
Abstract
T-cell receptor (TCR)-engineered T cells are a novel option for adoptive cell therapy used for the treatment of several advanced forms of cancer. Work using TCR-engineered T cells began more than two decades ago, with numerous preclinical studies showing that such cells could mediate tumor lysis and eradication. The success of these trials provided the foundation for clinical trials, including recent clinical successes using TCR-engineered T cells to target New York esophageal squamous cell carcinoma (NY-ESO-1). These successes demonstrate the potential of this approach to treat cancer. In this review, we provide a perspective on the current and future applications of TCR-engineered T cells for the treatment of cancer. Our summary focuses on TCR activation and both pre-clinical and clinical applications of TCR-engineered T cells. We also discuss how to enhance the function of TCR-engineered T cells and prolong their longevity in the tumor microenvironment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
It was shown as early as 1976 that interleukin-2 (IL-2), regarded as a T-cell growth factor, induced T-cell proliferation without loss of effector function in vitro (Morgan et al., 1976). It is now known that the cytokine IL-2 is crucial for sustained clonal expansion of responding T cells. A study by Rosenberg et al. demonstrated that syngeneic tumor-infiltrating lymphocytes (TILs) could undergo expansion in the presence of IL-2. Adoptive transfer of these TILs to murine models was shown to lead to regression in lung and liver tumors (Rosenberg et al., 1986). Subsequently, adoptive cell therapy using autologous TILs has become one of the most effective approaches to induce long-lasting regression in patients with metastatic melanoma (Rosenberg et al., 1988; Dudley et al., 2002; Scanlan et al., 2002; Rosenberg et al., 2011; Pilon-Thomas et al., 2012; Radvanyi et al., 2012; Besser et al., 2013). The presence of TILs has also been associated with improved prognosis in other cancer types, including ovarian, colon, and breast cancer (Clemente et al., 1996; Sato et al., 2005; Galon et al., 2006; Loi, 2013).
Early studies found that TILs isolated from melanoma patients recognized two non-mutated melanoma melanocyte differentiation proteins: MART-1 and gp100 (Kawakami et al., 1994a; Kawakami et al., 1994b). MART-1 and gp100 proteins are often expressed by melanocytes in the skin, eye, and ear. However, many patients who present complete cancer regression did not have a toxic response after treatment with TILs targeting MART-1 or gp100. This demonstrated that it is the antigen-specific T cells in TILs that are crucial for cancer regression. There are however several hurdles to purifying the amount of antigen-specific T cells necessary to be used as a therapy: (1) it is difficult to isolate tumor-specific T cells from many cancer patients; (2) it takes considerable time to obtain a therapeutic amount of tumor-specific T cells. With the introduction of T-cell receptor (TCR) engineering technologies, it became possible to produce antigen-specific T cells. Treatment with engineering, tumor antigen-specific T cells has demonstrated significant clinical successes in patients with metastatic melanoma, colorectal carcinoma, synovial sarcoma, and multiple myeloma (Morgan et al., 2006; Johnson et al., 2009; Parkhurst et al., 2011; Robbins et al., 2011; Rapoport et al., 2015; Robbins et al., 2015) (Fig. 1). Tumor antigen-specific TCR gene-engineered T cells are therefore considered as a potentially “off-the-shelf” treatment for cancer patients.

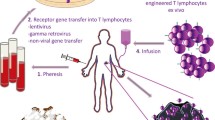
Process of TCR-engineered T cells therapy. T cells are isolated from patient blood or tumor tissue. TCR α and β chains are then isolated from single T-cell clones and inserted into a lentivirus or retrovirus vector. T cells isolated from the peripheral blood of the patient can be modified with the lentivirus or retrovirus vector to encode the desired TCRαβ sequences. These modified T cells are then expanded in vitro to obtain sufficient numbers for treatment and re-infusion back into the patient
THE FOUNDATION OF TCR ACTIVATION
TCR recognition of pMHC molecules
TCR is expressed on the surface of T cells and consists of two distinct protein chains. In the majority of mature T cells, the TCR consists of α and β chains, although there is a smaller population of T cells in which the TCR consists of γ and δ chains. Antigen recognition by the αβTCR is central to the function of the adaptive immune system. αβTCR bind to the peptide major histocompatibility complex (pMHC) on the surface of antigen-presenting cells. The interaction between an αβTCR and a pMHC is highly specific owing to the fact that T cells are able to distinguish between rare foreign pMHCs and the abundant self pMHC molecules (Germain and Stefanova, 1999). CD8+ T cells play an important role in the adaptive immune response in cancer patients and are activated by TCR recognition of specific peptide epitopes. These peptide epitopes are largely generated from endogenous proteins that are presented by MHC class I proteins on the surface of tumor cells (Phan and Rosenberg, 2013). MHC class I proteins are membrane proteins that are expressed on almost all nucleated cells. They are encoded by several families of human leukocyte antigen (HLA-A, B, and C) genes (Brown et al., 2014). Expression of HLA genes can be upregulated by interferon (IFN) signaling, but the expression is often notably down-regulated in tumors. The degree of down-regulation correlates with immune evasion and disease progression in patients with cancer (Agrawal and Kishore, 2000; Leone et al., 2013). T cell activation requires translation of pMHC antigen binding to the TCR and then on to intracellular signaling pathways (Zhang and Bevan, 2011; Obst, 2015; Pageon et al., 2016). Ex vivo and in vivo studies demonstrate that the dose of antigen presented determines the nature of cytokine expression in T cells (Corse et al., 2011; Tkach et al., 2014).
TCR signaling transduction
Naïve T cells undergo clonal expansion of between 10 and 20 rounds of cell division after activation by TCR/pMHC interaction (Zhang and Bevan, 2011; Obst, 2015). Compared with naïve T cells, antigen-stimulated T cells substantially increase antigen responsiveness via a process termed “functional avidity maturation” (Margulies, 2001; Slifka and Whitton, 2001). Studies have found that antigen-stimulated T cells exhibit greater proliferation and cytokine production than naïve T cells (Akbar et al., 1988; Byrne et al., 1988; Sanders et al., 1989; Sallusto et al., 1999). T cells recognize antigen-MHC complexes through the TCR-CD3 cluster. After interaction between the MHC and TCR, several classes of protein are then recruited to the plasma membrane by activated receptors to participate in signal propagation (Fig. 2). Phospholipase C-γ1 (PLC-γ1) cleaves molecules of membrane phospholipid phosphatidylinositol bisphosphate (PIP2), into inositol triphosphate (IP3), and diacylglycerol (DAG). The interaction of IP3 with its receptors in the endoplasmic reticulum upregulates the level of Ca2+ in the cytosol, activating the Ca2+-binding protein calmodulin. This subsequently regulates nuclear factor of activated T cells (NFAT) proteins. Additionally, DAG activates the Ras/extracellular regulating kinase (Erk) pathway, modulating the nuclear factor Fos. Through all of these interacting signaling pathways, T cells are activated, releasing numerous cytokines and chemokines, including IFN-γ, Granzyme B, and IL-2 (Abraham and Weiss, 2004; Smith-Garvin et al., 2009).

Schematic demonstration of TCR signaling. T cells recognize pMHC complexes through the TCR-CD3 cluster. Several classes of proteins are then recruited to the plasma membrane by the activated receptors and participate in signal propagation. Phospholipase C-γ1 (PLC-γ1) cleaves molecules of the membrane phospholipid phosphatidylinositol bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). The interaction of IP3 with its receptors in the endoplasmic reticulum up-regulates the level of Ca2+ in the cytosol, further activating the Ca2+-binding protein calmodulin. NFAT, as a nuclear factor of activated T cells, is regulated by the calcium pathway. DAG is primarily involved in the activation of the Ras/Erk pathway. T cells are activated though these signaling pathways, releasing IFN-γ, Granzyme B, IL-2, and so on
The function of T lymphocytes is largely regulated by TCR signaling. Studies have shown that initial TCR signaling via p38 leads to successive induction of Vitamin D receptor (VDR) and PLC-γ1, both of which are required for classical TCR signaling and T cell activation (von Essen et al., 2010). Genetically blocking TCR internalization inhibits T cell expansion, demonstrating that TCR signaling is required for T cell proliferation. TCR internalization was also required for sustained signaling and activation of key metabolic pathways, including the mechanistic target of rapamycin (mTOR) (Willinger et al., 2015). Acting to control T cell activation, T cell anergy is a tolerance mechanism in which lymphocytes are intrinsically functionally-inactivated following antigen encounter. This phenomenon is often observed in the tumor microenvironment. Zheng et al. found that early growth response protein 2 (Egr2) was necessary for in vivo anergy induction when using antigen-induced and tumor-induced anergy models. Egr2 is therefore considered an essential transcriptional regulator of T cell anergy (Zheng et al., 2012). The activation status of T cells and the level of immune response are further controlled by various co-stimulatory (CD28, inducible T cell co-stimulator (ICOS), and OX40) and co-inhibitory (cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), programmed death 1 (PD-1)) molecules (Keir et al., 2008; Chen and Flies, 2013). For example, in the absence of the immune checkpoint protein PD-1, the fusion antibody B7H1-Ig can augment T cell proliferation and immune response. This demonstrates that it is possible to intervene and augment or down-regulate immune response based on B7H1-mediated pathways (Deng et al., 2014).
APPLICATION OF TCR-ENGINEERED T LYMPHOCYTES
A great deal of scientific studies have shown that TCR-engineered T cells can target and kill cancer cells expressing appropriate antigens (Parkhurst et al., 2011; Cohen et al., 2015; Kageyama et al., 2015; Rapoport et al., 2015; Stronen et al., 2016). However, for treatment to be feasible, it is necessary to first enrich TCR-gene modified antigen specific T cells in vitro. Adoptive cell therapy with these engineered cells would be a precise therapy as it targets antigens expressed on cancer cells present in the patient. We will next examine the application of this therapy for the treatment of cancer.
Selection of an appropriate antigen
Cancer cells can express proteins during development that are different from those found in untransformed cells. Certain antigens that are more frequently expressed by similar tumors are appealing candidates for therapies utilizing immune recognition. These antigens can be unique or shared. Shared antigens are divided into tumor differentiation antigens, over-expressed antigens, and shared tumor-specific antigens (http://cancerimmunity.org/peptide/). Many shared tumor-specific antigens, encoded by cancer-germline genes, have been identified. These antigens can induce an immune responses and are promising candidate targets for use in vaccination or T cell therapy, such as melanoma-associated antigen (MAGE)-A3 (Zhang et al., 2003), MAGE-A4 (Zhang et al., 2002), and New York esophageal squamous cell carcinoma (NY-ESO)-1 (van der Bruggen et al., 1994; Valmori et al., 2000; Van Der Bruggen et al., 2002). Many studies have also reported that tumor differentiation antigens and overexpressed antigens can evoke T cell responses, including MART-1, gp100, carcino-embryonicantigen (CEA), and p53 (Kawakami et al., 1995; Kawashima et al., 1998; Barfoed et al., 2000). In addition to shared antigens, unique antigens also have potential to be used as a targeted treatment. Unique antigens are abnormal proteins that are only expressed by tumor cells. Viral associated antigens found in some cancers can be used to produce antigen-specific T cells, human papilloma virus for example (Draper et al., 2015).
In the past 3 years, neoantigen has garnered much attention as a potential precision immunotherapy. Neoantigens are generated from somatic point mutations in tumor tissues that are absent in normal tissue. Whole genome or exome sequencing can be applied to identify optimal neoantigen candidates for personalized cancer treatment. RNA sequencing can be performed to examine expression and predict whether a neoepitope will be presented by the MHC to be recognized by T cells (Robbins et al., 2013; van Rooij et al., 2013; Brown et al., 2014). In 2016, a study found that mutation-reactive T cells could be enriched from donor-derived T cells and used as an effective therapy for the treatment of patients with metastatic cancer (Prickett et al., 2016; Stronen et al., 2016). CD4+ and CD8+ T lymphocytes have been shown to target epitopes arising from epigenetic, transcriptional, translational, and post-translational alterations of tumor cells (Coulie et al., 2014). More recently, technological breakthroughs have shown that numerous endogenous mutant cancer proteins are unique to tumor cells. These can be processed into peptides and presented on the surface of tumor cells leading to these cells being recognized in vivo as “non-self” or foreign by the immune system. Targeting highly specific neoantigens would enable immune cells to distinguish cancer cells from normal cells and avoid the risk of autoimmunity (Bobisse et al., 2016). Neoantigens therefore represent ideal targets for successful immunotherapy.
Recent exciting results have demonstrated that TILs responding to patient neoantigens can be detected at much higher frequencies than other types (Robbins et al., 2013; van Rooij et al., 2013; Linnemann et al., 2015). Several studies have also found that monoclonal antibodies directed against CTLA-4 are particularly effective at treating cancers with a high burden of somatic mutation (Snyder et al., 2014; Van Allen et al., 2015). In lung and bladder cancer patients treated with pembrolizumab, an antibody targeting PD-1, the non-synonymous mutation burden strongly associates with clinical efficacy (Powles et al., 2014; Rizvi et al., 2015). Isolation and reinfusion of neoantigen-specific T cells may be required to mediate tumor regression without inducing on-target but off-tumor toxicities (Klebanoff et al., 2016). The best currently available technology to obtain large amounts of neoantigen specific T cells is to insert TCR sequences targeting identified neoantigens into T cells in vitro. For example, transgenic CD4+ lymphocytes that recognize a mutant tumor-specific neoantigen ERBB2 protein induced sustained tumor regression in a patient with cholangiocarcinoma (Tran et al., 2014). These findings indicated that it may be feasible to develop treatments based on the adoptive transfer of TCR-engineered T cells sorted with tetramers bearing mutated epitopes that recognize autologous peripheral T cells (Cohen et al., 2015; Gros et al., 2016). Further interesting results revealed that CD8+PD-1+ cell populations from PBMCs and TILs had lymphocytes targeting patient specific neoantigens (Gros et al., 2016; Pasetto et al., 2016). However, another study found that the lack of a defined neoantigen resulted in tumor cell resistance in a transplantable tumor model (Matsushita et al., 2012). Whether the neoantigen repertoire in human tumors is stable and therefore consistently targetable is currently unclear. Verdegaal et al. designed a study to observe the landscape of neoantigen dynamics and reveal any detectable stability. Their data demonstrated that specific T cell-recognized neoantigens could be lost by either reduced transcript expression or complete loss of the mutant allele (Verdegaal et al., 2016). Cancer immunotherapy with neoantigen specific T cells should therefore aim to exploit the adaptive capacity of the immune system. Based on these promising results, it may be possible that, in the near future, neoantigen specific T cells could be used as a novel strategy to develop personalized therapies to treat cancer.
Candidate target antigens that are used for TCR-engineered T cell treatment require three features if they are to be utilized: (1) they must be selectively expressed in tumors and not in normal tissues (tumor specificity); (2) they are related to oncogenesis (tumor addiction); (3) they are able to evoke a T-cell response (immunogenicity) (Debets et al., 2016). Broadly speaking, choosing an appropriate antigen is the first and most important step to determine the effectiveness of TCR-engineered T cells.
Identification of TCR sequences
Identifying TCR sequences is inherently difficult because each T cell contains its own unique TCR that recognizes a distinct set of pMHC molecules. Several methods have been developed to identify TCR sequences from single T cells (Fig. 3). Culturing of T cell clones is the canonical method to identify TCR sequence. Briefly, CD8+ or CD4+ T cells are purified from peripheral blood mononuclear cells (PBMCs). Serial dilutions allow single T cells being seeded into individual wells of 96-well plates. Finally, these single cells are propagated, generating T-cell clones. The TCR α chain and β chain from these T cell clones are identified and sequenced (Nishimura et al., 1994; Zhang et al., 2010). Using this method, several antigen-specific T cells have been identified that effectively recognize relevant antigens, including tumor antigens. The efficacies of these tumor-specific TCRs to treat cancer have been tested in clinical trials using engineered TCRs (Morgan et al., 2006; Robbins et al., 2015). NY-ESO-1 specific TCR-T cells are the most thoroughly examined and their therapeutic potential has been tested in synovial cell sarcoma, melanoma, and myeloma (Robbins et al., 2011; Rapoport et al., 2015; Robbins et al., 2015).

Three typical procedures to obtain TCR sequence. Antigen responsive T cells are isolated, specific TCR genes are identified by single clone derived-cDNA sequencing (I), single cell sequencing (II) or paired sequencing of bulk DNA (III)
Recently, single cell RT-PCR and pairSEQ methods have been developed to more rapidly identify TCR sequences. Single cell RT-PCR allows the transcriptomes of thousands of cells to be processed simultaneously (Wu et al., 2014; Redmond et al., 2016). This method can identify the unique TCR and the paired α and β heterodimer of each T cell and has been successfully used to identify paired α and β chains from 91 naïve CD4+ T helper cells in mice (Mahata et al., 2014). PairSEQ technology can leverage the diversity of TCR sequences to accurately identify many TCR α and β chain sequences in a single high throughput experiment. Howie et al. used this technology to pair hundreds of thousands of TCR α and β chain sequences from PBMCs isolated from two healthy donors, as well as thousands of sequences from TILs in nine pairs of matched tumor and blood samples (Howie et al., 2015). Pasetto et al. applied PairSEQ technology to identify TCR sequences of T cells derived from 12 fresh metastatic melanomas TIL. This study successfully identified several sequences that showed reactivity against tumor antigens (Pasetto et al., 2016). In summary, PairSEQ is an exciting high throughout technology that can be used to identify the TCR sequences of TILs. This information can then be used to engineer T cells that express antigen specific TCRs. These new technologies have great potential to boost development of TCR-engineered T cell therapy.
Preclinical studies
Following the identification of tumor antigen-targeting TCRs and their introduction into T cells, it is necessary to perform functional assessment to analyze the sensitivity of T cell responses towards the cognate peptides or autologous tumor cells (Dembic et al., 1986; Kessels et al., 2001). Targeting shared tissue differentiation antigens, such as MART-1, gp100 and CEA, will likely come with the price of toxicity to normal cells in critical organs. It is therefore also necessary to evaluate off-tumor toxicities of TCR-engineered T cells. Although some antigens are not widely expressed, such as the cancer-testis NY-ESO-1 and MAGE families that are expressed on tumor tissue, fetal tissue, and adult testes but not on other normal adult tissues, the safety and affinity of these TCR-engineered T cells should still be assessed. Parkhurst et al. developed a mouse model to isolate CEA-reactive TCRs from splenocytes and perform functional assessment. In this study, they proved that the modified CEA-reactive TCRs were good candidates for future gene therapy and also showed the power of selected amino acid substitutions in the antigen-binding regions of TCR to enhance TCR reactive affinity (Parkhurst et al., 2009). Kunert et al. isolated 10 TCR sequences against four MAGE-C2 epitopes from melanoma patients and designed a set of experiments to evaluate TCR-transgenic T cell function (Kunert et al., 2016). Two MAGE-A3 specific TCRs were isolated from PBMCs of two melanoma patients after MAGE-A3 vaccination. These TCRs recognized MAGE-A3 peptides presented by HLA-DPB1*04:01. The specificity and affinity of these two TCRs were compared and it was found that 6F9 TCR specifically recognized MAGE-A3, but not other members of the MAGE-A family in the context of HLA-DPB1*04:01. The 6F9 TCR was selected for potential TCR gene therapy targeting MHC class II-restricted MAGE-A3 (Yao et al., 2016). An additional issue is that many modified T cells circulating in patients do not have any therapeutic effect because they possess decreased retroviral transgene expression (Kohn et al., 1998). Some studies have reported possible methods to improve TCR gene transfer and to provide a stable system for immunotherapy. Fujio et al. used two independent monocistronic retrovirus vectors to generate ovalbumin (OVA)-specific TCR-T cells. These cells showed a remarkable response to antigen (Fujio et al., 2000). Additionally, a lentiviral vector carrying a bidirectional promoter was used in the Bobisse et al. study. This gene delivery system demonstrated increased transfer efficiency, suggesting lentiviral vectors may be a valid tool for TCR expression in immunotherapy (Bobisse et al., 2009). These preclinical experiments can help guide the application of TCR-T cells in clinical trials but it is essential that new TCRs targeting tumor antigens are tested for their affinity, toxicity, and safety.
Clinical trials
Adoptive immunotherapy using TCR-engineered T cells has become an important strategy for cancer therapy (Rosenberg and Restifo, 2015) and recent clinical trials have provided encouraging results (Table 1). It was first reported that MART-1 TCR modified lymphocytes could mediate tumor regression in humans in 2006 (Morgan et al., 2006). Clinical trials of MART-1 TCR-engineered T cells in 2009 and 2014 also demonstrated this phenomenon (Johnson et al., 2009; Chodon et al., 2014). Johnson et al. showed that 19% patients treated with gp100 TCR-engineered T cells experience an objective antitumor response (Johnson et al., 2009). In addition to differentiation antigens, clinical trials have also examined cancer-testis antigens, such as MAGE-A3 and NY-ESO-1. In clinical trials using a TCR targeting HLA-A*0201-restricted NY-ESO-1 antigen, objective responses were observed in more than 50% of patients with synovial cell sarcoma, melanoma, and myeloma (Robbins et al., 2011; Rapoport et al., 2015; Robbins et al., 2015). Kageyama et al. conducted a clinical trial examining TCR-modified T cells with a HLA-A*2402-restricted MAGE-A4 in the treatment of esophageal cancer. These TCR-modified T cells could be detected in vivo for a prolonged period of time and three patients present minimal tumor lesions for more than 27 months (Kageyama et al., 2015). These clinical trials demonstrate that there can be dramatic tumor regression using TCR-engineered T cells therapy. This has elicited considerable enthusiasm, although it must be noted that most of these clinical trials used only a small number of cancer patients. Additionally, although there has been great progress in adoptive cell therapy with TCR-engineered T cells, some unexpected toxicities have occurred. In a clinic trial using TCR-engineered T cells targeting metastatic colorectal cancer and a high avidity CEA-reactive TCR, all three patients developed severe transient inflammatory colitis due to the TCR reacting to CEA-expressing normal colon epithelium cells (Although one patient had an objective regression of cancer metastatic to the lung and liver) (Parkhurst et al., 2011). In another study, two patients died of cardiogenic shock after infusion with T cells engineered with a TCR against HLA-A*01-restricted MAGE-A3. The artificially modified MAGE-A3 TCR had 4 substitutions in the alpha chain of the CDR2 region and retained the wild type sequences in the beta chain to increase the TCR affinity. This affinity-enhanced TCR may have recognized an epitope derived from an unrelated protein expressed by normal cardiac tissue, but the parental MAGE-A3-specific TCR may also have expanded in the patient without cardiac toxicity through natural thymic selection processes (Linette et al., 2013). A further study also resulted in two patients lapsing into comas and subsequently dying after treatment with autologous MAGE-A3 TCR engineered-T cells. In this study, the modified T cells also recognized an MAGE-A12-derived epitope that was detected in human brain (Morgan et al., 2013). Potential cross-reactivity makes it essential to carefully evaluate the affinity of TCRs and select the appropriate antigens for safe clinical application of TCR-engineered T cells.
IMPROVING THE FUNCTION OF TCR-ENGINEERED T CELLS IN TUMOR MICROENVIRONMENT
Improving the function of TCR-engineered T cells is critical to overcome inhibitory factors within the tumor microenvironment and elicit tumor regression. Many efforts to enhance antigen reactivity and circumvent T cell tolerance have focused on increasing TCR signal strength and generating highly functional T cells. Immune checkpoint proteins, such as PD-1 and CTLA-4, can prevent the activation of T cells in immune system. Blocking the PD-1 pathway has been shown to improve the function of TILs and enhance antitumor immunity (Herbst et al., 2014; Tumeh et al., 2014). It is therefore likely that the PD-1/PD-L1 signaling pathway is a major negative feedback regulator of antigen responsiveness (Okazaki et al., 2013; Honda et al., 2014). Other evidence has implicated PD-1 signaling in modulating the phosphoinositide3-kinase (PI3K), AKT and RAS pathways and cell cycle control (Parry et al., 2005; Patsoukis et al., 2012a; Patsoukis et al., 2012b). TCR-engineered T cells expressing a high level of the inhibitory receptor PD-1 reduced their functional activity (Perez et al., 2015). The efficacy of NY-ESO-1 TCR-engineered T cells was augmented when used in combination with anti-PD-1 antibody (Moon et al., 2016). In addition, cancer cells can influence the local microenvironment by releasing extracellular signals, promoting tumor angiogenesis, and inducing peripheral immune tolerance. Conversely, immune cells in the microenvironment can affect the growth and evolution of cancer cells. Several recombinant cytokines are routinely used in the treatment of cancer, especially IL-2. This cytokine stimulates the growth, differentiation, and survival of antigen-specific T cells and has been used as monotherapy for several different cancer types, including melanoma (Dillman et al., 2012; Vacchelli et al., 2013).
In contrast to the immune checkpoint proteins and immunosuppressive cytokines described above, the number of antigen specific T cells can be affected by chemokines in the tumor microenvironment. Blocking CXCR3, the receptor for the chemokine ligand CXCL9/10, impairs the accumulation of STAT3 deficient CD8+ T cells in tumor sites (Yue et al., 2015). Another study demonstrated that the chemokine CXCL10 maintains the effector T cell population (Harris et al., 2012). The chemokine CCL17 produced by CD8α+ dendritic cells attracted naïve cytotoxic T cells expressing the chemokine receptor CCR4 (Semmling et al., 2010). Research using chimeric antigen receptor (CAR) transgenic T cells suggests that chemokines enhance the trafficking of T cells to the tumors. CAR-T cells have shown great successes in clinical trials treating leukemia and lymphoma, although there are still issues when using them with solid tumors, such as the low numbers of T cells present at the tumor site. However, the number of GD2-CAR transgenic T cells increased in tumors after co-modification with CCR2b (Craddock et al., 2010). Migration of CAR-T lymphocytes also improved by forced expression of CCR4. The functionality of these cells was not impeded by transgenic expression of CCR4 (Di Stasi et al., 2009). Considering that chemokine receptor-armed CAR-T cells exhibit enhanced tumor infiltration, improving other transgenic TCR-T cell lines with the addition of chemokine receptors may bring better clinical outcomes. In addition, TCR-T cells only recognize intracellular tumor antigens present on the cell surface by MHC molecules, while CAR-T cells can recognize tumor antigens expressed on the tumor-cell surface independent of MHC restriction and antigen processing. A recent study demonstrating the benefits of this approach created CD8+ T cells expressing two additional receptors; a gp100 antigen-specific TCR and a melanoma-associated chondroitin sulfate proteoglycan specific CAR (Uslu et al., 2016). These T cells using combined recognition pathways showed greater efficacy by by-passing the mechanisms by which tumor cells escape immune recognition. In conclusion, TCR-engineered T cells therapy, in combination with drugs targeting chemokines, cytokines, and immune checkpoint proteins, may obtain better clinical responses in future treatments.
FUTURE PROSPECTIVE
There has been considerable progress in adoptive cell therapies using TCR-engineered T cells, a highly personalized cancer therapy. There are still some questions that remain to be answered: (1) How can the inhibitory factors present in the tumor microenvironment be overcome; (2) Is it possible to improve TCR-engineered T cell longevity at the tumor site in vivo; (3) Can an effective cocktail of TCR-engineered T cells, including different types of antigen-specific T cells targeting different antigenic epitopes, be identified.
Although some antibodies or recombinant cytokines can be used with TCR-engineered T cells, other currently unidentified factors still exist in tumor microenvironment that may also affect outcome. Another issue is that, once being activated, naïve T cells rapidly proliferate and differentiate into effector T cells and memory T cells after TCR-pMHC interaction. Although these differentiated effector T cells can produce a variety of effector molecules, these cells show high expression of exhaustion markers and rapid progression to cell death. To solve the problem of T cell exhaustion and prolong an effective immune response, some options may be feasible. One such approach is to alter metabolic pathways to enhance engineered T cells persistence. It has been shown that mTOR signals, AMPK-α1 signals, and IL-7 signals support the development of memory CD8+ T cells (Rolf et al., 2013; Cui et al., 2015). Based on these observations, it may be necessary to produce long-lived memory-like TCR-engineered T cells expressing metabolic associated molecules.
Verdegaal et al. observed dynamic changes in neoantigens from two patients with stage IV melanoma and found that expression of T cell recognized neoantigens reduced or even lost at the tumor site. This suggests that patients have an improved clinical response if infused with multiple T cell lines with engineered TCRs that recognize different neoantigens (Verdegaal et al., 2016). For example, a recent study demonstrated that T cells engineered with TCRs of the ten most abundant CD8+PD-1+ clonotypes from a TIL had reactivity against cancer germline antigens and neoantigens (Pasetto et al., 2016). In the future, different types of TCRs may be obtained by culturing CD8+PD-1+ T cells isolated from patient and could be a novel strategy to develop personalized cancer therapies. T cells co-expressing TCR and CAR also open a new avenue for the design of multifunctional tumor-specific T cells to be used in adoptive transfer (Uslu et al., 2016). For the potential of these therapies to be met, new and accurate approaches will need to be developed.
References
Abraham RT, Weiss A (2004) Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol 4:301–308
Agrawal S, Kishore MC (2000) MHC class I gene expression and regulation. J Hematother Stem Cell Res 9:795–812
Akbar AN, Terry L, Timms A, Beverley PC, Janossy G (1988) Loss of CD45R and gain of UCHL1 reactivity is a feature of primed T cells. J Immunol 140:2171–2178
Barfoed AM, Petersen TR, Kirkin AF, Thor Straten P, Claesson MH, Zeuthen J (2000) Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365-73 wild type peptide loaded on dendritic cells in vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand J Immunol 51:128–133
Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, Kubi A, Shoshani N, Zikich D, Ohayon Y et al (2013) Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res 19:4792–4800
Bobisse S, Rondina M, Merlo A, Tisato V, Mandruzzato S, Amendola M, Naldini L, Willemsen RA, Debets R, Zanovello P et al (2009) Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res 69:9385–9394
Bobisse S, Foukas PG, Coukos G, Harari A (2016) Neoantigen-based cancer immunotherapy. Ann Transl Med 4:262
Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA (2014) Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res 24:743–750
Byrne JA, Butler JL, Cooper MD (1988) Differential activation requirements for virgin and memory T cells. J Immunol 141:3249–3257
Chen L, Flies DB (2013) Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 13:227–242
Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, Ng C, Avramis E, Seja E, Villanueva A et al (2014) Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res 20:2457–2465
Clemente CG, Mihm MC Jr, Bufalino R, Zurrida S, Collini P, Cascinelli N (1996) Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 77:1303–1310
Cohen CJ, Gartner JJ, Horovitz-Fried M, Shamalov K, Trebska-McGowan K, Bliskovsky VV, Parkhurst MR, Ankri C, Prickett TD, Crystal JS et al (2015) Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J Clin Invest 125:3981–3991
Corse E, Gottschalk RA, Allison JP (2011) Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol 186:5039–5045
Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 14:135–146
Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, Foster AE (2010) Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 33:780–788
Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, Kaech SM (2015) IL-7-induced glycerol transport and TAG synthesis promotes memory CD8+ T cell longevity. Cell 161:750–761
Debets R, Donnadieu E, Chouaib S, Coukos G (2016) TCR-engineered T cells to treat tumors: Seeing but not touching? Semin Immunol 28:10–21
Dembic Z, Haas W, Weiss S, McCubrey J, Kiefer H, von Boehmer H, Steinmetz M (1986) Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature 320:232–238
Deng R, Cassady K, Li X, Yao S, Zhang M, Racine J, Lin J, Chen L, Zeng D (2014) B7H1/CD80 interaction augments PD-1-dependent T cell apoptosis and ameliorates graft-versus-host disease. J Immunol 194:560–574
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B (2009) T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113:6392–6402
Dillman RO, Barth NM, VanderMolen LA, Mahdavi K, McClure SE (2012) Should high-dose interleukin-2 still be the preferred treatment for patients with metastatic melanoma? Cancer Biother Radiopharm 27:337–343
Draper LM, Kwong ML, Gros A, Stevanovic S, Tran E, Kerkar S, Raffeld M, Rosenberg SA, Hinrichs CS (2015) Targeting of HPV-16+ epithelial cancer cells by TCR gene engineered T cells directed against E6. Clin Cancer Res 21:4431–4439
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM et al (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854
Fujio K, Misaki Y, Setoguchi K, Morita S, Kawahata K, Kato I, Nosaka T, Yamamoto K, Kitamura T (2000) Functional reconstitution of class II MHC-restricted T cell immunity mediated by retroviral transfer of the alpha beta TCR complex. J Immunol 165:528–532
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Germain RN, Stefanova I (1999) The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu Rev Immunol 17:467–522
Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM et al (2016) Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nate Med 22:433–438
Harris TH, Banigan EJ, Christian DA, Konradt C, Tait Wojno ED, Norose K, Wilson EH, John B, Weninger W, Luster AD et al (2012) Generalized Levy walks and the role of chemokines in migration of effector CD8+ T cells. Nature 486:545–548
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN et al (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515:563–567
Honda T, Egen JG, Lammermann T, Kastenmuller W, Torabi-Parizi P, Germain RN (2014) Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 40:235–247
Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS et al. (2015). High-throughput pairing of T cell receptor alpha and beta sequences. Sci Transl Med 7,301ra131
Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR et al (2009) Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 114:535–546
Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, Sugino S, Ueda S, Ishikawa T, Kokura S et al (2015) Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res 21:2268–2277
Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, Miki T, Rosenberg SA (1994a) Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci USA 91:3515–3519
Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, Yannelli JR, Adema GJ, Miki T, Rosenberg SA (1994b) Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci USA 91:6458–6462
Kawakami Y, Eliyahu S, Jennings C, Sakaguchi K, Kang X, Southwood S, Robbins PF, Sette A, Appella E, Rosenberg SA (1995) Recognition of multiple epitopes in the human melanoma antigen gp100 by tumor-infiltrating T lymphocytes associated with in vivo tumor regression. J Immunol 154:3961–3968
Kawashima I, Hudson SJ, Tsai V, Southwood S, Takesako K, Appella E, Sette A, Celis E (1998) The multi-epitope approach for immunotherapy for cancer: identification of several CTL epitopes from various tumor-associated antigens expressed on solid epithelial tumors. Hum Immunol 59:1–14
Keir ME, Butte MJ, Freeman GJ, Sharpe AH (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26:677–704
Kessels HW, Wolkers MC, van den Boom MD, van der Valk MA, Schumacher TN (2001) Immunotherapy through TCR gene transfer. Nat Immunol 2:957–961
Klebanoff CA, Rosenberg SA, Restifo NP (2016) Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 22:26–36
Kohn DB, Hershfield MS, Carbonaro D, Shigeoka A, Brooks J, Smogorzewska EM, Barsky LW, Chan R, Burotto F, Annett G et al (1998) T lymphocytes with a normal ADA gene accumulate after transplantation of transduced autologous umbilical cord blood CD34+ cells in ADA-deficient SCID neonates. Nat Med 4:775–780
Kunert A, van Brakel M, van Steenbergen-Langeveld S, da Silva M, Coulie PG, Lamers C, Sleijfer S, Debets R (2016) MAGE-C2-specific TCRs combined with epigenetic drug-enhanced antigenicity yield robust and tumor-selective T cell responses. J Immunol 197(6):2541–2552
Leone P, Shin EC, Perosa F, Vacca A, Dammacco F, Racanelli V (2013) MHC class I antigen processing and presenting machinery: organization, function, and defects in tumor cells. J Natl Cancer Inst 105:1172–1187
Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ et al (2013) Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122:863–871
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE et al (2015) High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21:81–85
Loi S (2013) Tumor-infiltrating lymphocytes, breast cancer subtypes and therapeutic efficacy. OncoImmunology 2:e24720
Mahata B, Zhang X, Kolodziejczyk AA, Proserpio V, Haim-Vilmovsky L, Taylor AE, Hebenstreit D, Dingler FA, Moignard V, Gottgens B et al (2014) Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep 7:1130–1142
Margulies DH (2001) TCR avidity: it’s not how strong you make it, it’s how you make it strong. Nat Immunol 2:669–670
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK et al (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482:400–404
Moon EK, Ranganathan R, Eruslanov E, Kim S, Newick K, O’Brien S, Lo A, Liu X, Zhao Y, Albelda SM (2016) Blockade of Programmed Death 1 Augments the Ability of Human T Cells Engineered to Target NY-ESO-1 to Control Tumor Growth after Adoptive Transfer. Clin Cancer Res 22:436–447
Morgan DA, Ruscetti FW, Gallo R (1976) Selective in vitro growth of T lymphocytes from normal human bone marrows. Science 193:1007–1008
Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP et al (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314:126–129
Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM et al (2013) Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 36:133–151
Nishimura MI, Kawakami Y, Charmley P, O’Neil B, Shilyansky J, Yannelli JR, Rosenberg SA, Hood L (1994) T-cell receptor repertoire in tumor-infiltrating lymphocytes. Analysis of melanoma-specific long-term lines. J Immunother Emphasis Tumor Immunol 16:85–94
Obst R (2015) The timing of T cell priming and cycling. Front Immunol 6:563
Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T (2013) A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol 14:1212–1218
Pageon SV, Tabarin T, Yamamoto Y, Ma Y, Bridgeman JS, Cohnen A, Benzing C, Gao Y, Crowther MD, Tungatt K et al (2016) Functional role of T-cell receptor nanoclusters in signal initiation and antigen discrimination. Proc Natl Acad Sci USA 113(37):E5454–E5463
Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosenberg SA (2009) Characterization of genetically modified T-cell receptors that recognize the CEA:691-699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. Clin Cancer Res 15:169–180
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM et al (2011) T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19:620–626
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25:9543–9553
Pasetto A, Alena G, Robbins PF, Deniger DC, Prickett TD, Matus-Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR et al (2016) Tumor- and neoantigen-reactive T-cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunology Research 4(9):734–743
Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA (2012a) Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal 5(230):ra46
Patsoukis N, Sari D, Boussiotis VA (2012b) PD-1 inhibits T cell proliferation by upregulating p27 and p15 and suppressing Cdc25A. Cell Cycle 11:4305–4309
Perez C, Jukica A, Listopad JJ, Anders K, Kuhl AA, Loddenkemper C, Blankenstein T, Charo J (2015) Permissive expansion and homing of adoptively transferred T cells in tumor-bearing hosts. Int J Cancer 137:359–371
Phan GQ, Rosenberg SA (2013) Adoptive cell transfer for patients with metastatic melanoma: the potential and promise of cancer immunotherapy. Cancer Control 20:289–297
Pilon-Thomas S, Kuhn L, Ellwanger S, Janssen W, Royster E, Marzban S, Kudchadkar R, Zager J, Gibney G, Sondak VK et al (2012) Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J Immunother 35:615–620
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng SL et al (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515:558–562
Prickett TD, Crystal JS, Cohen CJ, Pasetto A, Parkhurst MR, Gartner JJ, Yao X, Wang R, Gros A, Li YF et al (2016) Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol Res 4:669–678
Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, Wu R, Lizee G, Mahoney S, Alvarado G et al (2012) Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res 18:6758–6770
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, Badros AZ, Garfall A, Weiss B, Finklestein J et al (2015) NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 21:914–921
Redmond D, Poran A, Elemento O (2016) Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med 8(1):80
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124–128
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL et al (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29:917–924
Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E et al (2013) Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 19:747–752
Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM et al (2015) A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21:1019–1027
Rolf J, Zarrouk M, Finlay DK, Foretz M, Viollet B, Cantrell DA (2013) AMPKalpha1: a glucose sensor that controls CD8 T-cell memory. Eur J Immunol 43:889–896
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68
Rosenberg SA, Spiess P, Lafreniere R (1986) A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 233:1318–1321
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA et al (1988) Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 319:1676–1680
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR et al (2011) Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17:4550–4557
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712
Sanders ME, Makgoba MW, June CH, Young HA, Shaw S (1989) Enhanced responsiveness of human memory T cells to CD2 and CD3 receptor-mediated activation. Eur J Immunol 19:803–808
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C et al (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102:18538–18543
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT (2002) Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 188:22–32
Semmling V, Lukacs-Kornek V, Thaiss CA, Quast T, Hochheiser K, Panzer U, Rossjohn J, Perlmutter P, Cao J, Godfrey DI et al (2010) Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat Immunol 11:313–320
Slifka MK, Whitton JL (2001) Functional avidity maturation of CD8(+) T cells without selection of higher affinity TCR. Nat Immunol 2:711–717
Smith-Garvin JE, Koretzky GA, Jordan MS (2009) T cell activation. Annu Rev Immunol 27:591–619
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189–2199
Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J et al (2016) Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352:1337–1341
Tkach KE, Barik D, Voisinne G, Malandro N, Hathorn MM, Cotari JW, Vogel R, Merghoub T, Wolchok J, Krichevsky O et al (2014) T cells translate individual, quantal activation into collective, analog cytokine responses via time-integrated feedbacks. Elife 3:e01944
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS et al (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–645
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571
Uslu U, Schuler G, Dörrie J, Schaft N (2016) Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp Dermatol 25:872–879
Vacchelli E, Eggermont A, Fridman WH, Galon J, Zitvogel L, Kroemer G, Galluzzi L (2013) Trial Watch: Immunostimulatory cytokines. OncoImmunology 2:e24850
Valmori D, Dutoit V, Lienard D, Rimoldi D, Pittet MJ, Champagne P, Ellefsen K, Sahin U, Speiser D, Lejeune F et al (2000) Naturally occurring human lymphocyte antigen-A2 restricted CD8+ T-cell response to the cancer testis antigen NY-ESO-1 in melanoma patients. Cancer Res 60:4499–4506
Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM et al (2015) Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350:207–211
van der Bruggen P, Bastin J, Gajewski T, Coulie PG, Boel P, De Smet C, Traversari C, Townsend A, Boon T (1994) A peptide encoded by human gene MAGE-3 and presented by HLA-A2 induces cytolytic T lymphocytes that recognize tumor cells expressing MAGE-3. Eur J Immunol 24:3038–3043
Van Der Bruggen P, Zhang Y, Chaux P, Stroobant V, Panichelli C, Schultz ES, Chapiro J, Van Den Eynde BJ, Brasseur F, Boon T (2002) Tumor-specific shared antigenic peptides recognized by human T cells. Immunol Rev 188:51–64
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El Atmioui D et al (2013) Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31:e439–e442
Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H et al (2016) Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536:91–95
von Essen MR, Kongsbak M, Schjerling P, Olgaard K, Ødum N, Geisler C (2010) Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat Immunol 11:344–349
Willinger T, Staron M, Ferguson SM, De Camilli P, Flavell RA (2015) Dynamin 2-dependent endocytosis sustains T-cell receptor signaling and drives metabolic reprogramming in T lymphocytes. Proc Nat Acad Sci 112:4423–4428
Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF et al (2014) Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 11:41–46
Yao X, Lu YC, Parker LL, Li YF, El-Gamil M, Black MA, Xu H, Feldman SA, van der Bruggen P, Rosenberg SA et al (2016) Isolation and characterization of an HLA-DPB1*04: 01-restricted MAGE-A3 T-Cell Receptor for Cancer Immunotherapy. J Immunother 39:191–201
Yue C, Shen S, Deng J, Priceman SJ, Li W, Huang A, Yu H (2015) STAT3 in CD8+ T cells inhibits their tumor accumulation by downregulating CXCR3/CXCL10 axis. Cancer Immunol Res 3:864–870
Zhang N, Bevan MJ (2011) CD8(+) T cells: foot soldiers of the immune system. Immunity 35:161–168
Zhang Y, Stroobant V, Russo V, Boon T, van der Bruggen P (2002) A MAGE-A4 peptide presented by HLA-B37 is recognized on human tumors by cytolytic T lymphocytes. Tissue Antigens 60:365–371
Zhang Y, Chaux P, Stroobant V, Eggermont AM, Corthals J, Maillere B, Thielemans K, Marchand M, Boon T, Van Der Bruggen P (2003) A MAGE-3 peptide presented by HLA-DR1 to CD4+ T cells that were isolated from a melanoma patient vaccinated with a MAGE-3 protein. J Immunol 171:219–225
Zhang Y, Liu Y, Moxley KM, Golden-Mason L, Hughes MG, Liu T, Heemskerk MH, Rosen HR, Nishimura MI (2010) Transduction of human T cells with a novel T-cell receptor confers anti-HCV reactivity. PLoS Pathog 6:e1001018
Zheng Y, Zha Y, Driessens G, Locke F, Gajewski TF (2012) Transcriptional regulator early growth response gene 2 (Egr2) is required for T cell anergy in vitro and in vivo. J Exp Med 209:2157–2163
ACKNOWLEDGEMENTS
This study was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81171986, and 81271815), Research Grant from the Ministry of Public Health (No. 201501004), the National Key Research and Development Program of China (2016YFC1303501), Major Science and Technology Projects of Henan Province (1611003101000).
ABBREVIATIONS
CAR, chimeric antigen receptor; CDR, complementarity determining region; CEA, carcino-embryonic antigen; CTLA-4, cytotoxic T lymphocyte-associated antigen-4; DAG, diacylglycerol; Egr2, early growth response protein 2; Erk, extracellular regulating kinase; HLA, human leukocyte antigen; ICOS, inducible T cell costimulator; IFN, interferon; IL-2, interleukin-2; IP3, inositol triphosphate; MAGE, melanoma-associated antigen; MART, melanoma-associated antigen recognized by T cells; MHC, major histocompatibility complex; mTOR, the mechanistic target of rapamycin; NFAT, nuclear factor of activated T cells; NY-ESO-1, New York esophageal squamous cell carcinoma; OVA, ovalbumin; PD-1, programmed death 1; PBMC, peripheral blood mononuclear cell; PIP2, phosphatidylinositol bisphosphate; PLC-γ1, phospholipase C-γ1; TCR, T cell receptor; TIL, tumor infiltrating lymphocyte; VDR, Vitamin D receptor.
COMPLIANCE WITH ETHICS GUIDELINES
Yu Ping, Chaojun Liu, and Yi Zhang declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ping, Y., Liu, C. & Zhang, Y. T-cell receptor-engineered T cells for cancer treatment: current status and future directions. Protein Cell 9, 254–266 (2018). https://doi.org/10.1007/s13238-016-0367-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-016-0367-1