
Abstract
Cancer stem cells (CSCs), a subpopulation of cancer cells with ability of initiating tumorigenesis, exist in many kinds of tumors including breast cancer. Cancer stem cells contribute to treatment resistance and relapse. Conventional treatments only kill differentiated cancer cells, but spare CSCs. Combining conventional treatments with therapeutic drugs targeting to CSCs will eradicate cancer cells more efficiently. Studying the molecular mechanisms of CSCs regulation is essential for developing new therapeutic strategies. Growing evidences showed CSCs are regulated by non-coding RNA (ncRNA) including microRNAs and long non-coding RNAs (lncRNAs), and histone-modifiers, such as let-7, miR-93, miR-100, HOTAIR, Bmi-1 and EZH2. Herein we review the roles of microRNAs, lncRNAs and histone-modifiers especially Polycomb family proteins in regulating breast cancer stem cells (BCSCs).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The concept of cancer stem cells (CSCs) has been generally accepted since leukaemia cancer stem cells were discovered by John E. Dick in 1994 (Lapidot et al., 1994). CSCs maintain CSCs pool or generate more CSCs via self-renewal, and generate non-CSCs progenies forming the differentiated cancer cells via differentiation. CSCs also initiate tumor formation, required critical cell amount for tumor formation in xenografts is reduced compared to non-CSCs. It has been reported that CSCs exist in many kinds of tumors, including breast cancer, lung cancer, leukaemia, glioblastoma, colon cancer, live cancer and so on (Visvader and Lindeman, 2008), meanwhile, some molecular markers have been used to separate CSCs from total cancer cell population, such as ALDH (Ginestier et al., 2007) and CD24−CD44+ (Al-Hajj et al., 2003) for BCSCs, CD90 (Yang et al., 2008) and CD133 (Ma et al., 2007) for liver cancer stem cells, ABCB5 and CD271+ for melanoma cancer stem cells (Schatton et al., 2008), CD133 for brain tumor stem cells (Singh et al., 2004). CSCs are heterogenous, for example, BCSCs and colon cancer stem cells include at least two types of CSCs identified with different molecular markers (Liu et al., 2014). BCR-ABL1 lymphoblastic leukaemia contains multiple genetically distinct leukaemia stem cell sub-clones (Notta et al., 2011).
It has revealed that some signaling pathways play critical roles in regulating the self-renewal and differentiation of CSCs. Wnt signaling is important for CSC self-renewal, for example, Wnt/β-catenin signaling is activated in poor differentiated basal-like breast cancer with worse overall survival (Khramtsov et al., 2010). Constitutive activation of Wnt signaling caused by the mutation of tumor suppressor APC leads to breast cancer stem cell (BCSC) expansion. In Her2+ breast cancer inhibition of Wnt signaling represses tumor initiation and metastasis (Monteiro et al., 2014; Schade et al., 2013). These suggest deregulated Wnt signaling promotes the expansion of CSCs. Hedgehog (HH) signaling promotes glioma growth by stimulating self-renewal of CD133+ glioma CSCs, and increases chemotherapeutic agent resistance (Clement et al., 2007). Hedgehog signaling also maintain CSCs in breast cancer and myeloid leukaemia (Liu et al., 2006; Zhao et al., 2009). Notch signaling is also important in regulating CSCs (Pannuti et al., 2010). For example, Notch-GFP reporter system has been used to separate CSCs from lung adenocarcinoma and breast cancer, GFP+ cancer cells could differentiate into GFP− cancer cells, and have strong tumor initiation capacity (D’Angelo et al., 2015; Hassan et al., 2013). Notch signaling induces deacetylase sirtuin 2 (SIRT2) to deacetylate and activate ALDH1A1 and then increases BCSCs (Zhao et al., 2014). Apart from these signaling pathways, TGF-β, IL6/JAK/STAT3, NF-κB signaling and other signaling pathways also play critical role in regulating CSCs, and they sometimes cross-talk with each other in the regulation.
With the development of epigenetics, histone-modifying enzymes and ncRNAs have been found to play vital roles in regulating CSCs. In this review, we focus on the research progress about ncRNAs and histone-modifiers in regulating BCSCs.
BREAST CANCER STEM CELLS
In 2003, Clarke and his colleagues isolated putative BCSCs as ESA+CD44+CD24− cell population (Al-Hajj et al., 2003). As few as 200 ESA+CD44+CD24− cells were capable to generate tumor in vivo, whereas a 100-fold more cells without these markers isolated from the same tumors were non-tumorigenic. In addition, the secondary tumors resemble the phenotype (morphology and ESA/CD44/CD24 expression profile) of the initial tumor and the tumorigenic ESA+CD44+CD24− tumor cells could be serially passaged at least four passages in vivo. Subsequent studies employed several methodologies adapted from stem cell research to isolate or investigate BCSCs, including side population (SP) assay, ALDEFLUOR assay and sphere assay. The SP assay is based on the ability of stem cells to exclude DNA dye such as Hoechst 33342 by membrane transporters, and the SP has been shown to contain the most tumorigenic population within breast cancer cell line when being transplanted in vivo (Dontu et al., 2003). The Aldefluor assay represents a group of enzymes catalyzing the oxidation of aldehydes. In malignant mammary epithelium, cells with high Aldehyde dehydrogenase (ALDH) activity were associated with the greatest self-renewal and differentiation abilities both in vitro and in vivo, and positive ALDH immunostaining in breast carcinomas correlated with poor prognosis (Ginestier et al., 2007). Mammary stem/progenitor cells are able to survive in serum-free and anchorage-independent conditions in the form of spheroids (Dontu et al., 2003). BCSCs were also enriched when grown as non-adherent spheroids in vitro (Ponti et al., 2005). Interestingly, the two tumor initiating populations (ALDH+ cells and ESA+CD44+CD24− cells) only showed limited overlapping (Box 1) (Ginestier and Wicha, 2007). Similar finding was also demonstrated by the other group that breast cancers may contain tumor initiating cells displaying different cell surface markers (Wright et al., 2008). Despite the heterogeneity of BCSCs, these cells are usually associated with therapy resistance and tumor relapse, the two main obstacles in cancer treatment. Therefore, understanding the biology of CSCs will help the development of new therapeutic approaches to target CSCs, leading to more effective therapies and ultimate cure for cancer.
BREAST CANCER STEM CELLS (BCSCs) ARE REGULATED BY microRNAs
MicroRNAs (miRNAs) regulate targeted mRNAs through a combination of translational repression and mRNA destabilization. The biogenesis of miRNAs has been summarized in details by V. Narry Kim (Kim et al., 2009). Studies have shown microRNAs regulate cells proliferation, invasion, metastasis and angiogenesis in both solid tumors and leukemia (Nicoloso et al., 2009). miR-29 promotes hepatocellular carcinoma cell apoptosis by targeting Mcl-1 and Bcl-2 (Xiong et al., 2010). miR-10b initiates tumor invasion and metastasis by targeting RHOC in breast cancer (Ma et al., 2008).
In recent years, miRNAs have been studied in BCSCs intensively (Table 1). We have shown that let-7a is downregulated in mammosperes in comparison to differentiated cancer cells utilizing miRNA array analysis; let-7a is also lower in BCSCs marked by CD24−CD44+ than non-CD24−CD44+ cells, and let-7a overexpression suppressed the mammosphere formation and tumor initiation. Further analysis reveals let-7a suppresses self-renewal of BCSCs in part by targeting H-Ras, and promotes cellular differentiation by targeting HMGA2 (Yu et al., 2007). Let-7 is also regulated by some signaling pathways, e.g., Wnt-β-catenin pathway activates Lin28 which suppress let-7 biogenesis by inducing urdylation of precursor let-7 (pre-let-7) at its 3′ end and then represses let-7 to expand CSCs (Cai et al., 2013; Heo et al., 2008). Some protein methyltransferases not only catalyze methylation of histones, but also nonhistone proteins. For example, SET7/9 which catalyzes monomethylation of histone 3 also catalyzes methylation of Lin28A at K135 to promote Lin28 accumulation in nucleus, and increases the stability and pri-let-7-binding ability of Lin28 (Kim et al., 2014), suggesting epigenetic proteins can regulate CSCs.
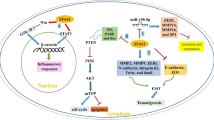
miR-200 family, including miR-200a, miR-200b, miR-200c, miR-141 and miR-429, are reported as tumor suppressors. They repress EMT by targeting ZEB1 and ZEB2. miR-200c suppresses BCSCs through targeting BMI1 (Shimono et al., 2009). miR-200b inhibits BCSCs by targeting SUZ12, H3K27me3 of E-cadherin and other genes (Iliopoulos et al., 2010). Inhibition of miR-141 increases both CD44+ and CK5+ cells by targeting Stat5a and progesterone receptor (PR), and enhances the abilities of mammosphere formation and tumor initiation. miR-200 family could be regulated by some signaling pathways, for example, abnormal expression of AKT1 and AKT2 causes dysregulation of miR-200 family to regulate epithelial-mesenchymal transition (EMT) and CSCs self-renewal (Iliopoulos et al., 2009). Besides ZEB1 and ZEB2, some Polycomb group (PcG) proteins are also targets of miR-200. The role of some Polycomb family proteins will be introduced later in the text (Fig. 1).

Let-7 and miR-200 inhibit BCSCs. Wnt-β-catenin could regulate BCSCs not only by regulating Lin28, but also by other proteins. Lin28 can be activated by NF-κB and SET7/9. Lin28 inhibits let-7. AKT1 and AKT2 suppress miR-200, and also activate NF-κB to regulate BCSCs. miR-22 inhibits TET family proteins which can activate miR-200, and lncRNA-ATB also inhibits miR-200
Recently, Pier Paolo Pandolfi and colleagues found miR-22 promotes the EMT, tumor invasion and metastasis of normal and cancer mammary stem cells by targeting TET1, TET2 and TET3; miR-22 overexpression enhances some stemness and EMT-related genes expression, such as BMI1, ZEB1 and ZEB2. TET1, TET2 and TET3 could inhibit the demethylation of miR-200 promoter. So miR-22 promotes BCSCs by suppressing miR-200 expression, suggesting DNA demethylases could regulate BCSCs.
miR-93 plays different roles in BCSCs come from different subtypes of breast cancer. miR-93 inhibits CSCs and initiates mesenchymal-epithelial transition (MET) in basal type of breast cancer cells such as SUM159 by targeting AKT3, SOX4 and STAT3. However, it promotes BCSCs in luminal type of breast cancer such as MCF-7, suggesting the dual roles of miR-93 in regulating BCSCs is dependent on the state of cellular differentiation, but the mechanism is yet to be elucidated (Liu et al., 2012).
In addition to BCSCs, dysregulation of miRNAs is also found in other types of CSCs. In colon cancer stem cells, the function of miR-34a depends on its expression levels: High miR-34a expression suppresses Notch signaling pathway and promotes differentiation of CSCs; low miR-34a expression promotes Notch signaling pathway and maintains CSCs phenotype (Bu et al., 2013). CD44 is a marker of prostate cancer stem cells, miR-34a inhibits CSCs and cancer metastasis by targeting CD44 (Liu et al., 2011). miR-218 inhibits glioma cancer stem cells by targeting BMI1 (Tu et al., 2013). More and more studies showed that miRNAs can potentially be used for tumor therapy by being linked to therapeutic vectors, such as nanoparticles.
lncRNAs PLAY POTENTIAL ROLES IN REGULATING BREAST CANCER AND CANCER STEM CELLS
lncRNAs (long non-coding RNAs) are >200 nt non-coding RNA. A great number of lncRNAs have been discovered, but only a few lncRNAs have been well studied by now. Recently, lncRNAs have been studied in many cancers. For example, lncRNA-ATB activated by TGF-β induces EMT in hepatocellular carcinoma, breast cancer and colon cancer (Yuan et al., 2014). It not only function as a competing endogeneous RNA (ceRNA) competitively binding to miR-200 family to upregulate their targets and induces EMT, but also binds to IL11 mRNA, increasing the stability of IL11 and causing autocrine induction of IL11 to activate STAT3 pathway, which plays a vital role in regulating BCSCs. lncRNA-ATB may regulate BCSCs by regulating miR-200 family and STAT3.
Chemokine CCL21 binds to its receptor CCR7 to induce the phosphorylation of GLI2 mediated by citron (CIT) kinase, phosphorylation of GLI2 activates the target genes of GLI. lncRNA BCAR4 is required for transcription activation of GLI2 target genes. RNA pull-down and mass spectrometry analysis reveals that BCAR4 interacts with SNIP1 and PNUTS. When BCAR4 interacts with SNIP1, SNIP1 releases the suppression of p300, and p300 acetylates GLI2 target gene promoters marked H3K18ac and promotes gene transcription. The acetylated H3K18 can be recognized by PNUTS, and interact with it to activate the phosphatase activity of PP1 to maintain hypophosphorylation level of RNA Pol II Ser5 at gene promoter regions. BCAR4 could induce the activation of GLI’s target genes and promotes breast cancer metastasis, especially triple-negative breast cancer (Xing et al., 2014). The target genes of GLI plays pivotal role in BCSCs. These suggest that BCAR4 may regulate BCSCs, which is to be demonstrated with further studies.
Furthermore, lncRNA lncTCF7 regulated self-renewal of hepatocellular carcinoma stem cells demonstrated by tumorsphere formation ability in vitro and tumor initiating frequency in vivo. lncTCF7 recruits the SWI/SNF complex to bind to TCF7 promoter and activate TCF7 expression, and TCF7 activates Wnt pathway to expand hepatocellular carcinoma stem cells (Wang et al., 2015). lncRNA-ROR is a modulator of cell reprogramming and pluripotency. In breast cancer, lncRNA-ROR induces EMT and promotes metastasis, lncRNA-ROR overexpression increases the percentage of CD24−CD44+ cell population and mammosphere numbers. Further analysis reveals that it can act as a ceRNA of miR-205 which targets the EMT inducer ZEB2 and blocks the degradation of ZEB2 to promote EMT (Hou et al., 2014).
HOTAIR has been studied for many types of cancers (Zhang et al., 2014b). In breast cancer HOTAIR promotes cancer metastasis (Gupta et al., 2010). It can act as a scaffold to bring two epigenetic protein complexes. The 5′ domain of HOTAIR binds to Ploycomb repressive complex 2 (PRC2), and the 3′ domian binds to the LSD1/CoREST/REST complex. HOTAIR can regulate the function of epigenetic complex and causes chromatin state change (Tsai et al., 2010). HOTAIR also regulates BCSCs, and microarray analysis reveals HOTAIR overexpression upregualtes the genes related to stemness and EMT, such as CD44, STAT3, ALDH2, ZEB1 and VIM, but the tumor initiating frequency in vivo assay are needed to demonstrate the role of HOTAIR in regulating BCSCs further (Padua Alves et al., 2013).
With the progression of studies about lncRNAs, more and more lncRNAs will been demonstrated in modulating CSCs. lncRNAs represent a new type of CSCs regulator by regulating miRNAs, mRNAs and other lncRNAs. The study of lncRNAs will improve the understanding of novel molecular regulation of CSCs, and lncRNAs can function as targets for novel therapies and as prognosis factors.
HISTONE-MODIFIERS, ANOTHER VITAL REGULATOR OF BREAST CANCER STEM CELLS (BCSCs)
Histone H2A, H2B, H3 and H4 is modified by histone modifying enzymes, including histone acetyltransferases, histone deacetylases, histone methyltransferase and histone demethylases. Histone modifying enzymes play a role in the regulation of transcription by modulating the state of chromatin, and they also cross-talk with each other (Portela and Esteller, 2010). Studies about the roles of histone modifying enzymes in breast cancer and other cancers have been reported (Patani et al., 2011). H3K4 demethylase Jarid1B/KDM5B is amplified and overexpressed in breast cancer to promote cell proliferation. RNA-seq and ChIP-seq analysis revealed the binding sites of Jarid1B are significantly enriched in the promoters and enhancers of luminal-high genes than those of basal-high genes, indicating it is a luminal lineage-driving oncogene (Yamamoto et al., 2014). H3K9me2 methyltransferase G9a interacts with Snail and DNA methyltransferase, recruits them to the promoter of E-cadherin for DNA methylation and promotes EMT in breast cancer (Dong et al., 2012). Coactivator-associated arginine methyltransferase 1 (CARM1) methylates BAF155 at R1064. Methylation of BAF155 promotes breast cancer invasion and metastasis in vivo and in vitro. Chromatin immunoprecipitation (ChIP) analysis demonstrates CARM1 control the expression of genes in the c-myc pathway (Wang et al., 2014).
NYD1/KDM2B
H3K36me1/2 and H3K4me3 demethylase NYD1/KDM2B plays an important role in ES and iPS. It is directly regulated by pluripotency factors OCT4 and SOX2, and it interacts with the core subunits of Ploycomb repressive complex 1 (PRC1), such as Ring1B and recruits PRC1 to the CpG island of promoter of genes which control the differentiation, resulting in the inhibition of differentiation genes. This also suggests KDM2B can function as a Polycomb group repressive element (PRE) to inhibit differentiation gene expression (He et al., 2013). KDM2B also inhibits let-7 and miR-101 to induce upregulation of EZH2 and the levels of H3K27me3 in the sites of Ink4a-Arf-Ink4b. So KDM2B can function as an oncogene (Kottakis et al., 2011; Tzatsos et al., 2011). In breast cancer, downreguation of KDM2B inhibits anchorage-dependent and -independent growth, arrests cell cycle in G1 and promotes apoptosis, and CSCs were also reduced. These results suggest KDM2B is an oncogene, not only promoting breast cancer but also maintaining BCSCs. Basal marker and luminal marker analysis showed KDM2B is required for the maintenance of the myoepithelial/luminal progenitor cell phenotype of basal breast cancer cells. Western blot analysis indicates Polycomb group (PcG) proteins, SUZ12, EZH2, RING1B and BMI1, are downregulated upon KDM2B knockdown, but they are not the direct downstream of KDM2B. KDM2B binds to the sites encoding miR-200 family, miR-101, miR-181 and miR-203. SUZ12, EZH2, RING1B and BMI1 are the direct targets of these miRNAs, and KDM2B inhibits BCSCs through repressing these miRNAs and upregulation of the core subunits of PcG proteins (Kottakis et al., 2014). In clinical samples, KDM2B expression has a negative correlation with these miRNAs, but has a positive correlation with these core subunits of PcG proteins. So PcG proteins have prominent role in BCSCs. RING1B is an E3 ubiquitin ligase for H2AK119, but the role of RING1B in breast stem cells have to be studied further in BCSCs.
PcG proteins are divided into two main subfamilies of complexes: PRC1 and PRC2. The targets of PcG proteins are highly enriched for transcription factors of signaling pathways involved in development and disease. With the new algorithms emerging, more targets of PcG proteins will be identified, including lncRNAs and miRNAs. The core subunits of PRC2 catalyze H3K27me3 in target sites, and H3K27me3 recruits PRC1 to inhibit target gene expression (Kerppola, 2009; Simon and Kingston, 2009). In addition to this mechanism, RYBP1-PRC1 complexes mediate H2A ubiquitylation at the Polycomb target sites also suppress the expression of targets independent on PRC2 and H3K27me3(Tavares et al., 2012).
EZH2 and SUZ12
EZH2 and SUZ12 belong to PRC2. EZH2 is a histone methyltransferase and catalyzes H3K27me3, and SUZ12 stimulates the activity of EZH2. EZH2 plays critical roles in embryonic stem cells, adult stem cells and cancer. For example, in mammary gland, EZH2 maintains luminal progenitor cell self-renewal (Michalak et al., 2013). EZH2 is also an oncogene and the downstream of the pRB-E2F pathway. EZH2 is essential for proliferation and amplified in many primary cancers, and is inhibited by AKT through phosphorylation at Ser21 (Cha et al., 2005). EZH2 can be used as a molecular marker for precancerous diagnosis, and EZH2 overexpression in histologically normal breast epithelium increases the risk of developing cancer. Furthermore, EZH2 is upregulated in breast cancer, and high EZH2 levels are associated with aggressive breast cancer. Kaplan-Meier analysis of metastasis-free survival and overall survival show the survival rate is lower in patients with high EZH2 (Kleer et al., 2003). It has been well-known that hypoxia promotes CSCs, such as glioma stem cells (Li et al., 2009), colorectal cancer stem cells (Yeung et al., 2011) and BCSCs (Conley et al., 2012). Chun-Ju Chang and colleagues found hypoxia induces the expression of EZH2 to expand BCSCs, and EZH2 overexpression increases the number of SP and CD24−CD44+ cells. But the mechanism that hypoxia regulating EZH2 has to be elucidated. EZH2 inhibits the expression of tumor suppressor RAD51 which participates in DNA repair leading to genomic instability and increases some oncogene expression such as RAF1 which activates ERK and Wnt-β-catenin pathway to promote cancer cell survival and proliferation, and expands BCSCs (Chang et al., 2011). Notch signaling pathway maintains the stemness of cancer stem cells, induces EMT transition and promotes chemoresistance (Pannuti et al., 2010). In clinical samples, the expression of EZH2 and Notch1 are positively correlated. EZH2 knockdown inhibits the onset and growth of xenografts derived from triple-negative breast cancer, and the opposite phenotype emerges when EZH2 is overexpressed. EZH2 overexpression activates Notch1 signaling activity to promote BCSC self-renewal. Further analysis indicates EZH2 regulates Notch transcriptional activity depending on direct binding to the promoter of Notch1 rather than its histone methyltransferase activity. In glioblastoma stem cells, EZH2 activates STAT3 signaling and promotes tumorigenicity (Kim et al., 2013). Activation of STAT3 also promotes BCSCs, and intronic RNAs also mediate regulation of epigenetic targets (Guil et al., 2012). Recently, Hae-Yun Jung and colleagues find PAF (PCNA-associated factor) interacts with β-catenin to recruit EZH2 and form a transcriptional complex, and this complex specifically transactivates the target genes of Wnt signaling, suggesting EZH2 expands BCSCs by activating Wnt pathway (Jung et al., 2013), which indicates EZH2 may be a central protein, and is regulated by several key signaling pathways which regulate CSCs. Further studies are needed to reveal the regulatory mechanism.
SUZ12 promotes the silencing of Hox gene, cell proliferation and embryogenesis (Cao and Zhang, 2004; Pasini et al., 2004). The mutations of SUZ12 often are found in some cancer, for example, SUZ12 mutations cause the malignant transformation of peripheral nerve sheath tumors (Zhang et al., 2014c). SUZ12 knockdown inhibits mammosphere formation ability of BCSCs, and suppresses CD44. SUZ12 is also a direct target miR-200b which is a BCSC suppressor. miR-200b inhibits BCSCs through SUZ12 partly, but the molecular mechanism of SUZ12 regulating BCSCs is yet to be elucidated.
BMI1
BMI1 which a canonical component of PRC1, is a co-factor for E3 ubiquitin ligase and compact polynucleosomes. Growing evidences suggest BMI1 plays a vital role in regulating self-renewal of normal and cancer stem cells. BMI1 stimulates the self-renewal of normal and leukaemic stem cells (Lessard and Sauvageau, 2003), and enhances self-renewal of hematopoietic stem cells (Iwama et al., 2004). Bmi1 promotes neural stem self-renewal by repressing the p16 and p19 senescence pathways (Lessard and Sauvageau, 2003), but other studies have shown BMI1 controls neural stem self-renewal through p21-Rb pathway (Christopher A. Fasano et al., 2007). BMI1 is a regulator of prostate stem cell self-renewal and malignant transformation (Lukacs et al., 2010). BMI1 is a marker for intestinal stem cells, and a BMI1 inhibitor has been found to inhibit colorectal cancer stem cells (Kreso et al., 2014; Yan et al., 2012). In BCSCs, BMI1 is a target of miR-200 family and miR-128, which regulate BCSCs by targeting BMI1. BMI1 is also regulated by some signalings, such as Hedgehog (Liu et al., 2006). Activation of Hedgehog signaling pathway promotes self-renewal in both mammary stem/progenitor cells and BCSCs, and downregulation of BMI1 eliminates this effect. BMI1 also activates Wnt signaling pathway by repressing WNT inhibitors, Dickkopf (DKK), to activate WNT pathway. BMI1 also auto-activates itself. c-Myc is a target of WNT, and also an activator of BMI1. BMI1, DKK1, WNT and c-Myc form a positive feedback loop to promote BCSCs (Cho et al., 2013). So BMI1 plays a critical role in self-renewal of BCSCs (Fig. 2). Recently, Xu and colleagues reported that a lncRNA FAL1 is overexpressed in some types of cancers including breast cancer, and it interacts with BMI1-PRC1 complex to enhance its stabilization by blocking its proteasomal degradation, which influences the ubiquitylation levels of H2AK119 to epigenetically repress genes expression such as CDKN1A (Neven et al., 2014). PRC1 binds to the target sites through PREs, and PREs may be ncRNAs or proteins, such as Jarid2, KDM2B and HOTAIR (Schwartz and Pirrotta, 2013). These suggest BMI1 can cross-talk with ncRNAs.

BMI1 regulates BCSC. BMI1 is activated by Hedgehog, Notch and Wnt pathway, and it is inactivated by miR-200 family and miR-128. There may be some other microRNAs and lncRNAs regulated BMI1
Histone-modifiers are a new frontier for drug discovery, and lots of small molecular compound inhibitors have been developed for disease therapies, among which some have been investigated in Phase III of clinical trials (Arrowsmith et al., 2012). The research of histone-modifiers in depth in regulating BCSCs will benefit the survival rate of patients with breast cancer and other type cancers.
CONCLUDING REMARKS
In this review, we mainly summarize the role of ncRNA and histone-modifiers in regulating BCSCs. More and more non-coding RNAs have been been found to regulate CSCs, and the new function and mechanism of histone-modifiers is also getting clearer.
miRNAs are ncRNAs with short sequence, and they can be covalently conjugated with nanovectors easily. The tumor cells will be eradicated when the miRNAs or antisense nucleotide of specific miRNAs can be delivered to tumor cells by nanovectors. So, the development of nanocarriers of the drugs is important. lncRNAs are newly identified regulators of CSCs, and the regulation mechanisms are getting clearer by investigating more and more lncRNAs. lncRNAs can also function as therapeutic targets. For example, inhibiting lncRNA BCAR4 with locked nucleic acid (LNA)-based antisense oligonucleotides suppressed the metastasis of breast cancer (Xing et al., 2014).
CSCs could not be cultured like embryonic stem cells in undifferentiated state in vitro to date, and flow cytometry with specific markers is the main method for CSC separation. Some important technologies used to study mechanism, such as immunoprecipitation and ChIP-seq, require a larger number of cells, which is not suitable for cancer stem cell research. But new technologies such as single cell RNA-sequencing (Hou et al., 2012) and ChIP-seq of 500 cells (Lara-Astiaso et al., 2014), will bring new hope for CSC research. For example, epigenetic regulator proteins play an important role in the regulation of CSCs, but the mechanism of regulation has still not been well studied due to the small CSCs number, single cell RNA-sequencing and ChIP-seq of 500 cells will eventually solve this problem. The promises and challenges of single cell RNA-sequencing have been reviewed by Stegle, O. and colleagues (Stegle et al., 2015).
The interaction among signaling pathways, ncRNAs and histone-modifiers plays a vital role in regulating CSCs. They can cross-talk with each other. Fully understanding the network of signaling pathways, ncRNAs and histone-modifiers will be helpful for tumor therapies.
References
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF (2003) Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 100:3983–3988
Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M (2012) Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov 11:384–400
Bu P, Chen KY, Chen JH, Wang L, Walters J, Shin YJ, Goerger JP, Sun J, Witherspoon M, Rakhilin N et al (2013) A microRNA miR-34a-regulated bimodal switch targets Notch in colon cancer stem cells. Cell Stem Cell 12:602–615
Cai WY, Wei TZ, Luo QC, Wu QW, Liu QF, Yang M, Ye GD, Wu JF, Chen YY, Sun GB et al (2013) The Wnt-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci 126:2877–2889
Cao R, Zhang Y (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15:57–67
Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, Ping B, Otte AP, Hung MC (2005) Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 310:306–310
Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC (2011) EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell 19:86–100
Cho JH, Dimri M, Dimri GP (2013) A positive feedback loop regulates the expression of polycomb group protein BMI1 via WNT signaling pathway. J Biol Chem 288:3406–3418
Clement V, Sanchez P, de Tribolet N, Radovanovic I, i Altaba R (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol 17:165–172
Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS (2012) Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA 109:2784–2789
D’Angelo RC, Ouzounova M, Davis A, Choi D, Tchuenkam SM, Kim G, Luther T, Quraishi AA, Senbabaoglu Y, Conley SJ et al (2015). Notch reporter activity in breast cancer cell lines identifies a subset of cells with stem cell activity. Mol Cancer Ther
Deng L, Shang L, Bai S, Chen J, He X, Trevino RM, Chen S, Li X, Meng X, Yu B et al (2014) microRNA100 inhibits self-renewal of breast cancer stem-like cells and breast tumor development. Cancer Res 74:6648–6660
Dong CF, Wu YD, Yao J, Wang YF, Yu YH, Rychahou PG, Evers BM, Zhou BP (2012) G9a interacts with Snail and is critical for snail-mediated E-cadherin repression in human breast cancer. J Clin Investig 122:1469–1486
Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS (2003) In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 17:1253–1270
Fasano CA, Dimos JT, Ivanova NB, Lowry N, Lemischka IR, Temple S (2007) shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell 1:13
Finlay-Schultz J, Cittelly DM, Hendricks P, Patel P, Kabos P, Jacobsen BM, Richer JK, Sartorius CA (2014) Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene 0
Ginestier C, Wicha MS (2007) Mammary stem cell number as a determinate of breast cancer risk. Breast Cancer Res 9:109
Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu SL et al (2007) ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 1:555–567
Guil S, Soler M, Portela A, Carrere J, Fonalleras E, Gomez A, Villanueva A, Esteller M (2012) Intronic RNAs mediate EZH2 regulation of epigenetic targets. Nat Struct Mol Biol 19:664–670
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL et al (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464:1071–1076
Gwak JM, Kim HJ, Kim EJ, Chung YR, Yun S, Seo AN, Lee HJ, Park SY (2014) MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Treat 147:39–49
Han M, Liu M, Wang Y, Mo Z, Bi X, Liu Z, Fan Y, Chen X, Wu C (2012a) Re-expression of miR-21 contributes to migration and invasion by inducing epithelial-mesenchymal transition consistent with cancer stem cell characteristics in MCF-7 cells. Mol Cell Biochem 363:427–436
Han M, Wang Y, Liu M, Bi X, Bao J, Zeng N, Zhu Z, Mo Z, Wu C, Chen X (2012b) MiR-21 regulates epithelial-mesenchymal transition phenotype and hypoxia-inducible factor-1alpha expression in third-sphere forming breast cancer stem cell-like cells. Cancer Sci 103:1058–1064
Han ML, Liu MR, Wang YM, Chen X, Xu JL, Sun Y, Zhao LY, Qu HB, Fan YM, Wu CY (2012c) Antagonism of miR-21 reverses epithelial-mesenchymal transition and cancer stem cell phenotype through AKT/ERK1/2 inactivation by targeting PTEN. PLoS ONE 7:e39520
Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS (2013) Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res 19:1972–1980
He J, Shen L, Wan M, Taranova O, Wu H, Zhang Y (2013) Kdm2b maintains murine embryonic stem cell status by recruiting PRC1 complex to CpG islands of developmental genes. Nat Cell Biol 15:373–384
Heo I, Joo C, Cho J, Ha M, Han J, Kim VN (2008) Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell 32:276–284
Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D et al (2012) Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell 148:873–885
Hou P, Zhao Y, Li Z, Yao R, Ma M, Gao Y, Zhao L, Zhang Y, Huang B, Lu J (2014) LincRNA-ROR induces epithelial-to-mesenchymal transition and contributes to breast cancer tumorigenesis and metastasis. Cell Death Dis 5:e1287
Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EYHP, Lee WH (2011) miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene 30:2463–2474
Iliopoulos D, Polytarchou C, Hatziapostolou M, Kottakis F, Maroulakou IG, Struhl K, Tsichlis PN (2009) MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Sci Signal 2:r62
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K (2010) Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell 39:761–772
Isobe T, Hisamori S, Hogan DJ, Zabala M, Hendrickson DG, Dalerba P, Cai S, Scheeren F, Kuo AH, Sikandar SS et al (2014) miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 3:e01977
Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, Ema H, Kamijo T, Katoh-Fukui Y, Koseki H et al (2004) Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21:843–851
Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD, Park JI (2013) PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell 52:193–205
Ke J, Zhao Z, Hong SH, Bai S, He Z, Malik F, Xu J, Zhou L, Chen W, Martin-Trevino R et al (2015) Role of microRNA221 in regulating normal mammary epithelial hierarchy and breast cancer stem-like cells. Oncotarget 6:3709
Kerppola TK (2009) Polycomb group complexes–many combinations, many functions. Trends Cell Biol 19:692–704
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH (2010) Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol 176:2911–2920
Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10:126–139
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, Oh YT, Kim H, Rheey J, Nakano I et al (2013) Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 23:839–852
Kim SK, Lee H, Han K, Kim SC, Choi Y, Park SW, Bak G, Lee Y, Choi JK, Kim TK et al (2014) SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell 15:735–749
Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF et al (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100:11606–11611
Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN (2011) FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell 43:285–298
Kottakis F, Foltopoulou P, Sanidas I, Keller P, Wronski A, Dake BT, Ezell SA, Shen Z, Naber SP, Hinds PW et al (2014) NDY1/KDM2B functions as a master regulator of polycomb complexes and controls self-renewal of breast cancer stem cells. Cancer Res 74:3935–3946
Kreso A, van Galen P, Pedley NM, Lima-Fernandes E, Frelin C, Davis T, Cao LX, Baiazitov R, Du W, Sydorenko N et al (2014) Self-renewal as a therapeutic target in human colorectal cancer. Nat Med 20:29–36
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Cacerescortes J, Minden M, Paterson B, Caligiuri MA, Dick JE (1994) A cell initiating human acute myeloid-leukemia after transplantation into Scid mice. Nature 367:645–648
Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S et al (2014) Immunogenetics. Chromatin state dynamics during blood formation. Science 345:943–949
Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423:255–260
Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE et al (2009) Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell 15:501–513
Li Q, Eades G, Yao Y, Zhang Y, Zhou Q (2014) Characterization of a stem-like subpopulation in basal-like ductal carcinoma in situ (DCIS) lesions. J Biol Chem 289:1303–1312
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS (2006) Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 66:6063–6071
Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S et al (2011) The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 17:211–215
Liu S, Patel SH, Ginestier C, Ibarra I, Martin-Trevino R, Bai S, McDermott SP, Shang L, Ke J, Ou SJ et al (2012) MicroRNA93 regulates proliferation and differentiation of normal and malignant breast stem cells. PLoS Genet 8:e1002751
Liu SL, Cong Y, Wang D, Sun Y, Deng L, Liu YJ, Martin-Trevino R, Shang L, McDermott SP, Landis MD et al (2014) Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep 2:78–91
Lukacs RU, Memarzadeh S, Wu H, Witte ON (2010) Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 7:682–693
Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, Zheng BJ, Guan XY (2007) Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 132:2542–2556
Ma L, Teruya-Feldstein J, Weinberg RA (2008) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer (vol 449, p. 682, 2007). Nature 455:256
Michalak EM, Nacerddine K, Pietersen A, Beuger V, Pawlitzky I, Cornelissen-Steijger P, Wientjens E, Tanger E, Seibler J, van Lohuizen M et al (2013) Polycomb group gene Ezh2 regulates mammary gland morphogenesis and maintains the luminal progenitor pool. Stem Cells 31:1910–1920
Monteiro J, Gaspar C, Richer W, Franken PF, Sacchetti A, Joosten R, Idali A, Brandao J, Decraene C, Fodde R (2014) Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis 35:2–13
Neven E, De Schutter TM, Dams G, Gundlach K, Steppan S, Buechel J, Passlick-Deetjen J, D’Haese PC, Behets GJ (2014) A magnesium based phosphate binder reduces vascular calcification without affecting bone in chronic renal failure rats. PLoS ONE 9:e107067
Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA (2009) MicroRNAs—the micro steering wheel of tumour metastases. Nat Rev Cancer 9:293–302
Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE (2011) Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469:362–367
Okuda H, Xing F, Pandey PR, Sharma S, Watabe M, Pai SK, Mo YY, Iiizumi-Gairani M, Hirota S, Liu Y et al (2013) miR-7 suppresses brain metastasis of breast cancer stem-like cells by modulating KLF4. Cancer Res 73:1434–1444
Padua Alves C, Fonseca AS, Muys BR, de Barros ELBR, Burger MC, de Souza JE, Valente V, Zago MA, Silva WA Jr (2013) Brief report: the lincRNA Hotair is required for epithelial-to-mesenchymal transition and stemness maintenance of cancer cell lines. Stem Cells 31:2827–2832
Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, Miele L (2010) Targeting Notch to target cancer stem cells. Clin Cancer Res 16:3141–3152
Park EY, Chang E, Lee EJ, Lee HW, Kang HG, Chun KH, Woo YM, Kong HK, Ko JY, Suzuki H et al (2014) Targeting of miR34a-NOTCH1 axis reduced breast cancer stemness and chemoresistance. Cancer Res 74:7573–7582
Pasini D, Bracken AP, Jensen MR, Denchi EL, Helin K (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23:4061–4071
Patani N, Jiang WG, Newbold RF, Mokbel K (2011) Histone-modifier gene expression profiles are associated with pathological and clinical outcomes in human breast cancer. Anticancer Res 31:4115–4125
Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG (2005) Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res 65:5506–5511
Portela A, Esteller M (2010) Epigenetic modifications and human disease. Nat Biotechnol 28:1057–1068
Schade B, Lesurf R, Sanguin-Gendreau V, Bui T, Deblois G, O’Toole SA, Millar EKA, Zardawi SJ, Lopez-Knowles E, Sutherland RL et al (2013) beta-Catenin signaling is a critical event in ErbB2-mediated mammary tumor progression. Cancer Res 73:4474–4487
Schatton T, Murphy GF, Frank NY, Yamaura K, Waaga-Gasser AM, Gasser M, Zhan Q, Jordan S, Duncan LM, Weishaupt C et al (2008) Identification of cells initiating human melanomas. Nature 451:345–349
Schwartz YB, Pirrotta V (2013) A new world of polycombs: unexpected partnerships and emerging functions. Nat Rev Genet 14:853–864
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E et al (2009) Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 138:592–603
Simon JA, Kingston RE (2009) Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10:697–708
Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB (2004) Identification of human brain tumour initiating cells. Nature 432:396–401
Song SJ, Poliseno L, Song MS, Ala U, Webster K, Ng C, Beringer G, Brikbak NJ, Yuan X, Cantley LC et al (2013) MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 154:311–324
Stegle O, Teichmann SA, Marioni JC (2015) Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 16:133–145
Takahashi RU, Miyazaki H, Takeshita F, Yamamoto Y, Minoura K, Ono M, Kodaira M, Tamura K, Mori M, Ochiya T (2015) Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat Commun 6:7318
Tang W, Yu F, Yao H, Cui X, Jiao Y, Lin L, Chen J, Yin D, Song E, Liu Q (2014) miR-27a regulates endothelial differentiation of breast cancer stem like cells. Oncogene 33:2629–2638
Tavares L, Dimitrova E, Oxley D, Webster J, Poot R, Demmers J, Bezstarosti K, Taylor S, Ura H, Koide H et al (2012) RYBP-PRC1 complexes mediate H2A ubiquitylation at polycomb target sites independently of PRC2 and H3K27me3 (vol 148, p. 664, 2012). Cell 149:1647–1648
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329:689–693
Tu Y, Gao X, Li G, Fu H, Cui D, Liu H, Jin W, Zhang Y (2013) MicroRNA-218 inhibits glioma invasion, migration, proliferation, and cancer stem-like cell self-renewal by targeting the polycomb group gene Bmi1. Cancer Res 73:6046–6055
Tzatsos A, Paskaleva P, Lymperi S, Contino G, Stoykova S, Chen Z, Wong KK, Bardeesy N (2011) Lysine-specific demethylase 2B (KDM2B)-let-7-enhancer of zester homolog 2 (EZH2) pathway regulates cell cycle progression and senescence in primary cells. J Biol Chem 286:33061–33069
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8:755–768
Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW et al (2014) CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25:21–36
Wang Y, He L, Du Y, Zhu P, Huang G, Luo J, Yan X, Ye B, Li C, Xia P et al (2015) The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 16:413–425
Wright MH, Robles AI, Herschkowitz JI, Hollingshead MG, Anver MR, Perou CM, Varticovski L (2008) Molecular analysis reveals heterogeneity of mouse mammary tumors conditionally mutant for Brca1. Mol Cancer 7:29
Xing Z, Lin A, Li C, Liang K, Wang S, Liu Y, Park PK, Qin L, Wei Y, Hawke DH et al (2014) lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 159:1110–1125
Xiong YJ, Fang JH, Yun JP, Yang J, Zhang Y, Jia WH, Zhuang SM (2010) Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 51:836–845
Yamamoto S, Wu ZH, Russnes HG, Takagi S, Peluffo G, Vaske C, Zhao X, Vollan HKM, Maruyama R, Ekram MB et al (2014) JARID1B Is a luminal lineage-driving oncogene in breast cancer. Cancer Cell 25:762–777
Yan KS, Chia LA, Li XN, Ootani A, Su J, Lee JY, Su N, Luo YL, Heilshorn SC, Amieva MR et al (2012) The intestinal stem cell markers Bmi1 and Lgr5 identify two functionally distinct populations. Proc Natl Acad Sci USA 109:466–471
Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST (2008) Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 13:153–166
Yeung TM, Gandhi SC, Bodmer WF (2011) Hypoxia and lineage specification of cell line-derived colorectal cancer stem cells. Proc Natl Acad Sci USA 108:4382–4387
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J et al (2007) let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131:1109–1123
Yu F, Deng H, Yao H, Liu Q, Su F, Song E (2010) Mir-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 29:4194–4204
Yu FY, Jiao Y, Zhu YH, Wang Y, Zhu JD, Cui XY, Liu YJ, He YH, Park EY, Zhang HY et al (2012) MicroRNA 34c gene down-regulation via DNA methylation promotes self-renewal and epithelial-mesenchymal transition in breast tumor-initiating cells. J Biol Chem 287:465–473
Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF, Liu F, Pan W, Wang TT, Zhou CC et al (2014) A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell 25:666–681
Zhang Y, Eades G, Yao Y, Li Q, Zhou Q (2012) Estrogen receptor alpha signaling regulates breast tumor-initiating cells by down-regulating miR-140 which targets the transcription factor SOX2. J Biol Chem 287:41514–41522
Zhang H, Cai K, Wang J, Wang X, Cheng K, Shi F, Jiang L, Zhang Y, Dou J (2014a) MiR-7, inhibited indirectly by lincRNA HOTAIR, directly inhibits SETDB1 and reverses the EMT of breast cancer stem cells by downregulating the STAT3 pathway. Stem Cells 32:2858–2868
Zhang JS, Zhang PJ, Wang L, Piao HL, Ma L (2014b) Long non-coding RNA HOTAIR in carcinogenesis and metastasis. Acta Biochim Biophys Sin 46:1–5
Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL et al (2014c) Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46:1170–1172
Zhao C, Chen A, Jamieson CH, Fereshteh M, Abrahamsson A, Blum J, Kwon HY, Kim J, Chute JP, Rizzieri D et al (2009) Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458:776–779
Zhao D, Mo Y, Li MT, Zou SW, Cheng ZL, Sun YP, Xiong Y, Guan KL, Lei QY (2014) NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J Clin Investig 124:5453–5465
Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, Gong C, Chen J, Su F, Zhang Y et al (2011) Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res 17:7105–7115
COMPLIANCE WITH ETHICS GUIDELINES
Suling Liu, Zhiju Zhao, Shu Li and Erwei Song declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
ABBREVIATIONS
BCSCs, breast cancer stem cells; CARM1, coactivator-associated arginine methyltransferase 1; ceRNA, competing endogeneous RNA; ChIP, chromatin immunoprecipitation; CSCs, cancer stem cells; EMT, epithelial-mesenchymal transition; lncRNAs, long non-coding RNAs; MET, mesenchymal-epithelial transition; miRNAs, microRNAs; ncRNA, non-coding RNA; PcG, Polycomb group; PR, progesterone receptor; PRC2, Ploycomb repressive complex 2; SIRT2, sirtuin 2; SP, side population.
Author information
Authors and Affiliations
Corresponding author
Additional information
Zhiju Zhao and Shu Li have contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhao, Z., Li, S., Song, E. et al. The roles of ncRNAs and histone-modifiers in regulating breast cancer stem cells. Protein Cell 7, 89–99 (2016). https://doi.org/10.1007/s13238-015-0199-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-015-0199-4