
ABSTRACT
Voltage-gated sodium (Nav) channels are indispensable membrane elements for the generation and propagation of electric signals in excitable cells. The successes in the crystallographic studies on prokaryotic Nav channels in recent years greatly promote the mechanistic investigation of these proteins and their eukaryotic counterparts. In this paper, we mainly review the progress in computational studies, especially the simulation studies, on these proteins in the past years.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
As one of the fundamental elements in the membrane of excitable cells, voltage-gated sodium (Nav) channels are critical in the generation and propagation of electrical signals in both nerves and muscles (Hille, 2001), and therefore have become therapeutic targets for many neurological disorders, including epilepsy, migraine, neurodegenerative diseases and neuropathic pain (Clare et al., 2000; Dib-Hajj et al., 2010; Mantegazza et al., 2010). Nav channels are members of the voltage-gated ion channels (VGICs), a superfamily of proteins that allow the cross-membrane permeation of various ions under the control of the cross-membrane voltage (Ertel et al., 2000; Catterall et al., 2005; Gutman et al., 2005). Many VGIC superfamily members, particularly the voltage-gated cation channels (VGCCs) that control the transport of Na+, K+ and Ca2+ ions (named as Nav, Kv and Cav channels respectively), adopt similar structural topologies and mechanisms (Hille, 2001; Catterall et al., 2005). The physiological roles and mechanisms of VGCCs have been extensively studied in history, even in the absence of structural information. Among these previous researches, we want to particularly mention two famous biophysical models which have been generally accepted because of their agreement with enormous experimental observations: the Hodgkin-Huxley model and the knock-on model. The former perfectly explains the relationship between VGCCs and the electrical signaling in excitable cells (Hodgkin and Huxley, 1952) while the latter describes the mechanism of ion permeation in these channels (Hodgkin and Keynes, 1955). Moreover, the electrophysiological studies over the past half of century indicate two key characteristics of VGCCs: ion selectivity and voltage gating (Favre et al., 1996; Sontheimer et al., 1996; Sun et al., 1997; Catterall, 2000), both of which are essential for the normal electrical signaling. The former requires the channel to selectively allow the permeation of its target cation in the presence of various other ions, and the latter enables the channel to open and close in response to the variation of cross-membrane voltage.
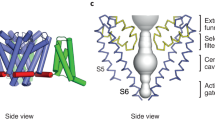
In the late 1990s, the potassium channels became the breakthrough point for the structural studies on VGICs (Doyle et al., 1998; Jiang et al., 2002; Jiang et al., 2003; Long et al., 2005; Long et al., 2007). Unlike the Kv channels that are assembled from four identical subunits, the corresponding four structural units are joined to form a single polypeptide of more than 2000 amino acid residues in eukaryotic Nav channels, a structural organization that brings tremendous additional difficulty on their structural determination. The earliest Nav structure was purified from the electric organ of the eel Electrophorus electricus and was determined by helium-cooled cryo electron microscopy (Sato et al., 2001) with low resolution (19 Å). Almost at the same time, a prokaryotic Nav channel was found to possess similar characteristic of ion selection with its eukaryotic counterpart (Ren et al., 2001). This observation encouraged crystallographic studies on the prokaryotic Nav channels, since they follow the same homotetrameric architecture as potassium channels (Yu and Catterall, 2004; Payandeh and Minor, 2015). A large family of bacterial Nav channels were identified and the high-resolution structures of several orthologs were sequentially determined in recent years (Payandeh et al., 2011; McCusker et al., 2012; Payandeh et al., 2012; Zhang et al., 2012; Shaya et al., 2014). According to the structural insights from these prokaryotic homologs, all Nav channels are supposed to consist of four transmembrane domains (TDs), each of which adopts a unique amino acid sequence (Catterall et al., 2005). Every TD contains six transmembrane helices (TMs), which are named from S1 to S6 sequentially. TMs S1–S4 form the voltage-sensing domain (VSD) that is responsible for perceiving the changes of cross-membrane voltage, while TMs S5–S6 as well as the intervening P-loops and half helices from the four TDs jointly constitute the pore domain (PD) that allows the ion permeation (Fig. 1). Particularly notably, the P-loop from each TD contribute 6–7 residues to form a geometrically constricted region at the extracellular side of the PD, which is mainly responsible for ion selection according to mutagenesis analysis (Favre et al., 1996; Schlief et al., 1996; Sun et al., 1997) and is therefore named as the selectivity filter (SF).

The structural organization of VGCCs. Only the opposing two TDs are shown to facilitate visual recognition. The PD (purple) and VSDs (brown) are connected by a half helix (cyan) between TMs S4 and S5. The SF region within the PD is colored in red. Ligands can bind at both PD and VSDs, as shown by the black arrows
Despite the success in the structural biology, the mechanistic illustration on the ion selection, voltage gating and ion permeation for Nav channels are still lacking, since each crystal structure only reflects one static conformation and therefore cannot describe the structural dynamics that is required for the proper functioning of proteins. As a complementary tool, computer simulations, particularly the molecular dynamics (MD) simulations and the associated theoretical calculations, can overcome such deficiencies and have therefore become essential tools in the investigation of a wide range of chemical and biological systems (Karplus and Petsko, 1990; Karplus and McCammon, 2002). As an example, the pioneering simulation studies on potassium channels performed by Roux and coworkers even anticipated some results that were validated in the subsequent crystallographic studies (Roux and MacKinnon, 1999; Berneche and Roux, 2001). In the past decade, progress in hardware development, improvement in simulation methodologies and refinement of interaction potentials have rendered the possibility to model increasingly complex processes that were intractable previously (Karplus and Kuriyan, 2005). Nowadays, with the maturation of simulation protocols (Phillips et al., 2005; Hess et al., 2008; Brooks et al., 2009; Case et al., 2012), MD simulations about ion channels have gradually reached satisfying agreement with the experimental data (Allen et al., 2006; Gordon et al., 2013), although most of the prominent achievements were obtained for the potassium channels (Roux and MacKinnon, 1999; Shrivastava and Sansom, 2000; Berneche and Roux, 2001; Noskov et al., 2004; Noskov and Roux, 2006; Jensen et al., 2010; Delemotte et al., 2011; Jensen et al., 2012). With the successful determination of bacterial Nav structures in the past years, several groups have conducted MD simulations and proposed models to illustrate the molecular mechanism of Nav channels. In this review, we will go over the progress of these simulation studies in the following four categories: ion binding and permeation, ion selectivity, voltage gating and ligand binding.
ION BINDING AND PERMEATION
Quickly after the first Nav structure (NavAb) was unveiled in 2011, an MD simulation was performed to investigate the interaction between Na+ ions and this protein (Carnevale et al., 2011). The NavAb structure is believed to represent a pre-active state, since its PD is completely closed to cytosol although the four VSDs have reached the active (or “up”) state (Payandeh et al., 2011). The overall protein structure was stable during the 140 ns equilibrium simulation, with the cytoplasmic entrance of the PD remaining closed to the cytoplasmic ions. However, the periplasmic Na+ ions quickly entered the SF and bound at two favorable sites, which were named as SHFS and SCEN respectively. Interestingly, Na+ ions bound at these sites are coordinated by the oxygen atoms from both protein and water. Such an asymmetric binding pattern with incomplete dehydration is different from the observations in the potassium channels where the K+ ions are completely dehydrated and form strong coordination with eight symmetrically positioned carbonyl oxygen atoms of the protein (Zhou et al., 2001).
The unique ion binding behavior of the sodium channels was later confirmed by a series of subsequent simulations (Corry and Thomas, 2012; Furini and Domene, 2012; Qiu et al., 2012; Chakrabarti et al., 2013; Boiteuxa et al., 2014). According to both single-ion and multi-ion profiles of the potential of mean force (PMF) evaluated along the ion permeation pathway within the SF (Corry and Thomas, 2012; Furini and Domene, 2012; Qiu et al., 2012), several favorable ion binding sites were rigorously identified and were named as SHFS, SCEN and SIN in the order from the periplasmic entrance to the central cavity (Fig. 2). Moreover, in all equilibrium simulations, the protein structures were stable and the periplasmic Na+ ions quickly entered the unoccupied SF together with coordinated water molecules. The ion initially arrived at the site SHFS and then permeated further to the more favorable site SCEN, where the ion could either transit backward to SHFS intermittently or move forward to SIN and even the central cavity within the nanosecond timescale of simulations (Furini and Domene, 2013). The site SHFS was named to reflect the high field strength, since it is surrounded by the carboxylate groups from four Glu residues (or the EEEE motif). Notably, this site is of great interest since the corresponding residues in all eukaryotic homologs are highly conserved (as Asp, Glu, Lys and Ala respectively, or the DEKA motif) and have been reported as the key residues to determine the ion selectivity (Catterall, 2000). Despite the electrostatic repulsion between the acidic residues at SHFS in NavAb, the simulations indicated that at least three of the Glu residues should be deprotonated in order to maintain the efficient ion permeation (Boiteuxa et al., 2014; Furini et al., 2014). In the absence of cations, these Glu side groups uniformly point towards the periplasm to minimize the unfavorable repulsion (Chakrabarti et al., 2013). After the cation arrives at this site, one or two Glu side groups may swing to a partially downward orientation (Boiteuxa et al., 2014) to facilitate ion diffusion.

The ion binding sites identified in the SF of Na v channels. (A) The SF region is labeled by a cyan frame. (B) The schematic ion binding sites in NavAb. (C) The schematic ion binding sites in NavRh. (D) Aligned amino acid sequences for the SF region in various prokaryotic Nav channels as well as eukaryotic Nav and Cav channels. The four structural units in the eukaryotic channels are labeled as I, II, III and IV, respectively
With the determination of more Nav structures, some groups began to simulate these orthologous proteins. The NavRh structure was claimed to represent the inactive state, since the side chains of four Ser residues occlude the ion permeation pathway in the SF of the crystal structure (Zhang et al., 2012). In the simulation, however, the collapsed SF region quickly became ion accessible by the self-adjustment of side chains (Zhang et al., 2013). Although the NavMs crystal structure lacks the VSDs, its PD exhibits a continuous pore that geometrically allows ion permeation (McCusker et al., 2012), which enables the evaluation of conductance of the channel from MD simulations. The electric current and the conductance estimated in the subsequent simulations agreed well with the values obtained from electrophysiological experiments (Ulmschneider et al., 2013), which further reinforces the power of MD simulation in the study of ion channels. In spite of the minor difference in details, simulations on both orthologs echoed the observations in the simulations of NavAb: the SF contains multiple ion binding sites and cations bind in an asymmetrical manner with incomplete dehydration (Ulmschneider et al., 2013; Zhang et al., 2013).
After the identification of ion binding sites, the ion permeation patterns were investigated in numerous simulations (Carnevale et al., 2011; Furini and Domene, 2012; Qiu et al., 2012; Chakrabarti et al., 2013; Ke et al., 2013; Stock et al., 2013; Ulmschneider et al., 2013; Zhang et al., 2013; Bagnéris et al., 2014; Boiteuxa et al., 2014). Nearly all studies indicated the presence of weak coupling between the 2–3 permeating Na+ ions accommodated within the SF. This permeation mechanism is different from that of the potassium channels where K+ ions move in a highly concerted manner, and is therefore proposed as a loosely coupled knock-on mechanism. Since the closed conformation of NavAb hinders the observation of continuous ion transport, Klein and Treptow as well as their coworkers modeled its open conformation based on a homologous structure from the potassium channel (Amaral et al., 2012). Their subsequent simulations suggested that Na+ ions permeate in pairs within the SF for most of the inward ion flows and that these ion pairs transit among five major configurations (Stock et al., 2013). According to the PMF calculation, the energy barrier for the overall permeation process of such a two-ion mode is ~3 kcal/mol (Stock et al., 2013). However, a third ion could participate in the outward ion flows, which leads to an average occupancy of 2.3 ions within the SF (Stock et al., 2013). Similarly, Wallace and coworkers simulated the continuous inward ion conduction in the open NavMs structure with a constant electric field applied across the membrane, and found that on average 1.8 Na+ ions are involved in the ion permeation within the SF (Ulmschneider et al., 2013). These simulations supplemented the weakly coupled knock-on mechanism with molecular details. Apart from the weakened coupling between permeating ions, the water flux through the sodium channels is uncorrelated with ion flux (Ulmschneider et al., 2013), in sharp contrast to the potassium channels where the rate of ionic flow limits the maximum water flux through the channel (Saparov and Pohl, 2004; Jensen et al., 2010).
ION SELECTIVITY
Selective conduction of Na+ ions is mainly conferred by the SF region, which is highly conserved among all known Nav channels (Catterall et al., 2005). The mechanism of ion selection has been theoretically investigated decades before, in the absence of any crystal structures. Eisenman ascribed the ion selection to the field strength of the ion-coordinating groups and predicted that partitioning of the better solvated Na+ ions into the SF of potassium channel is unfavorable (Eisenman and Horn, 1983; Hille, 2001). With the great success of crystallographic studies on ion channels, the potassium channels soon became a good prototype for comprehending the mechanism of selectivity for the various ions. The SF regions of potassium channels are geometrically confined so that the ions have to be completely dehydrated when entering this region. The de-solvation of cations is energetically compensated by the coordination with symmetrically distributed carbonyl oxygen atoms donated by the P-loop residues. Naïve structural analysis suggested that the K+ ions are snugly coordinated by eight carbonyl oxygen atoms at the most favorable distance and that both the number of coordinating groups and the coordinating distances in the crystal structure render the site’s preference of K+ binding (Doyle et al., 1998; Zhou et al., 2001). Since the structural dynamics were completely neglected in such naïve analysis, Roux and coworkers then conducted MD simulations on the KcsA channel and evaluated the difference between ion binding affinities using the free energy perturbation (FEP) method (Noskov et al., 2004). According to their calculations, the electrostatic repulsion between the carbonyl groups partially restrains the structural fluctuation of the ion binding sites within the SF, and therefore prevents coordinating groups from interacting with Na+ ions at shorter and more favorable distances (Noskov et al., 2004). In numerous following studies, people tended to explain the ion selectivity in terms of the number and field strength of coordinating groups by assuming that they form a homogeneous coordinating environment (Noskov et al., 2004; Bostick and Brooks, 2007; Fowler et al., 2008).
The emergence of high-resolution crystal structures for the sodium channels broke the theoretical foundation of such analysis, both because the sodium channels use sidechain atoms rather than the backbone carbonyl groups to coordinate ions and because the ion coordinating groups are heterogeneously distributed. Furini and Domene evaluated the 1D PMF profiles of Na+ and K+ ions along the permeation pathway in the SF of NavAb. Although the two types of ions bind at similar sites (SCEN and SHFS), the free energy barrier for K+ permeation is 2–3 kcal/mol higher, which relatively disfavors the K+ conduction in kinetics. Such a small difference in the free energy barrier agrees with the mild Na+/K+ selectivity of the NavAb channel (Furini and Domene, 2012). Corry and Thomas proposed an alternative hypothesis according to their simulations on NavAb, by assuming that a water molecule bridged between the ion and the carboxylate group of one Glu residue at the site SHFS confines the pore size at this site and therefore prohibits the passage of the larger K+ ions (Corry and Thomas, 2012). The subsequent studies examined the equilibrium behavior of NavAb by performing MD simulations in microseconds and reported more evenly distributed free energy landscape for Na+ ions (Chakrabarti et al., 2013; Boiteuxa et al., 2014). The free energy barrier between various binding modes are effectively reduced by the downward swinging of the Glu side groups at the site SHFS, a phenomenon that was neglected in previous simulations with a large negative internal voltage applied across the membrane (Stock et al., 2013; Ulmschneider et al., 2013). As pointed out in previous theoretical studies, reasonable structural fluctuations are required for the mechanistic understanding of ion selection (Allen et al., 2004), and the interplay between the attractive ion-ligand and repulsive ligand-ligand interactions jointly govern the Na+/K+ selectivity in the flexible binding sites (Noskov and Roux, 2006).
Besides the interference from the outward flow of cytoplasmic K+ ions, the periplasmic Ca2+ ions may compete with Na+ ions for the inward permeation through the sodium channels. Therefore, the selection of Na+ vs. Ca2+ ions in Nav channels is of particular interest, since Ca2+ ions frequently act as secondary messengers and their cross-membrane leaking may cause cell disorder. Interestingly, eukaryotic Ca2+ channels have the EEEE motif at the constriction site (similar to NavAb) but they strongly prefer Ca2+ over Na+ ions (Sather and McCleskey, 2003). Dudev and Lim investigated this phenomenon using a reduced model system and proposed the protein matrix as the decisive factor for Ca2+ vs. Na+ selectivity in the SF of calcium channels (Dudev and Lim, 2012). On the other hand, simulations on NavAb suggested that Ca2+ translocation lacks the knock-on mechanism for efficient permeation although they can favorably bind at sites SCEN and SHFS (Ke et al., 2013). In the SF of NavRh, the site SHFS is composed of four Ser residues and Glu residues are located three residues away on the periplasmic side to constitute a site called IonEX (Fig. 2C and 2D). Our simulations on NavRh suggested that the extremely strong binding preference of Ca2+ ions at IonEX renders a huge free energy barrier (~10 kcal/mol) which prevents the Ca2+ ions from further moving towards the site SCEN (Zhang et al., 2013), an explanation consistent with the experimental observation that periplasmic Ca2+ ions block the Na+ flux in NavRh (Zhang et al., 2012).
Note that most of these proposed mechanisms for ion selectivity are applicable to the prokaryotic Nav channels only, since eukaryotic channels have a highly conserved DEKA motif rather than the EEEE motif at the position equivalent to the site SHFS of NavAb and since the DEKA motif has been identified to be the major determinant of the ion selection in most eukaryotic Nav channels (Favre et al., 1996; Catterall, 2000). Lipkind and Fozzard generated a homology model for the eukaryotic Nav channels in the lack of any available structures for sodium channels. Based on the extensive experimental data and their MD simulations on this structural model, they proposed that electrostatic competition between the alkali cations and the residue pair of Glu and Lys (E and K in the DEKA motif) finely tuned the preference of ion binding at SHFS towards Na+ ions in eukaryotic Nav channels (Lipkind and Fozzard, 2008). Subsequently, Dudev and Lim investigated the factors controlling Na+/K+ selectivity in sodium channels using reduced models and proposed that the architecture, chemical composition and physicochemical properties of the SF in eukaryotic sodium channels jointly contribute to the Na+/K+ selectivity (Dudev and Lim, 2010). After the structural determination of NavRh, we modeled the structure of eukaryotic Nav channels by mutating the Ser residues at the constriction site in NavRh to form a DEKA motif. By conducting MD simulations on this structural model and evaluating the 2D PMF profiles of ion binding at this site, we successfully identified the essential roles of Lys and Asp/Glu in determining the Na+/K+ selectivity. The positively charged Lys repels the cation to bind at the position sandwiched between the two carboxylate groups of Asp and Glu, a location where the cations are coordinated by these carboxylate oxygen atoms at short distances that are favorable for Na+ binding (Xia et al., 2013). In a recent work, Dudev and Lim discussed the evolution of eukaryotic VGCCs with their ion preference converted from Ca2+-favored to Na+-selective (Dudev and Lim, 2014).
VOLTAGE GATING
VGCCs needs to open and close in response to the variation of cross-membrane potentials and the perception on voltages is accomplished by the VSDs, which are composed of TMs S1–S4 in each TD (Yu and Catterall, 2004). Once the changes in voltage is sensed by the VSDs, these domains experience large-scale conformational change among the resting, active and inactive states, which subsequently triggers the PD structure to transit between the open and closed states (Hodgkin and Huxley, 1952; Isacoff et al., 2013). The combination of the conformational changes of both the VSD and PD following the voltage change is called the voltage gating, which is indispensable for the initiation and propagation of the action potentials in mammals. The TM S4 has been supposed to be the key element for voltage sensing. This helix carries 4–5 highly conserved positively charged residues (called gating charges), which tend to move in the extracellular direction and generate gating current when the membrane is depolarized (Kuzmenkin et al., 2004; Blanchet and Chahine, 2007; DeCaen et al., 2009; Yarov-Yarovoy et al., 2012; Zhang and Yan, 2013). Among the numerous models proposed to describe the movement of S4 helix, the most famous one is the sliding-helix mechanism (Catterall, 2010), which stated that in response to the membrane depolarization, the S4 helix slides within the membrane towards the extracellular side by the sequential formation and break of ion pairs composed of the gating charges on S4 and acidic residues on S1–S3 (Vargas et al., 2012). A conserved Phe residue in the hydrophobic constriction site (HCS) and two negative charges have been implicated as a gating charge transfer center in Kv channels (Tao et al., 2010). Nevertheless, the VSDs of the VGCCs frequently form an hourglass-shaped structure within the membrane that essentially functions as a voltage-dependent Arg side-chain transporter (Payandeh and Minor, 2015).
The structural determination of prokaryotic and eukaryotic Kv channels (KvAP and Kv1.2/Kv2.1 chimera channel) (Jiang et al., 2003; Long et al., 2007) stimulated the interests of computational biologists to simulate their voltage gating process. Delemotte et al. conducted MD simulations on the VSD of Kv1.2 and analyzed effect of residue mutation on the gating behavior (Delemotte et al., 2010). Subsequently, Jensen et al. simulated the whole protein (VSD + PD) for milliseconds and directly observed the complete protein conformational change in the hyperpolarization and depolarization conditions (Jensen et al., 2012). They then proposed a detailed mechanistic model to illustrate the molecular events occurred during the inactivation and activation processes of Kv channels. In a recent work, Delemotte et al. found the reaction pathway for the first step of structural transition during the activation of the VSDs in Kv1.2 and rigorously evaluated the PMF profile along the reaction coordinate (Delemotte et al., 2015). Their calculations were consistent with the phenomenological models adopted to illustrate the observations and measurements in physiological experiments.
The voltage gating in Nav channels has been lagging behind for a long time, because of the late emergence of high-resolution structures. In the absence of the structural information for Nav channels, Catterall and coworkers took the KvAP and Kv1.2 structures as templates and used the Rosetta program to model the structure basis for gating charge movement in the VSD of the NaChBac channels during activation (DeCaen et al., 2008; DeCaen et al., 2009; Yarov-Yarovoy et al., 2012). Since the structural determination of NavAb, which is believed to reflect a pre-active conformational state (Payandeh et al., 2011), Klein and Treptow as well as their coworkers have conducted a series of simulations to investigate the gating mechanism of Nav channels. Benefited from their previous success in the simulation of Kv channels (Treptow and Tarek, 2006; Delemotte et al., 2011), they modeled the activated-open and resting-closed states of NavAb using the experimental and simulated conformations of Kv1.2 as templates, and simulated the structural transition of NavAb from the activated-closed state (in the crystal structure) towards these two states (Amaral et al., 2012). In another work, they modeled the structure of NaChBac based on NavAb and simulated the conformational change of this model structure upon activation (Barber et al., 2012).
LIGAND BINDING
The identification of the different subtypes in pathophysiology of Nav channels has provided a rational basis for selective intervention in clinic treatment and some drugs have shown therapeutic value in Nav channelopathies (Ragsdale and Avoli, 1998; Mantegazza et al., 2010; Payandeh et al., 2012; Bagal et al., 2013; McCormack et al., 2013; Bagnéris et al., 2014). Many pathogenic Nav channels have been intensively investigated by in vitro mutagenesis and electrophysiological experiments. On the other hand, an array of highly potent and selective neurotoxins have proven the lethal effects by destroying the normal functional modulation of Nav channels, including pore blockage, over-stabilization of the opened pore as well as alteration on the movement of voltage sensors and on the gating mechanism (Hille, 2001; Catterall et al., 2005; Dib-Hajj et al., 2010; Stevens et al., 2011; Knapp et al., 2012; Moreau et al., 2014; Thottumkara et al., 2014; Kalia et al., 2015; Payandeh and Minor, 2015).
Computational studies, including MD simulations, Brownian dynamics and molecular docking, have become new tools to study the roles of various subtypes of Nav channels in pathophysiology at an atomistic level (Gordon et al., 2013). Even before the systematic crystallographic studies on VGICs, Lipkind and Fozzard had developed a reasonable molecular model to supposedly describe the binding pockets for tetrodotoxin (TTX) and saxitoxin (STX) in eukaryotic Nav channels (Lipkind and Fozzard, 1994). At present, some toxin-channel interactions can be investigated at atomistic levels using the available high-resolution structures of the prokaryotic Nav channels, by considering that the SF region is structurally stable during the evolution of Nav channels (Tikhonov and Zhorov, 2012). Although some bacterial Nav channels are resistant to TTX (Ren et al., 2001), MD simulations indicated that μ-conotoxins is effective in inhibiting the NavAb structure (Chen and Chung, 2012a), which was further confirmed in the electrophysiological experiment (Stevens et al., 2012). In the subsequent simulations, Chen and Chung identified the functional surface of β-toxins to interact with Nav channels (Chen and Chung, 2012b). This computationally predicted function surface appeared to overlap with that of α-toxins determined experimentally (Gordon et al., 2007). In a later work, they used MD simulations to investigate the interaction between TTX and a model structure of Nav1.4, and found that TTX may occlude the channel entrance by forming a network of hydrogen bonds in the outer lumen of the SF (Chen and Chung, 2014), consistent with the experimental evidence that the charged outer ring of the SF is critical in the TTX blockage (Penzotti et al., 1998). Recently, Allen and coworkers conducted microseconds of MD simulations to comprehensively analyze the interactions between lipophilic drugs (benzocaine and phenytoin) and NavAb (Boiteuxa et al., 2014). They finally identified two different drug-access pathways, which may provide insight into mechanistic studies on the Nav channel inhibition and may assist future drug development.
OUTLOOK
Computer simulations have experienced impressively rapid development in the past decade. On one hand, the conventional simulation time has risen from nanoseconds to microseconds or even milliseconds, due to the improvement in both computer hardware and algorithms. On the other hand, numerous new methods have been developed to facilitate overcoming the energetic barrier and therefore more efficient sampling in the conformational space (Piccinini et al., 2008). We could only list a few here: accelerated molecular dynamics (Hamelberg et al., 2004) and metadynamics (Laio and Gervasio, 2008) to enhance the conformational sampling, the STRING method (Maragliano et al., 2006) to find the minimal free energy path (MFEP) during the structural transition, the orthogonal space random walk (Zheng et al., 2008) and adaptive umbrella sampling (Bartels and Karplus, 1998) to allow the identification of an ensemble of reaction pathways and reliable free-energy estimation, etc. Improvements in both facets jointly enable the observation of large-scale conformational changes that were unmanageable previously, particularly the voltage gating process of Nav channels. Moreover, simulations with the polarizable force field (Jiang et al., 2011) has been available to consider the charge transfer between ion and proteins during the transport process, which could further improve the accuracy of observations and calculations on the ion selectivity and the ion permeation mechanism. The molecular mechanisms of Nav channels are expected to be better illustrated in the future with these new techniques.
Despite the above progresses, the simulation studies highly depend on the crystallographic achievement. In the absence of high-resolution structures, the simulations have to start from a modeled structure, which eventually weakens the reliability of the simulation observations. The simulation studies on Nav channels are awaiting the determination of more structures, especially eukaryotic structures. The eukaryotic Nav channels have the unique DEKA motif in the SF that is absent in the prokaryotic counterparts. Consequently, the molecular mechanisms of ion permeation and ion selection in eukaryotic Nav channels cannot be accurately illustrated from the simulations on their prokaryotic homologs. On the other hand, the structural determination of other conformational states, especially the resting state conformation, will greatly facilitate the simulation studies on the voltage gating mechanism. All currently available structures have their VSDs maintained at the “up” position. Therefore, a resting state conformation is required for a comprehensive understanding on the overall cycle of conformational change. The presence of representative structures for both the active and resting states will greatly improve the efficiency of free energy analysis for the activation and de-activation processes. In addition, the computational drug development on Nav channels requires the determination of structures from higher-level organisms, especially mammals. In summary, with more structures of Nav channels solved in the near future, computer simulations are expected to play more important roles in connecting the molecular details and macroscopic experimental measurements for these channels.
References
Allen TW, Andersen OS, Roux B (2004) On the importance of atomic fluctuations, protein flexibility, and solvent in ion permeation. J Gen Physiol 124:679–690
Allen TW, Andersen OS, Roux B (2006) Molecular dynamics—potential of mean force calculations as a tool for understanding ion permeation and selectivity in narrow channels. Biophys Chem 124:251–267
Amaral C, Carnevale V, Klein ML, Treptow W (2012) Exploring conformational states of the bacterial voltage-gated sodium channel NavAb via molecular dynamics simulations. Proc Natl Acad Sci USA 109:21336–21341
Bagal S, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI et al (2013) Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem 56:593–624
Bagnéris C, DeCaen PG, Naylor CE, Pryde DC, Nobeli I, Clapham DE, Wallace BA (2014) Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc Natl Acad Sci USA 111(23):8428–8433
Barber AF, Carnevale V, Raju SG, Amaral C, Treptow W, Klein ML (2012) Hinge-bending motions in the pore domain of a bacterial voltage-gated sodium channel. Biochim Biophys Acta 1818:2120–2125
Bartels C, Karplus M (1998) Probability distributions for complex systems: adaptive umbrella sampling of the potential energy. J Phys Chem B 102:865–880
Berneche S, Roux B (2001) Energetics of ion conduction through the K+ channel. Nature 414:73–77
Blanchet J, Chahine M (2007) Accessibility of four arginine residues on the S4 segment of the Bacillus halodurans sodium channel. J Membr Biol 215:169–180
Boiteuxa C, Vorobyov I, French RJ, French C, Yarov-Yarovoy V, Allen TW (2014a) Local anesthetic and antiepileptic drug access and binding to a bacterial voltage-gated sodium channel. Proc Natl Acad Sci USA 111:13057–13062
Boiteuxa C, Vorobyov I, Allen TW (2014b) Ion conduction and conformational flexibility of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci USA 111:3454–3459
Bostick DL, Brooks CL III (2007) Selectivity in K+ channels is due to topological control of the permeant ion’s coordinated state. Proc Natl Acad Sci USA 104:9260–9265
Brooks BR, Brooks CL III, Mackerell AD Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S et al (2009) CHARMM: the biomolecular simulation program. J Comput Chem 30:1545–1614
Carnevale V, Treptow W, Klein ML (2011) Sodium ion binding sites and hydration in the lumen of a bacterial ion channel from molecular dynamics simulations. J Phys Chem Lett 2:2504–2508
Case D, Darden T, Cheatham T III, Simmerling C, Wang J, Duke R, Luo R, Walker R, Zhang W, Merz K (2012) AMBER 12, vol 1. University of California, San Francisco, p 3
Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26:13–25
Catterall WA (2010) Ion channel voltage sensors: structure, function, and pathophysiology. Neuron 67:915–928
Catterall WA, Goldin AL, Waxman SG (2005) International Union of Pharmacology. XLVII. Nomenclature and structure–function relationships of voltage-gated sodium channels. Pharmacol Rev 57:397–409
Chakrabarti N, Ing C, Payandeh J, Zheng N, Catterall WA, Pomès R (2013) Catalysis of Na+ permeation in the bacterial sodium channel NaVAb. Proc Natl Acad Sci USA 110:11331–11336
Chen R, Chung SH (2012a) Binding modes of muconotoxin to the bacterial sodium channel (NaVAb). Biophys J 102:483–488
Chen R, Chung SH (2012b) Conserved functional surface of antimammalian scorpion beta-toxins. J Phys Chem B 116:4796–4800
Chen R, Chung SH (2014) Mechanism of tetrodotoxin block and resistance in sodium channels. Biochem Biophys Res Commun 446:370–374
Clare JJ, Tate SN, Nobbs M, Romanos MA (2000) Voltage-gated sodium channels as therapeutic targets. Drug Discov Today 5:506–520
Corry B, Thomas M (2012) Mechanism of ion permeation and selectivity in a voltage gated sodium channel. J Am Chem Soc 134:1840–1846
DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, Catterall WA (2008) Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci USA 105:15142–15147
DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, Catterall WA (2009) Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci USA 106:22498–22503
Delemotte L, Treptow W, Klein ML, Tarek M (2010) Effect of sensor domain mutations on the properties of voltage-gated ion channels: molecular dynamics studies of the potassium channel Kv1.2. Biophys J 99:L72–L74
Delemotte L, Tarek M, Klein ML, Amaral C, Treptow W (2011) Intermediate states of the Kv1.2 voltage sensor from atomistic molecular dynamics simulations. Proc Natl Acad Sci USA 108:6109–6114
Delemotte L, Kasimova MA, Klein ML, Tarek M, Carnevale V (2015) Free-energy landscape of ion-channel voltage-sensor-domain activation. Proc Natl Acad Sci USA 112:124–129
Dib-Hajj SD, Cummins TR, Black JA, Waxman SG (2010) Sodium channels in normal and pathological pain. Annu Rev Neurosci 33:325–347
Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77
Dudev T, Lim C (2010) Factors governing the Na(+) vs K(+) selectivity in sodium ion channels. J Am Chem Soc 132:2321–2332
Dudev T, Lim C (2012) Why voltage-gated Ca2+ and bacterial Na+ channels with the same EEEE motif in their selectivity filters confer opposite metal selectivity. Phys Chem Chem Phys 14:12451–12456
Dudev T, Lim C (2014) Evolution of eukaryotic ion channels: principles underlying the conversion of Ca2+-selective to Na+-selective channels. J Am Chem Soc 136:3553–3559
Eisenman G, Horn R (1983) Ionic selectivity revisited—the role of kinetic and equilibrium processes in ion permeation through channels. J Membr Biol 76:197–225
Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L et al (2000) Nomenclature of voltage-gated calcium channels. Neuron 25:533–535
Favre I, Moczydlowski E, Schild L (1996) On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys J 71:3110–3125
Fowler PW, Tai K, Sansom MS (2008) The selectivity of K+ ion channels: testing the hypotheses. Biophys J 95:5062–5072
Furini S, Domene C (2012) On conduction in a bacterial sodium channel. PLoS Comput Biol 8:e1002476
Furini S, Domene C (2013) K(+) and Na(+) conduction in selective and nonselective ion channels via molecular dynamics simulations. Biophys J 105:1737–1745
Furini S, Barbini P, Domene C (2014) Effects of the protonation state of the EEEE motif of a bacterial NaD-channel on conduction and pore structure. Biophys J 106:2175–2183
Gordon D, Karbat I, Ilan N, Cohen L, Kahn R, Gilles N, Dong K, Stuhmer W, Tytgat J, Gurevitz M (2007) The differential preference of scorpion alpha-toxins for insect or mammalian sodium channels: implications for improved insect control. Toxicon 49:452–472
Gordon D, Chen R, Chung SH (2013) Computational methods of studying the binding of toxins from venomous animals to biological ion channels: theory and applications. Physiol Rev 93:767–802
Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stuhmer W et al (2005) International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 57:473–508
Hamelberg D, Mongan J, McCammon JA (2004) Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys 120:11919–11929
Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447
Hille B (2001) Ion channels of excitable membranes, vol 507. Sinauer, Sunderland
Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117:500–544
Hodgkin AL, Keynes RD (1955) The potassium permeability of a giant nerve fibre. J Physiol 128:61–88
Isacoff EY, Jan LY, Minor DL (2013) Conduits of life’s spark: a perspective on ion channel research since the birth of neuron. Neuron 80:658–674
Jensen MO, Borhani DW, Lindorff-Larsen K, Maragakis P, Jogini V, Eastwood MP, Dror RO, Shaw DE (2010) Principles of conduction and hydrophobic gating in K+ channels. Proc Natl Acad Sci USA 107:5833–5838
Jensen MO, Jogini V, Borhani DW, Leffler AE, Dror RO, Shaw DE (2012) Mechanism of voltage gating in potassium channels. Science 336:229–233
Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417:515–522
Jiang Y, Lee A, Chen J, Ruta V, Cadene M, Chait BT, MacKinnon R (2003) X-ray structure of a voltage-dependent K+ channel. Nature 423:33–41
Jiang W, Hardy DJ, Phillips JC, MacKerell AD, Schulten K, Roux B (2011) High-performance scalable molecular dynamics simulations of a polarizable force field based on classical Drude oscillators in NAMD. J Phys Chem Lett 2:87–92
Kalia J, Milescu M, Salvatierra J, Wagner J, Klint JK, King GF, Olivera BM, Bosmans F (2015) From foe to friend: using animal toxins to investigate ion channel function. J Mol Biol 427:158–175
Karplus M, Kuriyan J (2005) Molecular dynamics and protein function. Proc Natl Acad Sci USA 102:6679–6685
Karplus M, McCammon JA (2002) Molecular dynamics simulations of biomolecules. Nat Struct Mol Biol 9:646–652
Karplus M, Petsko GA (1990) Molecular dynamics simulations in biology. Nature 347:631–639
Ke S, Zangerl EM, Stary-Weinzinger A (2013) Distinct interactions of Na+ and Ca2+ ions with the selectivity filter of the bacterial sodium channel Na(V)Ab. Biochem Biophys Res Commun 430:1272–1276
Knapp O, McArthur JR, Adams DJ (2012) Conotoxins targeting neuronal voltage-gated sodium channel subtypes: potential analgesics? Toxins (Basel) 4:1236–1260
Kuzmenkin A, Bezanilla F, Correa AM (2004) Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J Gen Physiol 124:349–356
Laio A, Gervasio FL (2008) Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep Prog Phys 71:126601
Lipkind GM, Fozzard HA (1994) A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys J 66:1–13
Lipkind GM, Fozzard HA (2008) Voltage-gated Na channel selectivity: the role of the conserved domain III lysine residue. J Gen Physiol 131:523–529
Long SB, Campbell EB, Mackinnon R (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309:897–903
Long SB, Tao X, Campbell EB, MacKinnon R (2007) Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450:376–382
Mantegazza M, Curia G, Biagini G, Ragsdale DS, Avoli M (2010) Voltage-gated sodium channels as therapeutic targets in epilepsy and other neurological disorders. Lancet Neurol 9:413–424
Maragliano L, Fischer A, Vanden-Eijnden E, Ciccotti G (2006) String method in collective variables: minimum free energy paths and isocommittor surfaces. J Chem Phys 125:24106
McCormack K, Santos S, Chapman ML, Krafte DS, Marron BE, West CW, Krambis MJ, Antonio BM, Zellmer SG, Printzenhoff D et al (2013) Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci USA 110:E2724–E2732
McCusker EC, Bagneris C, Naylor CE, Cole AR, D’Avanzo N, Nichols CG, Wallace BA (2012) Structure of a bacterial voltage-gated sodium channel pore reveals mechanisms of opening and closing. Nat Commun 3:1102
Moreau A, Gosselin-Badaroudine P, Chahine M (2014) Biophysics, pathophysiology, and pharmacology of ion channel gating pores. Front Pharmacol 5:53
Noskov SY, Roux B (2006) Ion selectivity in potassium channels. Biophys Chem 124:279–291
Noskov SY, Berneche S, Roux B (2004) Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature 431:830–834
Payandeh J, Minor DL Jr (2015) Bacterial voltage-gated sodium channels (BacNas) from the soil, sea, and salt lakes enlighten molecular mechanisms of electrical signaling and pharmacology in the brain and heart. J Mol Biol 427:3–30
Payandeh J, Scheuer T, Zheng N, Catterall WA (2011) The crystal structure of a voltage-gated sodium channel. Nature 475:353–358
Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, Catterall WA (2012) Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486:135–139
Penzotti JL, Fozzard HA, Lipkind GM, Dudley SC Jr (1998) Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J 75:2647–2657
Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Piccinini E, Ceccarelli M, Affinito F, Brunetti R, Jacoboni C (2008) Biased molecular simulations for free-energy mapping: a comparison on the KcsA channel as a test case. J Chem Theory Comput 4:173–183
Qiu H, Shen R, Guo W (2012) Ion solvation and structural stability in a sodium channel investigated by molecular dynamics calculations. Biochim Biophys Acta 1818:2529–2535
Ragsdale DS, Avoli M (1998) Sodium channels as molecular targets for antiepileptic drugs. Brain Res Rev 26:16–28
Ren D, Navarro B, Xu H, Yue L, Shi Q, Clapham DE (2001) A prokaryotic voltage-gated sodium channel. Science 294:2372–2375
Roux B, MacKinnon R (1999) The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science 285:100–102
Saparov SM, Pohl P (2004) Beyond the diffusion limit: water flow through the empty bacterial potassium channel. Proc Natl Acad Sci USA 101:4805–4809
Sather WA, McCleskey EW (2003) Permeation and selectivity in calcium channels. Annu Rev Physiol 65:133–159
Sato C, Ueno Y, Asai K, Takahashi K, Sato M, Engel A, Fujiyoshi Y (2001) The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature 409:1047–1051
Schlief T, Schonherr R, Imoto K, Heinemann SH (1996) Pore properties of rat brain II sodium channels mutated in the selectivity filter domain. Eur Biophys J 25:75–91
Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, Minor DL (2014) Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J Mol Biol 426:467–483
Shrivastava IH, Sansom MSP (2000) Simulations of ion permeation through a potassium channel: molecular dynamics of KcsA in a phospholipid bilayer. Biophys J 78:557–570
Sontheimer H, Black JA, Waxman SG (1996) Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci 19:325–331
Stevens M, Peigneur S, Tytgat J (2011) Neurotoxins and their binding areas on voltage-gated sodium channels. Front Pharmacol 2:71
Stevens M, Peigneur S, Dyubankova N, Lescrinier E, Herdewijn P, Tytgat J (2012) Design of bioactive peptides from naturally occurring muconotoxin structures. J Biol Chem 287:31382–31392
Stock L, Delemotte L, Carnevale V, Treptow W, Klein ML (2013) Conduction in a biological sodium selective channel. J Phys Chem B 117:3782–3789
Sun YM, Favre I, Schild L, Moczydlowski E (1997) On the structural basis for size-selective permeation of organic cations through the voltage-gated sodium channel—effect of alanine mutations at the DEKA locus on selectivity, inhibition by Ca2+ and H+, and molecular sieving. J Gen Physiol 110:693–715
Tao X, Lee A, Limapichat W, Dougherty DA, MacKinnon R (2010) A gating charge transfer center in voltage sensors. Science 328:67–73
Thottumkara AP, Parsons WH, Du Bois J (2014) Saxitoxin. Angew Chem Int Ed Engl 53:5760–5784
Tikhonov DB, Zhorov BS (2012) Architecture and pore block of eukaryotic voltage-gated sodium channels in view of NavAb bacterial sodium channel structure. Mol Pharmacol 82:97–104
Treptow W, Tarek M (2006) Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys J 90:L64–L66
Ulmschneider MB, Bagneris C, McCusker EC, DeCaen PG, Delling M, Clapham DE, Ulmschneider JP, Wallace BA (2013) Molecular dynamics of ion transport through the open conformation of a bacterial voltage-gated sodium channel. Proc Natl Acad Sci USA 110:6364–6369
Vargas E, Yarov-Yarovoy V, Khalili-Araghi F, Catterall WA, Klein ML, Tarek M, Lindahl E, Schulten K, Perozo E, Bezanilla F et al (2012) An emerging consensus on voltage-dependent gating from computational modeling and molecular dynamics simulations. J Gen Physiol 140:587–594
Xia M, Liu H, Li Y, Yan N, Gong H (2013) The mechanism of Na(+)/K(+) selectivity in mammalian voltage-gated sodium channels based on molecular dynamics simulation. Biophys J 104:2401–2409
Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, Pan CY, Scheuer T, Baker D, Catterall WA (2012) Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci USA 109:E93–E102
Yu FH, Catterall WA (2004) The VGL-chanome: a protein superfamily specialized for electrical signaling and ionic homeostasis. Sci STKE 2004:re15
Zhang X, Yan N (2013) The conformational shifts of the voltage sensing domains between NavRh and NavAb. Cell Res 23:444
Zhang X, Ren W, DeCaen P, Yan C, Tao X, Tang L, Wang J, Hasegawa K, Kumasaka T, He J et al (2012) Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature 486:130–134
Zhang X, Xia MD, Li Y, Liu HH, Jiang X, Ren WL, Wu JP, DeCaen P, Yu F, Huang S et al (2013) Analysis of the selectivity filter of the voltage-gated sodium channel NavRh. Cell Res 23:409–422
Zheng L, Chen M, Yang W (2008) Random walk in orthogonal space to achieve efficient free-energy simulation of complex systems. Proc Natl Acad Sci USA 105:20227–20232
Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R (2001) Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 A resolution. Nature 414:43–48
ACKNOWLEDGEMENTS
We gratefully thank Mengdie Xia for her contribution in preparing the figures. This work was supported by the National Natural Science Foundation of China (Grant Nos. 31470033 and 31321062).
COMPLIANCE WITH ETHICS GUIDELINES
Yang Li and Haipeng Gong declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Li, Y., Gong, H. Theoretical and simulation studies on voltage-gated sodium channels. Protein Cell 6, 413–422 (2015). https://doi.org/10.1007/s13238-015-0152-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-015-0152-6