–534
Krencik R, Zhang SC (2011) Directed differentiation of functional astroglial subtypes from human pluripotent stem cells. Nat Protoc 6:1710–1717
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S et al (2016) Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352:1586–1590
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501:373–379
Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B (2010) The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci 30:16796–16808
Lee SM, Tole S, Grove E, McMahon AP (2000) A local Wnt-3a signal is required for development of the mammalian hippocampus. Development 127:457–467
Li L, Tian E, Chen X, Chao J, Klein J, Qu Q, Sun G, Sun G, Huang Y, Warden CD et al (2018) GFAP mutations in astrocytes impair oligodendrocyte progenitor proliferation and myelination in an hiPSC model of alexander disease. Cell Stem Cell 23(239–251):e236
Li W, Ghose S, Gleason K, Begovic A, Perez J, Bartko J, Russo S, Wagner AD, Selemon L, Tamminga CA (2015) Synaptic proteins in the hippocampus indicative of increased neuronal activity in CA3 in schizophrenia. Am J Psychiatry 172:373–382
Li Y, Muffat J, Omer A, Bosch I, Lancaster MA, Sur M, Gehrke L, Knoblich JA, Jaenisch R (2017) Induction of expansion and folding in human cerebral organoids. Cell Stem Cell 20(385–396):e383
Liu A, Niswander LA (2005) Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci 6:945–954
Liu Y, Liu H, Sauvey C, Yao L, Zarnowska ED, Zhang SC (2013) Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat Protoc 8:1670–1679
Maden M (2007) Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat Rev Neurosci 8:755–765
Madhavan M, Nevin ZS, Shick HE, Garrison E, Clarkson-Paredes C, Karl M, Clayton BLL, Factor DC, Allan KC, Barbar L et al (2018) Induction of myelinating oligodendrocytes in human cortical spheroids. Nat Methods 15:700–706
Madison JM, Zhou F, Nigam A, Hussain A, Barker DD, Nehme R, van der Ven K, Hsu J, Wolf P, Fleishman M et al (2015) Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry 20:703–717
Mansour AA, Goncalves JT, Bloyd CW, Li H, Fernandes S, Quang D, Johnston S, Parylak SL, Jin X, Gage FH (2018) An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 36:432–441
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, Beltrao-Braga P, Trujillo CA, Mendes APD, Padmanabhan K et al (2017) Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 22:820–835
Marchetto MC, Carromeu C, Acab A, Yu D, Yeo GW, Mu Y, Chen G, Gage FH, Muotri AR (2010) A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143:527–539
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, Amenduni M, Szekely A, Palejev D, Wilson M et al (2015) FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162:375–390
Maroof AM, Keros S, Tyson JA, Ying SW, Ganat YM, Merkle FT, Liu B, Goulburn A, Stanley EG, Elefanty AG et al (2013) Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 12:559–572
Mason I (2007) Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat Rev Neurosci 8:583–596
Mayer C, Hafemeister C, Bandler RC, Machold R, Batista Brito R, Jaglin X, Allaway K, Butler A, Fishell G, Satija R (2018) Developmental diversification of cortical inhibitory interneurons. Nature 555:457–462
Mertens J, Marchetto MC, Bardy C, Gage FH (2016) Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci 17:424–437
Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, Zheng Y, Diffenderfer KE, Zhang J, Soltani S et al (2015) Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527:95–99
Miller FD, Gauthier AS (2007) Timing is everything: making neurons versus glia in the developing cortex. Neuron 54:357–369
Miyoshi G, Hjerling-Leffler J, Karayannis T, Sousa VH, Butt SJ, Battiste J, Johnson JE, Machold RP, Fishell G (2010) Genetic fate mapping reveals that the caudal ganglionic eminence produces a large and diverse population of superficial cortical interneurons. J Neurosci 30:1582–1594
Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, Anderson DJ (2000) Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell 101:499–510
Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM et al (2016) Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 22:1358–1367
Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y (2015) Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep 10:537–550
Munoz-Sanjuan I, Brivanlou AH (2002) Neural induction, the default model and embryonic stem cells. Nat Rev Neurosci 3:271–280
Murai K, Sun G, Ye P, Tian E, Yang S, Cui Q, Sun G, Trinh D, Sun O, Hong T et al (2016) The TLX-miR-219 cascade regulates neural stem cell proliferation in neurodevelopment and schizophrenia iPSC model. Nat Commun 7:10965
Nave KA, Ehrenreich H (2014) Myelination and oligodendrocyte functions in psychiatric diseases. JAMA Psychiatry 71:582–584
Nestler EJ, Hyman SE (2010) Animal models of neuropsychiatric disorders. Nat Neurosci 13:1161–1169
Nicholas CR, Chen J, Tang Y, Southwell DG, Chalmers N, Vogt D, Arnold CM, Chen YJ, Stanley EG, Elefanty AG et al (2013) Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell 12:573–586
Nistor GI, Totoiu MO, Haque N, Carpenter MK, Keirstead HS (2005) Human embryonic stem cells differentiate into oligodendrocytes in high purity and myelinate after spinal cord transplantation. Glia 49:385–396
Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D et al (2017) Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci 20:753–759
Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD et al (2015) Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12:671–678
Pasca SP (2018) The rise of three-dimensional human brain cultures. Nature 553:437–445
Pasca SP (2019) Assembling human brain organoids. Science 363:126–127
Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S et al (2011) Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 17:1657–1662
Piao J, Major T, Auyeung G, Policarpio E, Menon J, Droms L, Gutin P, Uryu K, Tchieu J, Soulet D et al (2015) Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell 16:198–210
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C et al (2016) Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165:1238–1254
Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP et al (2017) Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545:48–53
Rasetti R, Mattay VS, White MG, Sambataro F, Podell JE, Zoltick B, Chen Q, Berman KF, Callicott JH, Weinberger DR (2014) Altered hippocampal-parahippocampal function during stimulus encoding: a potential indicator of genetic liability for schizophrenia. JAMA Psychiatry 71:236–247
Ricciardi S, Ungaro F, Hambrock M, Rademacher N, Stefanelli G, Brambilla D, Sessa A, Magagnotti C, Bachi A, Giarda E et al (2012) CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat Cell Biol 14:911–923
Rodrigues GMC, Gaj T, Adil MM, Wahba J, Rao AT, Lorbeer FK, Kulkarni RU, Diogo MM, Cabral JMS, Miller EW et al (2017) Defined and scalable differentiation of human oligodendrocyte precursors from pluripotent stem cells in a 3D culture system. Stem Cell Rep 8:1770–1783
Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, Takahashi J, Eiraku M, Sasai Y (2015) Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat Commun 6:8896
Santos R, Vadodaria KC, Jaeger BN, Mei A, Lefcochilos-Fogelquist S, Mendes APD, Erikson G, Shokhirev M, Randolph-Moore L, Fredlender C et al (2017) Differentiation of inflammation-responsive astrocytes from glial progenitors generated from human induced pluripotent stem cells. Stem Cell Rep 8:1757–1769
Sarkar A, Mei A, Paquola ACM, Stern S, Bardy C, Klug JR, Kim S, Neshat N, Kim HJ, Ku M et al (2018) Efficient generation of CA3 neurons from human pluripotent stem cells enables modeling of hippocampal connectivity in vitro. Cell Stem Cell 22(684–697):e689
Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, Kuret TJM, Liyanage M, Mansour AA, Jaeger BN et al (2019) Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci 22:243–255
Schizophrenia Working Group of the Psychiatric Genomics, C (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427
Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW et al (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, Fu T, Worringer K, Brown HE, Wang J et al (2019) Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci 22:374–385
Shao Z, Noh H, Bin Kim W, Ni P, Nguyen C, Cote SE, Noyes E, Zhao J, Parsons T, Park JM et al (2019) Dysregulated protocadherin-pathway activity as an intrinsic defect in induced pluripotent stem cell-derived cortical interneurons from subjects with schizophrenia. Nat Neurosci 22:229–242
Sharp J, Frame J, Siegenthaler M, Nistor G, Keirstead HS (2010) Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants improve recovery after cervical spinal cord injury. Stem Cells 28:152–163
Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, Krawisz A, Froehlich W, Bernstein JA, Hallmayer JF et al (2013) SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 503:267–271
Shi Y, Inoue H, Wu JC, Yamanaka S (2017) Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov 16:115–130
Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ (2012) Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 15(477–486):S471
Sloan SA, Darmanis S, Huber N, Khan TA, Birey F, Caneda C, Reimer R, Quake SR, Barres BA, Pasca SP (2017) Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron 95(779–790):e776
Stern S, Santos R, Marchetto MC, Mendes APD, Rouleau GA, Biesmans S, Wang QW, Yao J, Charnay P, Bang AG et al (2018) Neurons derived from patients with bipolar disorder divide into intrinsically different sub-populations of neurons, predicting the patients’ responsiveness to lithium. Mol Psychiatry 23:1453–1465
Sur M, Rubenstein JL (2005) Patterning and plasticity of the cerebral cortex. Science 310:805–810
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872
Tamminga CA, Stan AD, Wagner AD (2010) The hippocampal formation in schizophrenia. Am J Psychiatry 167:1178–1193
Tao Y, Zhang SC (2016) Neural subtype specification from human pluripotent stem cells. Cell Stem Cell 19:573–586
Tekin S, Cummings JL (2002) Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res 53:647–654
Tkachev D, Mimmack ML, Ryan MM, Wayland M, Freeman T, Jones PB, Starkey M, Webster MJ, Yolken RH, Bahn S (2003) Oligodendrocyte dysfunction in schizophrenia and bipolar disorder. Lancet 362:798–805
Vadodaria KC, Ji Y, Skime M, Paquola A, Nelson T, Hall-Flavin D, Fredlender C, Heard KJ, Deng Y, Le AT et al (2019) Serotonin-induced hyperactivity in SSRI-resistant major depressive disorder patient-derived neurons. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0363-y
Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474:380–384
Wang L, Xia J, Li J, Hagemann TL, Jones JR, Fraenkel E, Weitz DA, Zhang SC, Messing A, Feany MB (2018) Tissue and cellular rigidity and mechanosensitive signaling activation in Alexander disease. Nat Commun 9:1899
Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M et al (2013) Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell 12:252–264
Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13:753–760
Watanabe K, Kamiya D, Nishiyama A, Katayama T, Nozaki S, Kawasaki H, Watanabe Y, Mizuseki K, Sasai Y (2005) Directed differentiation of telencephalic precursors from embryonic stem cells. Nat Neurosci 8:288–296
Wen Z, Nguyen HN, Guo Z, Lalli MA, Wang X, Su Y, Kim NS, Yoon KJ, Shin J, Zhang C et al (2014) Synaptic dysregulation in a human iPS cell model of mental disorders. Nature 515:414–418
Wilson SW, Houart C (2004) Early steps in the development of the forebrain. Dev Cell 6:167–181
Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, Taubenschmid J, Hammerle M, Esk C, Bagley JA et al (2019) Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565:505–510
Windrem MS, Osipovitch M, Liu Z, Bates J, Chandler-Militello D, Zou L, Munir J, Schanz S, McCoy K, Miller RH et al (2017) Human iPSC glial mouse chimeras reveal glial contributions to schizophrenia. Cell Stem Cell 21(195–208):e196
Wonders CP, Anderson SA (2006) The origin and specification of cortical interneurons. Nat Rev Neurosci 7:687–696
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF et al (2018) Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 50:668–681
Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, Cakir B, Kim KY, Lombroso AP, Hwang SM et al (2017) Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 21(383–398):e387
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC et al (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211:1533–1549
Yang N, Zuchero JB, Ahlenius H, Marro S, Ng YH, Vierbuchen T, Hawkins JS, Geissler R, Barres BA, Wernig M (2013) Generation of oligodendroglial cells by direct lineage conversion. Nat Biotechnol 31:434–439
Yoon KJ, Nguyen HN, Ursini G, Zhang F, Kim NS, Wen Z, Makri G, Nauen D, Shin JH, Park Y et al (2014) Modeling a genetic risk for schizophrenia in iPSCs and mice reveals neural stem cell deficits associated with adherens junctions and polarity. Cell Stem Cell 15:79–91
Yu DX, Di Giorgio FP, Yao J, Marchetto MC, Brennand K, Wright R, Mei A, McHenry L, Lisuk D, Grasmick JM et al (2014) Modeling hippocampal neurogenesis using human pluripotent stem cells. Stem Cell Reports 2:295–310
Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920
Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C et al (2015) Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347:1138–1142
Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, Fair T, Yang N, Sudhof TC, Wernig M et al (2018) The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med 10:eaar4338
Zhang ZN, Freitas BC, Qian H, Lux J, Acab A, Trujillo CA, Herai RH, Nguyen Huu VA, Wen JH, Joshi-Barr S et al (2016) Layered hydrogels accelerate iPSC-derived neuronal maturation and reveal migration defects caused by MeCP2 dysfunction. Proc Natl Acad Sci U S A 113:3185–3190
Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, Zheng D, Lachman HM (2015a) MicroRNA profiling of neurons generated using induced pluripotent stem cells derived from patients with schizophrenia and schizoaffective disorder, and 22q11.2 Del. PLoS ONE 10:e0132387
Zhao T, Zhang ZN, Rong Z, Xu Y (2011) Immunogenicity of induced pluripotent stem cells. Nature 474:212–215
Zhao T, Zhang ZN, Westenskow PD, Todorova D, Hu Z, Lin T, Rong Z, Kim J, He J, Wang M et al (2015b) Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 17:353–359
ACKNOWLEDGMENTS
We thank M.L. Gage for editorial comments. M.W. was supported by a training grant from the California Institute for Regenerative Medicine. This study was supported by the Paul G. Allen Family Foundation, the National Institutes of Health U01# MH106882, the JPB Foundation, the Leona M. and Harry B. Helmsley Charitable Trust (#2017-PG-MED001), Annette Merle-Smith and the Lookout Foundation, and the G. Harold & Leila Y. Mathers Charitable Foundation to F.H.G.
ABBREVIATIONS
2D, two-dimensional; 3D, three-dimensional; A-P, anterior-posterior; ASD, autism spectrum disorder; AxD, Alexander disease; BMP, bone morphogenetic protein; CA, cornu ammonis; CGE, caudal ganglionic eminence; CNS, central nervous system; DG, dentate gyrus; DISC1, disrupted-in-schizophrenia 1; DKK, Dickkopf-related protein 1; D-V, dorsal-ventral; EB, embryoid bodies; EMP, erythromyeloid progenitors; hPSCs, human pluripotent stem cell; iPSCs, induced pluripotent stem cells; FGF, fibroblast growth factor; GABAergic, gamma-aminobutyric acid-ergic; GFAP, glial fibrillary acidic protein; hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell; MDS, Miller-Dieker syndrome; MGE, medial ganglionic eminence; NPC, neural progenitor cells; OPCs, oligodendrocyte progenitor cells; oRG, outer radial glia; oSVZ, outer subventricular zone; PROX1, prospero homeobox protein 1; PSD-95, postsynaptic density protein 95; PV, parvalbumin; RA, retinoic acid; S100β, S100 calcium-binding protein; SHH, sonic hedgehog; SVZ, subventricular zone; SZ, schizophrenia; TGF-β, transforming growth factor-β
Compliance with Ethics Guidelines
Meiyan Wang, Lei Zhang, and Fred H. Gage declare that they have no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, M., Zhang, L. & Gage, F.H. Modeling neuropsychiatric disorders using human induced pluripotent stem cells. Protein Cell 11, 45–59 (2020). https://doi.org/10.1007/s13238-019-0638-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-019-0638-8