
Abstract
G-protein coupled receptors (GPCRs) play essential roles in signal transduction from the environment into the cell. While many structural features have been elucidated in great detail, a common functional mechanism on how the ligand-binding signal is converted into a conformational change on the cytoplasmic face resulting in subsequent activation of downstream effectors remain to be established. Based on available structural and functional data of the activation process in class-A GPCRs, we propose here that a change in protonation status, together with proton transfer via conserved structural elements located in the transmembrane region, are the key elements essential for signal transduction across the membrane.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
G-protein coupled receptors (GPCRs) comprise the largest and most diversified family of signaling membrane proteins in eukaryotic cells (Rohrer and Kobilka, 1998). They are a major class of targets of therapeutic intervention (Insel et al., 2007). In response to ligand binding from the extracellular face, GPCRs change their conformations on the cytoplasmic face to affect downstream intracellular events (Rasmussen et al., 2011). Understanding the common mechanisms of agonist-induced GPCR activation will facilitate designs of various ligands that manipulate the properties GPCR proteins. Thus, studying the mechanism is of both theoretical as well as practical importance.
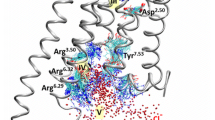
The majority of GPCRs (~85% (Tadevosyan et al., 2012)) belongs to the class-A subfamily. The structure of a class-A GPCR protein contains seven transmembrane (TM) helices (TMs 1–7) forming a TM core. Along the direction of the membrane normal, the TM core can be divided into three parts: The extracellular third is mainly responsible for ligand binding (Hanson and Stevens, 2009); the middle part is required for signal transduction (Zhang et al., 2013); and the intracellular third is responsible for interactions with downstream effectors (Rasmussen et al., 2011). To date, the precise function of the middle part remains enigmatic, yet this part contains most of the conserved structural elements of class-A GPCRs, including a major hydrogen-bond network (MHN) formed by the hydrophilic “2.50-cavity” and hydrophobic “middle-cavity” (Fig. 1) (Zhang et al., 2013; Katritch et al., 2013; Angel et al., 2009). These evolutionarily conserved elements are most likely to be the structural bases of a common, ligand independent, signal transduction mechanism of class-A GPCRs.

Conserved proton wire in the major hydrogen-bond network of α 2a AR. Key residues are shown as stick models, and water molecules and Na+ are shown in red and blue spheres, respectively. For clarity, only components along the major path of proton transfer are shown. Percentage conservativeness of the involved residues is included in parentheses. This figure is a modified version of a figure previously published by us (Zhang et al., 2013).
It has been proposed that interaction between protonated functional groups of the GPCR TM core and the ubiquitous, negative-inside, membrane potential plays a critical role in GPCR activation (Zhang et al., 2013). Open questions associated with such an activation mechanism include the following: What are the functional roles of conserved motifs in the activation mechanism? What is the status of GPCR protonation in its inactive (ground) state and in its active state? What do agonist and antagonist differ in terms of determining GPCR protonation? This mini-review addresses some of these questions, based mainly on the analysis of structural and functional data available for class-A GPCRs.
Protonation status
GPCR activation is associated with both a conformational change from the ground state conformation (CG) to the active conformation (CA) and the opening of the cytoplasmic face of the protein (Rasmussen et al., 2011). For such a large change in overall structure to occur, energy in one form or another would be required. On the one hand, the binding energy of an agonist is usually small (∆G = RTln([L]/Kd), where R represents the universal gas constant and T the temperature ~300K), especially when the ligand concentration is low in comparison with the dissociation constant (i.e. [L] ≈ Kd), excluding this option as a viable energy source. On the other hand, under the influence of the electrostatic field of the evolutionarily conserved membrane potential (typically ∆Ψ ≈ −100 mV), proton movement from theextracellular side to the cytoplasmic side could potentially provide sufficient energy for the conformational change, provided proper coupling can be established (Zhang et al., 2013). For each proton to move across the membrane, this energy is F∆Ψ ≈ 4RT (where F is the Faraday constant). In general, when combined with the high-energy barrier of the transition state, extra energy input into the signaling process would increase the signal-to-noise ratio and thus permit faithful yet low-noise responses to a variety of ligands of a wide range of binding affinities.
Proton-titratable residues (or functional groups) are essential for such regulated proton transfer. For a typical class-A GPCR, there are only two conserved acidic residues embedded in the TM region, namely D2.50 (as per B–W numbering (Ballesteros and Weinstein, 1995)) in the conserved MHN and D/E3.49 of the DRY motif, and they are the most likely candidates for protonation sites (Zhang et al., 2013). Since D/E3.49 (conserved in 94% class-A GPCRs, as estimated according to the online database 7TM Alignment Explorer (Van Durme et al., 2006)) is located downstream of D2.50 (92%) in the electrostatic field, the initial protonation site is likely to be D2.50; and the final destination is likely to be D/E3.49. In between these two residues, multiple functional groups of the MHN may provide a proton-relaying framework, including a cluster of ordered water molecules which can be protonated as well (Fig. 1). As long as a conformational change allows movement of the proton along the electrostatic field, such a change would be favored in the presence of membrane potential. Thus, proton transfer from D2.50 to D/E3.49 may generate energy that favors the CG-to-CA conformational change.
In all potential-bearing membranes, the equilibrium conformation of a charge-carrying membrane protein is the result of a balance between forces of electrostatic potential and a hydrophobic mismatch. The lipid bilayer functions like a belt around the structure of the GPCR protein. The location at which the belt interacts with a GPCR determines the conformation and functional status of the latter, including ligand potency and efficacy. From the protein point of view, in addition to non-specific hydrophobic interaction, a GPCR molecule is anchored within the lipid bilayer via a number of specific interactions, including the amphipathic helix-8 (H8), which fixes the position of TM7 relative to the membrane. These anchor positions may serve as pivots when the GPCR responds to external forces. Because of the different locations the two conserved acidic residues reside relative to these pivots, protonation events at D2.50 and D/E3.49 may result in distinct effects on GPCR conformation. In a more general sense, when a proton moves within the proton-relay path, different parts of the protein may experience forces and torques at different time points, a process that ultimately affects the dynamic properties of both the TM helices (including their positions and orientations relative to each other) and the overall conformation of the GPCR TM core as well.
Activation of class-A GPCR
On the basis of distinct micro-environments of D2.50 in the CG and CA states, we previously proposed a conformational switch that is centered around D2.50 (Zhang et al., 2013). In particular, a change in protonation status of D2.50 is the initial step in a cascade of conformational changes that results in the activation of the GPCR. The exact protonation status at D2.50, however, has remained an open question until now. Here, based on further structural analysis, we propose a mechanism by which the trigger for activation is not protonation of the GPCR per se, but the actual process of proton transfer from an inert position (D2.50) to an active position (the middle-cavity). This putative activation process will be described below (Fig. 2).

Schematic diagram of the putative GPCR activation process
-
(i)
D2.50 is protonated in the CG state. This assumption on the protonation status of D2.50 is based on the structural observation that D2.50 is deeply embedded in the middle of the TM core as well as a need for a proton source at the beginning of the activation process. Mutations at D2.50 (including D2.50 N) in a number of GPCRs inevitably result in abolishing GPCR activity (Bihoreau et al., 1993; Parent et al., 1996; Proulx et al., 2008; Strader et al., 1988; Ceresa and Limbird, 1994; Martin et al., 1999), suggesting that the ability of this position to switch between protonated and deprotonated status is essential for GPCR activation. Furthermore, sodium ion (Na+) wasfound to bind to D2.50 in several crystal structures of GPCRs in their CG states (Zhang et al., 2012; Liu et al., 2012), implying that D2.50 is able to attract positively charged ions. Thus, the absence of the Na+ very likely results in the protonation of D2.50 (or binding of a protonated water molecule H3O+). The bound proton most likely originates from the extracellular space, with its movement driven by the negative-inside membrane potential. The corresponding proton-binding energy may be utilized to put the GPCR into a permissive state ready for signaling. In the presence of a membrane potential, this protonation site is subjected to an electrostatic force, and this force can be balanced by the hydrophobic mismatch force established by the presence of the conserved amphipathic helix H8. In particular, since TMs 1, 2, and 7 form a rigid body (Zhang et al., 2013), the force applied onto D2.50 can be balanced by H8 via TM7.
-
(ii)
Agonist binding induces a characteristic change in the micro-environment of D2.50, including an “upshift” (i.e. towards the extracellular space) of TM3 relative to D2.50, which brings the conserved S3.39 (61%) to the vicinity of D2.50 (Zhang et al., 2013). In response to the approaching movement of S3.39, deprotonation occurs at D2.50. Since most D2.50N mutations show lower agonist affinity and become less active in response to agonist (Wilson et al., 2001), a protonated D2.50 appears to be incompatible with agonist binding, thus deprotonation is favored. In addition, in bacteriorhodopsin (a 7-TM, photon-catalyzed, proton pump), the moving of a threonine residue (T46) closer to an acidic residue (D96), from 3.0 Å to 1.85 Å, is believed to reduce the pKa of the latter by 5.5 units (Onufriev et al., 2003). This suggests that similar deprotonation may occur at D2.50 of the GPCR upon moving closer to the residue S3.39. In turn, this deprotonation event can stabilize the new conformation of S3.39 relative to D2.50.
-
(iii)
The released proton is transferred from the 2.50-cavity to a linearly aligned cluster of three water molecules in the middle-cavity (Fig. 1). Subsequently, one of the water molecules gets protonated (i.e. forming H3O+). Because of the negative-inside membrane potential and the deeply embedded positon of D2.50, the released proton is unlikely to move to the extracellular space. Furthermore, the proton transfer requires Y7.53 (96%) of the NPxxY motif, which is located between the two cavities. Using its side chain hydroxyl group, a tyrosine residue may function as a proton-relay intermediate. Y7.53F mutation has been shown to reduce GPCR activation (e.g. in AT2R), an effect similar to that of the D2.50N mutation (Marie et al., 1994). Other parts of theproton-wire are formed by ordered water molecules, which are precisely positioned by polar residues and mainchain atoms from highly conserved motifs (Zhang et al., 2013). In a typical class-A GPCR, the middle-cavity is located downstream of D2.50 in the electrostatic field, thus allowing the proton transfer to be powered by the membrane potential.
-
(iv)
Because the middle-cavity is formed mainly by conserved hydrophobic residues, including those from TM6 (Zhang et al., 2013), the protonated water molecule applies a force to the overall structure of the GPCR and TM6 in particular. The middle-cavity region was previously referred to as TM6-clamp (Hulme, 2013), implying its functional role in TM6 movement. Protonation of a buried water cluster has been shown to be important for functions of bacteriorhodopsin as well (Garczarek et al., 2005). In general, maintaining water molecules in a hydrophobic cavity inside a protein is energetically costly (Nucci et al., 2014), suggesting an important functional role of the middle-cavity. The hydrophobic interior of the middle-cavity provides insulation for the enclosed water cluster, separating it from the DRY pocket on the cytoplasmic side (Fig. 1). Such an insulation ensures both the provision of the protonation status of the water cluster and a focused electric field being applied on the protonated water molecule. Up to this point of the activation process, no major conformational change has occurred in the TM core region, except the relative movement between TMs 2 and 3 responsible for triggering the deprotonation of D2.50.
-
(v)
In response to the “downward” (i.e. towards the cytoplasm) electrostatic force mediated by the protonated water molecule H3O+, TM6 moves away from the rest of the protein, resulting in the release of the water molecules from the middle-cavity, including that protonated. Being a characteristic feature of the GPCR activation (Rasmussen et al., 2011), this TM6 outward movement (i.e. away from the rest of the TM core; by about 14 Å at its cytoplasmic tip) enlarges the DRY-cavity, partially by merging it with the middle-cavity. This movement both deepens and widens the DRY-cavity, thus enabling its interaction with downstream G-proteins. This step may correspond to the change from the Meta I to Meta II state observed during rhodopsin activation, which is reported to associate with the release of hydrogen-bonded water molecules (Angel et al., 2009; Mitchell and Litman, 1999). The putative favorable effect of membrane potential on TM6 movement may explain the observation of an incomplete conformational change in many crystal structures of “active” GPCRs (Warne et al., 2011; White et al., 2012; Xu et al., 2011), since in these crystals the membrane potential is absent. In vitro, where there is neither a membrane potential nor a hydrophobic mismatch, a charge-carrying GPCR would be in an equilibrium conformation different from that observed under in vivoconditions.
-
(vi)
Following the outward movement of TM6, a series of conformational changes occur in the DRY-cavity, as a consequence of which the CA state is stabilized. By rearrangement of the inter-helix packing between TMs 3 and 6, R3.50 (98%) is released from the D/E3.49-R3.50 salt-bridge bond, which is a signature event in GPCR activation (Rasmussen et al., 2011; White et al., 2012; Palczewski et al., 2000). In the CG state, D/E3.49 is deprotonated because of the proximate basic residue R3.50, while Y5.58 (90%) stabilizes the water cluster inside the middle-cavity. Upon the breaking of the salt-bridge, the pKa of D/E3.49 rises, and D/E3.49 gets protonated. Meanwhile, R3.50 switches its partner, forming a hydrogen-bond with Y5.58 (White et al., 2012). This D/E3.49-protonation state corresponds to the Meta IIbH+ state of rhodopsin (Lohse et al., 2014). In addition, in ca. 38% of GPCRs, the position 2.39 is occupied by a threonine residue. This T2.39 is located in the vicinity of D/E3.49 in the CG state (e.g. 2.7 Å in α2AAR/4EIY), but moves apart in the CA state (e.g. 4.2 Å in α2AAR/2YDV). Similar to the above-mentioned distance change between S3.39 and D2.50, the positional shift of T2.39 away from D/E3.49 may promote and stabilize protonation of the latter. D/E3.49 may pick up a proton either directly from the proton-wire of the MHN or from the cytoplasm. As long as the pKa of D/E3.49 is higher than the pH inside the DRY-cavity, protonation occurs spontaneously. In either case, the net result appears to be a proton transfer from D2.50 to D/E3.49. Since D/E3.49 is located at the end of the putative proton movement, a protonated D/E3.49 seems to stabilize the CA state under the influence of membrane potential rather than to drive the conformational change.
Additional issues related to protonation processes
In general, energy input would accelerate a molecular process in a particular direction, by overcoming energy barrier(s) of the transition-state(s). This is exemplified by the case of rhodopsin, where the energy input (~80RT at 600 nm wavelength) exceeds by far what is needed for the activation of a canonical class-A GPCRs (~4RT), namely by an order of magnitude. As a result, rhodopsin exhibits extremely high fidelity and reaction speed (Lohse et al., 2014). Furthermore, the kinetics of GPCR activation is usually much faster under in vivo conditions (~ 30 ms) than in vitro conditions (~30 s), indicating that factors critical for the conformational change during activation in the native environment are not available in purified or reconstituted systems (Lohse et al., 2014). In addition, in the presence of a membrane potential, the activation process is strongly favored in the forward direction. However, if deprotonation occurs (e.g. by dropping the proton into the cytoplasm at the end of the activation process), the equilibrium between the two states may shift. In principle, if the CA state were unstable thermodynamically, a GPCR could function as a proton transporter by cycling between the CG and CA states. Interestingly, the rate of an inverse agonist-induced deactivation is usually slower than that of an agonist-induced activation (Lohse et al., 2014). This intriguing observation could be explained as follows: First, the favorable energy input, which is associated with the interaction between the proton and membrane potential in the CG-to-CA activation process, is not available in the CA-to-CG deactivation process. Second, an inverse agonist may achieve its effect through an alternative pathway (e.g. by re-loading a proton from the extracellular side) rather than a backward movement of the proton.
A smooth proton-transfer is essential for the full activation of GPCR. An inverse agonist may prevent deprotonation of D2.50, thus completely blocking activation. In contrast, basal activity of class-A GPCRs may originate from spontaneous deprotonation at D2.50. In addition, in vivo biased activation may be the result of different extents of the proton movement. For instance, if ligand binding only allows a half-way proton transfer, this may result in only a partial rather than full exposure of the effector binding site. Thus, ligand binding not only determines the direction but also the extent of the conformational change.
As mentioned above, in several high-resolution crystal structures of inactive GPCRs, Na+ is found to bind with D2.50 (Zhang et al., 2012; Liu et al., 2012). If a positive charge alone could trigger activation, Na+ binding would have a similar effect as protonation. However, effects of a positive charge on overall conformational change may be realized only when the charge moves along the electrostatic field of the membrane potential so that its electrostatic energy is converted to conformational energy of the protein. It should be noted that there is significant difference between protonation and Na+ binding. A proton may be easily transferred among titratable groups including water molecules, whereas Na+ may not. The MHN of class-A GPCR seems to be unsuitable for Na+ transfer (considering the elegant structural details of a Na+ channel (Payandeh et al., 2011)). In other words, the energy barrier for Na+ transfer appears much higher than that of a proton. In addition, no proper Na+-binding site is found in the CA state. Thus, unlike protonation, Na+ seems unable to stabilize the CA state. In fact, Na+ binding inhibits the activation of GPCR, probably by competing with protonation at D2.50 (Martin et al., 1999).
Interestingly, class-B GPCRs also contain conserved (class-specific) polar residues in their TM cores (Wootten et al., 2013; Hollenstein et al., 2013; Siu et al., 2013), including proton-titratable residues (e.g. the H2.50b-E3.50b pair (as per Wootten numbering (Wootten et al., 2013))). These polar residues are likely to interact with buried water molecules, and together they form a hydrogen-bond network. Mutations of most of these conserved polar residues result in impaired activity or reduced cell-surface expression of the receptors (Wootten et al., 2013). Most of these conserved polar residues are clustered into two groups, one in an orthosteric ligand-binding cavity facing the extracellular side, and the other in a cavity facing the cytoplasmic side. In addition, there is a small cavity in the vicinity of N5.50b, which binds an antagonist in the CRF1 receptor crystal structure (Hollenstein et al., 2013). Physiological agonists of class-B GPCRs are usually peptide hormones (Hollenstein et al., 2014). The N-terminus of the ligand peptide are thought to insert into the orthosteric binding cavity, although alternative binding modes are also proposed (Beinborn, 2006). In parallel to what we propose for activation of class-A GPCRs, it is probable that, in class-B GPCRs, the ligand binding couples with deprotonation in the orthosteric ligand-binding cavity (e.g. at the conserved H6.52), and the released proton transfers into the N5.50b cavity (functionally similar to the middle cavity in class-A GPCRs), triggering the opening of the cytoplasmic side. The final destination of the proton is likely to be the H2.50b-E3.50b pair located in the cytoplasmic cavity of this class of GPCRs.
Conclusion Remarks
A fundamental question in GPCR activation concerns the mechanism that couples agonist binding at the extracellular side with the conformational opening at the cytoplasmic face. We hypothesize that it is the dynamics of protonation, rather than protonation per se, that affects the activation status of the class-A GPCR. During the activation process, a proton is translocated under the influence of membrane potential. In response, different parts of the GPCR experience forces at consecutive time points, determining the dynamics and kinetics of the activation process. Conserved motifs of class-A GPCRs, including D2.50, DRY, and NPxxY motifs, as well as a number of associated water molecules play essential roles in structure and functions of the proton-relay path.
References
Angel TE, Chance MR, Palczewski K (2009) Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc Natl Acad Sci USA 106(21):8555–8560
Ballesteros JA, Weinstein H (1995) Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 25:366–428
Beinborn M (2006) Class B GPCRs: A hidden agonist within ? Mol Pharmacol 70:1–4 (Relates to article by Dong, et al. FastForward 10 March 2006)
Bihoreau C et al (1993) Mutation of Asp74 of the rat angiotensin II receptor confers changes in antagonist affinities and abolishes G-protein coupling. Proc Natl Acad Sci USA 90(11):5133–5137
Ceresa BP, Limbird LE (1994) Mutation of an aspartate residue highly conserved among G-protein-coupled receptors results in nonreciprocal disruption of alpha 2-adrenergic receptor-G-protein interactions. A negative charge at amino acid residue 79 forecasts alpha 2A-adrenergic receptor sensitivity to allosteric modulation by monovalent cations and fully effective receptor/G-protein coupling. J Biol Chem 269(47):29557–29564
Garczarek F et al (2005) Proton binding within a membrane protein by a protonated water cluster. Proc Natl Acad Sci USA 102(10):3633–3638
Hanson MA, Stevens RC (2009) Discovery of new GPCR biology: one receptor structure at a time. Structure 17(1):8–14
Hollenstein K et al (2013) Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499(7459):438–443
Hollenstein K et al (2014) Insights into the structure of class B GPCRs. Trends Pharmacol Sci 35(1):12–22
Hulme EC (2013) GPCR activation: a mutagenic spotlight on crystal structures. Trends Pharmacol Sci 34(1):67–84
Insel PA et al (2007) Impact of GPCRs in clinical medicine: monogenic diseases, genetic variants and drug targets. Biochim Biophys Acta 1768(4):994–1005
Katritch V, Cherezov V, Stevens RC (2013) Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol 53:531–556
Liu W et al (2012) Structural basis for allosteric regulation of GPCRs by sodium ions. Science 337(6091):232–236
Lohse MJ, Maiellaro I, Calebiro D (2014) Kinetics and mechanism of G protein-coupled receptor activation. Curr Opin Cell Biol 27:87–93
Marie J et al (1994) Tyr292 in the seventh transmembrane domain of the AT1A angiotensin II receptor is essential for its coupling to phospholipase C. J Biol Chem 269(33):20815–20818
Martin S et al (1999) Pivotal role of an aspartate residue in sodium sensitivity and coupling to G proteins of neurotensin receptors. Mol Pharmacol 55(2):210–215
Mitchell DC, Litman BJ (1999) Effect of protein hydration on receptor conformation: decreased levels of bound water promote metarhodopsin II formation. Biochemistry 38(24):7617–7623
Nucci NV et al (2014) Role of cavities and hydration in the pressure unfolding of T4 lysozyme. Proc Natl Acad Sci USA 111:13846–13851
Onufriev A, Smondyrev A, Bashford D (2003) Proton affinity changes driving unidirectional proton transport in the bacteriorhodopsin photocycle. J Mol Biol 332(5):1183–1193
Palczewski K et al (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289(5480):739–745
Parent JL et al (1996) Mutation of an aspartate at position 63 in the human platelet-activating factor receptor augments binding affinity but abolishes G-protein-coupling and inositol phosphate production. Biochem Biophys Res Commun 219(3):968–975
Payandeh J et al (2011) The crystal structure of a voltage-gated sodium channel. Nature 475(7356):353–358
Proulx CD et al (2008) Mutational analysis of the conserved Asp2.50 and ERY motif reveals signaling bias of the urotensin II receptor. Mol Pharmacol 74(3):552–561
Rasmussen SG et al (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477(7366):549–555
Rohrer DK, Kobilka BK (1998) G protein-coupled receptors: functional and mechanistic insights through altered gene expression. Physiol Rev 78(1):35–52
Siu FY et al (2013) Structure of the human glucagon class B G-protein-coupled receptor. Nature 499(7459):444–449
Strader CD et al (1988) Conserved aspartic acid residues 79 and 113 of the beta-adrenergic receptor have different roles in receptor function. J Biol Chem 263(21):10267–10271
Tadevosyan A et al (2012) G protein-coupled receptor signalling in the cardiac nuclear membrane: evidence and possible roles in physiological and pathophysiological function. J Physiol 590(Pt 6):1313–1330
Van Durme J et al (2006) GRIS: glycoprotein-hormone receptor information system. Mol Endocrinol 20(9):2247–2255
Warne T et al (2011) The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature 469(7329):241–244
White JF et al (2012) Structure of the agonist-bound neurotensin receptor. Nature 490:508–513
Wilson MH, Highfield HA, Limbird LE (2001) The role of a conserved inter-transmembrane domain interface in regulating alpha(2a)-adrenergic receptor conformational stability and cell-surface turnover. Mol Pharmacol 59(4):929–938
Wootten D et al (2013) Polar transmembrane interactions drive formation of ligand-specific and signal pathway-biased family B G protein-coupled receptor conformations. Proc Natl Acad Sci USA 110(13):5211–5216
Xu F et al (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332(6027):322–327
Zhang C et al (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492:387–392
Zhang XC et al (2013) GPCR activation: protonation and membrane potential. Protein Cell 4(10):747–760
Acknowledgments
We thank Drs. Yongfang Zhao and Qiang Zhao for critical reading of the manuscript. We thank Dr. T. Juelich for linguistic assistance during the preparation of this manuscript. This work was supported by the National Basic Research Program (973 Program) (Nos. 2009CB918803 and 2011CB910301) and by the Chinese Academy of Sciences (XDB080203).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, X.C., Cao, C., Zhou, Y. et al. Proton transfer-mediated GPCR activation. Protein Cell 6, 12–17 (2015). https://doi.org/10.1007/s13238-014-0106-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0106-4