
Abstract
KDM5B is a histone H3K4me2/3 demethylase. The PHD1 domain of KDM5B is critical for demethylation, but the mechanism underlying the action of this domain is unclear. In this paper, we observed that PHD1KDM5B interacts with unmethylated H3K4me0. Our NMR structure of PHD1KDM5B in complex with H3K4me0 revealed that the binding mode is slightly different from that of other reported PHD fingers. The disruption of this interaction by double mutations on the residues in the interface (L325A/D328A) decreases the H3K4me2/3 demethylation activity of KDM5B in cells by approximately 50% and increases the transcriptional repression of tumor suppressor genes by approximately twofold. These findings imply that PHD1KDM5B may help maintain KDM5B at target genes to mediate the demethylation activities of KDM5B.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Covalent histone modifications, notably methylation, are reversible posttranslational modifications that play key roles in chromatin structure, gene transcription and the epigenetic state of the cell (Martin and Zhang, 2005; Mosammaparast and Shi, 2010). Six lysine residues, including histones H3 (i.e., H3K4, H3K9, H3K27, H3K36 and H3K79) and H4 (i.e., H4K20), can be mono-, di-, or trimethylated, and each modification is found in a stereotypical pattern with respect to the coding region of a gene and correlates with a different transcriptional outcome (Zhang and Reinberg, 2001; Margueron et al., 2005). As a general rule, the methylation of H3K9, H3K27 and H4K20 is associated with transcriptional repression, whereas the methylation of H3K4, H3K36 and H3K79 is related to transcriptional activation (Mosammaparast and Shi, 2010; Zhang and Reinberg, 2001). The methylation of H3K4 is a key regulator for RNA polymerase binding to an active gene (Sims et al., 2003) and of transcription factor binding within promoter elements (Szutorisz et al., 2005). One of the important aspects of H3K4 methylation is how this epigenetic mark is removed, thereby reducing the localization of RNA polymerase to the specific genes. The loss of H3K4 methylation appears to be a key step of differentiation (Mikkelsen et al., 2007). To date, two distinct classes of histone demethylases have been characterized. The first class includes two members, represented by LSD1 (lysine-specific demethylase 1, also known as KDM1A, which demethylates H3K4me1/2) and LSD2 (also called KDM1B or AOF1, which demethylates H3K4me2). Both members of this class use FAD as a cofactor through an amine-oxidation reaction to remove the lysine methyl group of H3K4me1/2 (Shi et al., 2004; Ciccone et al., 2009). The other class contains a JmjC (i.e., Jumonji C) domain to catalyze histone lysine demethylation assisted by two cofactors: Fe2+ ion and α-ketoglutarate (α-KG) (Chen et al., 2006; Cloos et al., 2006; Tsukada et al., 2006; Whetstine et al., 2006, Shi and Whetstine, 2007). Based on the sequence homology in the JmjC domain and the overall architecture of the associated motifs, JmjC domain-containing proteins have been classified into seven groups: JHDM1, PHF2/8, JARID, JHDM3/JMJD2, UTX/UTY, JHDM2 and JmjC domain only (Chen et al., 2006). Structural investigations of the members of this class, including JHDM1A, JHDM1D, JMJD2A and PHF8, have already been performed for their apo forms or in complex with the H3 peptide and α-KG substrates (Chen et al., 2006; Chen et al., 2007; Couture et al., 2007; Ng et al., 2007; Horton et al., 2010; Yang et al., 2010). However, no structure has been published for the members of the JARID1 sub-group that can specifically remove methyl groups of di- or tri-methylated H3K4.
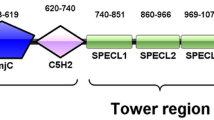
The members of the JARID1 subgroup are highly conserved from yeast to humans and contain a similar motif architecture, including JmjN, ARID (i.e., AT-rich interactive domain), JmjC, Zf-C5HC2 (i.e., zinc finger) and two or three PHD domains (denoted PHD1, PHD2 and PHD3 from the N-terminus to the C-terminus). A total of four members are found in mammals (Fig. 1A): JARID1A (also called RBP2 or KDM5A), JARID1B (also named PLU-1 or KDM5B), JARID1C (i.e., SMCX or KDM5C) and JARID1D (also known as SMCY orKDM5D). These members have been identified to be H3K4me2/3 demethylases (Christensen et al., 2007; Iwase et al., 2007; Klose et al., 2007; Lee et al., 2007; Tahiliani et al., 2007; Yamane et al., 2007). All of these proteins are key transcriptional co-repressors because they can remove the transcription-activating marker H3K4me3. KDM5B is involved in transcriptional repression and breast cancer cell proliferation (Yamane et al., 2007); thus, mechanistic studies on KDM5B demethylation are necessary and useful to understand the development of breast cancer. Notably, the deletion of the N-terminal PHD1 finger (i.e., PHD1KDM5B) of KDM5B results in the loss of enzymatic demethylase activity, implying that PHD1KDM5B is involved in H3K4me2/3 demethylation (Yamane et al., 2007). This observation is consistent with the fact that the N-terminal PHD1 finger of Lid (i.e., PHD1Lid), a homologue of KDM5B in Drosophila, is also required for the demethylase activity of H3K4me3, whereas the PHD2 and PHD3 of Lid are not (Li et al., 2010). However, the detailed mechanism underlying the function of PHD1KDM5B in the demethylation process remains unclear.

PHD1 KDM5B specifically binds to the tail of H3K4me0. (A) Mammalian KDM5 family members are highly similar in domain architecture and contain JmjN, ARID, Jmjc, zf-C5HC2 and PHD domains. (B) KDM5B variants described in the text. (C) In vitro binding assays for analysis of the binding of recombinant KDM5B variants to the unmodified histone H3K4 N-terminal tail. (D) PHD1KDM5B is sufficient for H3 tail binding. (E) The binding affinities of PHD1KDM5B to unmodified, mono-methylated, di-methylated or tri-methylated H3K4 peptides (residues 1–10) were measured through an ITC assay. The KD values are the means (± s.d.) of at least three experiments using varied peptide and protein concentrations
Recently, the NAD+-dependent PARylation on KDM5B by poly (ADP-ribose) polymerase (PARP-1) was reported to be able to regulate chromatin structure and transcription through a KDM5B-dependent pathway (Krishnakumar and Kraus, 2010). The demethylation inhibited by PARylation on KDM5B was confirmed by an in vitro H3 binding assay. Previously, PHD1Lid was reported to bind with unmodified H3K4me0 (Li et al., 2010). These observations indicate that KDM5B may bind to the H3 peptide, most likely to unmodified H3K4. Thus, in this study, we first tested the interactions of the unmodified H3K4 peptide with full-length KDM5B or its truncated variants through a biotin-labeled peptide binding assay. By sequentially depleting different regions in KDM5B-N, we found that PHD1 in KDM5B (i.e., PHD1KDM5B) can specifically bind to the unmodified histone H3. To probe the structural basis for this interaction, we determined a solution structure of PHD1KDM5B in complex with the unmodified H3K4 peptide. Through structural and biochemical data, we provide insights into the function of PHD1KDM5B in KDM5B-regulated demethylation and tumor-suppressor gene transcription.
Results
PHD1KDM5B specifically interacts with the unmodified H3K4me0 peptide
To investigate the function of PHD1KDM5B in the demethylation of H3K4me2/3 by KDM5B (as shown in Fig. 1B), the binding affinities of five recombinant KDM5B variants, namely KDM5B-N (N-terminal KDM5B), KDM5B-PC (only containing PHD1 and JmjC domains in KDM5B-N), KDM5B-N-△P (without the PHD1 domain in KDM5B-N), KDM5B-N-△C (without the JmjC domain in KDM5B), and KDM5B-N-△ZF (without the Zf-C5HC2 domain in KDM5B-N), were tested with a biotin-labeled unmodified H3 peptide (Fig. 1C). The results indicate that KDM5B-N interacts with the unmodified H3 peptide. Similar to the KDM5B-N variant, the KDM5B-PC, KDM5B-N-△C and KDM5B-N-△ZF variants bind to the unmodified H3K4 peptide, implying that the JmjC domain, the Zf-C5HC2 domain, and the JmjN-ARID domain are not involved in the interaction with the unmodified H3K4 peptide. However, the deletion of the N-terminal PHD1KDM5B domain in KDM5B-N (i.e., KDM5B-N-△P) significantly impaired the interaction between KDM5B-N and the H3K4me0 peptide. This result suggested that PHD1KDM5B primarily contributes to the binding of KDM5B to the H3K4me0 peptide. The glutathione S-transferase (GST) tag-fused PHD1KDM5B (306–360 aa, similarly hereinafter) protein binds to the unmodified H3 tail (Fig. 1D), further supporting the function of PHD1KDM5B in the specific binding to the unmodified H3. The methylation of H3R2 and H3K4 abolishes or weakens the binding affinity, suggesting that these two amino acids may be involved in the binding. In addition, the methylation of H3K9 does not notably inhibit this binding, which indicates that H3K9 does not participate in the binding. Moreover, an ITC analysis obtained the dissociation constants (KD) of 6.4 ± 0.6 μmol/L for PHD1KDM5B interacting with the unmodified H3K4 peptide, 25.6 ± 3.8 μmol/L for PHD1KDM5B interacting with mono-methylated H3K4me1, 80.0 ± 7.9 μmol/L for PHD1KDM5B interacting with dimethylated H3K4me2 and 103.7 ± 11.2 μmol/L for PHD1KDM5B interacting with tri-methylated H3K4me3 (Fig. 1E). These KDs revealed that H3K4 is highly involved in the binding and that the methylation of H3K4 inhibits this binding.
Solution structure of PHD1KDM5B in complex with H3K4me0
To understand the interaction of PHD1KDM5B with unmodified H3K4, we initially attempted to crystallize PHD1KDM5B in its free form and in complex with the H3K4me0 peptide. However, we only obtained the X-ray structure of the free PHD1KDM5B with a high resolution of 1.65 Å (Fig. S1) (Guo et al., 2011). Thus, we determined the solution structures of free PHD1KDM5B and of PHD1KDM5B in complex with the H3K4 peptide (1–10 aa) using multidimensional heteronuclear NMR spectroscopy (Fig. 2). To probe whether the electronic properties of histidines in solution are similar to those in the crystal state (two histidines, H335 and H344, are in the amino acid sequence of PHD1KDM5B; the H335 residue was suggested to ligate with a zinc ion in the X-ray crystal structure of free PHD1KDM5B) before structural determination, we analyzed the 1H-15N LR-HSQC spectra of PHD1KDM5B in the free and bound states (Fig. S2). This LR-HSQC experiment correlates the carbon-bound protons of the histidine rings with the imidazole nitrogen atoms and can unambiguously establish the tautomeric and protonation states of histidines in proteins (Pelton et al., 1993; Drohat et al., 1999). The characteristic upside-down L-shaped patterns of the peaks in the LR-HSQC spectra for the H335 and H344 residues and their well-separated 15N chemical shifts indicated that these two histidines are in the Nε2-H tautomeric form. Additionally, both histidines are in their neutral form under the NMR experimental conditions. The 15Nδ1 chemical shift of H335 is more deshielded than that of H344 in PHD1KDM5B, revealing the strong chelation of H335 to an electropositive Zn2+. This observation is identical to that observed in the X-ray structure of the free PHD1KDM5B.

Structure of PHD1 KDM5B with the unmodified H3 tail (1–10 aa). (A) An NMR titration assay was used to map the binding sites of the H3K4me0 peptide on PHD1KDM5B. The chemical shift of the PHD1KDM5B backbone atoms amide 1H and 15N after H3K4me0 peptide binding was calculated using the following equation: . (B) Ribbon representation of free PHD1KDM5B. To indicate the binding sites of the H3K4me0 peptide on PHD1KDM5B, the residues with different chemical shifts are marked in different colors. (C) Backbone atoms (N, Cα and C′) of the 20 superposed NMR structures of PHD1KDM5B (grey) in complex with the H3K4me0 peptide (yellow). (D) The ribbon representation of the structure of the complex highlights the secondary structural elements (protein, grey; peptide, yellow). The pink spheres represent zinc atoms. For clarity, only the representation of the zinc atoms in the lowest energy structure is shown in the ensemble. (E) Electrostatic potential surface representation of the PHD1KDM5B interaction with the H3K4me0 peptide (yellow). The residues in the H3 peptide are labeled. The orientations of the protein in figures (B) to (E) are identical. (F) The hydrogen-bond interactions between the backbone atoms of PHD1KDM5B and the H3K4me0 peptide. (G–K) Key protein-peptide side-chain interactions between A1, R2, T3, K4 and T6 in the H3 peptide and residues in PHD1KDM5B. The carbon atoms in the peptide and protein residues are shown in yellow and green, respectively. The nonpolar non-bonded interacting atoms are labeled with
The NMR solution structure of free PHD1KDM5B (Fig. 2B) was then determined using the program XPLOR-NIH (Kuszewski and Clore, 2000) with 670 NOEs, 10 hydrogen bonds and 106 dihedral angle constraints that were meaningful and acceptable (Table 1). The resulting structure was similar to that obtained by X-ray techniques with a backbone-atom RMSD of 0.48 Å, which was determined by superimposing the backbone Cα atoms in the secondary structural region (Fig. S1). Thus, in this paper, we only use the NMR structure of the free PHD1KDM5B for comparison with that of the complex. In contrast, the solution structure of PHD1KDM5B in complex with an unmodified H3K4me0 peptide (1–10 aa) was obtained through 1263 NOEs (including 150 observed intermolecular NOEs), 17 hydrogen bonds and 106 dihedral angle constraints. The PHD1KDM5B structures in both free and bound states adopt a ‘cross-braced’ topology of zinc ion coordinated residues that was identical to that of all structurally characterized PHD fingers. These states are well defined by NMR data; the RMSD of the free PHD1KDM5B and the PHD1KDM5B in complex with the H3 peptide were 1.18 ± 0.17 Å and 0.63 ± 0.14 Å for the heavy atoms in the secondary structural regions of the 20 lowest energy structures, respectively. By superimposing the backbone Cα atoms in the secondary structural region, the free and bound solution PHD1KDM5B structures have a backbone-atom RMSD value of 1.13 Å (Fig. S1). This RMSD indicates that the H3K4me0 peptide binding does not induce major conformational changes to the backbone of PHD1KDM5B.
In the structure of the PHD1KDM5B-H3K4me0 complex, the H3 peptide binds to the surface of the PHD1KDM5B as an anti-parallel β-sheet (residues 2–3 aa). This H3 β-sheet is shorter in this complex than in the complex of the PHD of human autoimmune regulator, (PHDAIRE) with unmethylated H3K4 (PDB code: 2KFT)(Chakravarty et al., 2009) and in the complex of the PHD finger of the BHC80 protein in the LSD1 co-repressor (PHDBHC80) with unmethylated H3K4 (PDB code: 2PUY)(Lan et al., 2007) (Fig. 2D). In the current structure of the complex, the backbone atoms of residues H3R2 and H3K4 in the unmethylated H3 peptide form hydrogen bonds with the backbone carbonyl oxygen and nitrogen atoms of L324 and L326 in PHD1KDM5B (Fig. 2F), respectively. The cognate PHD1KDM5B only contacts the first six residues of the H3 peptide, whereas H3K9 is completely exposed to solvent by extending its side chain away from the complex (Fig. 2E). This finding supports the concept that the methylation of H3K9 has no effect on the binding. The conformation of the side chain of H3R8 is also flexible. In the 20 final NMR structures for the complex, H3R8 does not contact the protein. This coincides with the fact that no intermolecular NOEs were observed between the H3R8 side chain and the protein.
Analysis of the interaction between PHD1KDM5B and the unmodified H3K4me0 peptide
According to the structural information provided by the structure of the complex of PHD1KDM5B with the unmodified H3K4 peptide, the H3K4me0 specificity of PHD1KDM5B is determined through the recognition of the residues in the H3 amino terminus, including H3A1, H3R2, H3T3, H3K4 and H3T6 (Fig. 2F–K). The H3A1 methyl group is anchored by intermolecular hydrogen bonds with the backbone carbonyl oxygen atoms of residues P347 and G349 (Fig. 2F, in which the H3A1 backbone nitrogen supplies two hydrogen bonds) and by nonpolar hydrophobic interactions between the H3A1 methyl group and the side chains of L325, P347 and W351 in PHD1KDM5B (Fig. 2G). These side chains are similar to those observed in the PHDAIRE-H3K4me0 complex and other complexes (Chakravarty et al., 2009; Lan et al., 2007, Li et al., 2006, Pena et al., 2006, Hu et al., 2011, Wang et al., 2011). These observations support our site-directed mutation studies for both the PHD1KDM5B protein and the H3K4me0 peptide (Table S1). On the one hand, both the W351A and L325A mutants of PHD1KDM5B have non-detectable binding affinities with the H3K4me0 peptide. On the other hand, removing the methyl group from H3A1 by changing alanine to glycine results in an approximately 50-fold decrease in the binding affinity of the unmodified H3 peptide to PHD1KDM5B (K H3K4-PHD1D = 6.4 ± 0.6 μmol/L and K H3 A1G-PHD1D = 304.9 ± 11.7 μmol/L). In the 1H-15N HSQC spectra, compared with that of free PHD1KDM5B, the binding to wild-type H3A1 (i.e., H3K4me0) produces a large shift in most of the cross-peaks of PHD1KDM5B, whereas the binding of the H3G1 variant does not induce this shift. This result indicates the important roles of the H3A1 methyl group in the interaction. One of the positively charged side-chain nitrogen atoms (Nη) of H3R2 forms a salt bridge with one of the negatively charged oxygen atoms in the side-chain of D328 (Fig. 2H). D328 mutations in PHD1KDM5B (the D328A mutant) or R2 mutations in the H3K4me0 peptide (H3 R2A or R2E mutants) remove the negative or positive charges in their side chains; therefore, the binding affinities were significantly reduced (K H3A1-PHD1 D328AD = 182.1 ± 21.9 μmol/L, the binding affinity decreased by 30-fold; K H3 R2A-PHD1D = 370.4 ± 52.1 μmol/L, the binding affinity decreased by 60-fold; and the KD value for the interaction of PHD1KDM5B to the H3 R2E mutant was too small to detect) (Table S1) compared with that of the wild-type PHD1KDM5B or the unmodified H3K4 peptide. These results were consistent with the observations mentioned for the above-described structures. In addition, the distance between the Cβ atom of L326 and the Cβ atom of H3R2 is 4.0 Å, indicating a hydrophobic interaction between the side chains of L326 and H3R2 (Fig. 2H).
The side-chain -OH group of H3T3 forms two additional hydrogen bonds with one of the side-chain carbonyl oxygen atoms and the backbone oxygen of E321 (Fig. 2I), further stabilizing the interaction between the H3 peptide and the PHD1KDM5B protein. Removing hydrogen bonds by adding mutations from E321 to A321 in PHD1KDM5B or from T3 to V3 in the H3 peptide lowers the binding affinities of the H3K4me0 peptide to PHD1KDM5B by approximately tenfold (K H3K4-PHD1 E321AD = 75.8 ± 5.7 μmol/L, K H3K4 T3V-PHD1D = 57.1 ± 2.2 μmol/L). The distances between the H3T6-OH group and the backbone carboxyl oxygen of D308 and Y310 are less than 4 Å (Fig. 2K), indicating that weak hydrogen bonds might form between these atoms. The presence of these weak hydrogen bonds is supported by the measured KD values of the mutants (K H3 T6V-PHD1D = 61.3 ± 1.6 μmol/L, the binding affinity decreased by approximately tenfold compared with that of the wild-type protein, which exhibits a K H3K4-PHD1D of 6.4 ± 0.6 μmol/L).
Moreover, the side-chain NH3+ group of H3K4 forms a rigid hydrogen bond with the side-chain carbonyl oxygen of D308 (Fig. 2J), which supports the results of the analysis of the structure of the complex and of the mutation studies (K H3 K4E-PHD1D is non-detectable, whereas K H3K4-PHD1 D308AD was measured as 41.7 ± 2.2 μmol/L). In addition to interacting with D308, the side chain of H3K4 also displays weak hydrophobic interactions with the aromatic ring of Y310 and the methyl groups of L326 (Fig. 2J). The distances between the Cγ or Cδ atom of the H3K4 side chain and the aromatic Cγ atom of Y310 or the methyl group of L326 in PHD1KDM5B are approximately 4.5 Å. The mutation of Y310 to F310 does not change the binding affinities of PHD1KDM5B to the unmodified H3 peptide (K H3K4-PHD1D = 6.4 ± 0.6 μmol/L and K H3K4-PHD1 Y310FD = 4.3 ± 0.2 μmol/L), suggesting that the-OH group in the side chain of residue Y310 may not be involved in the interaction. When Y310 is replaced by A310, the binding affinity between PHD1KDM5B and the H3 peptide decreased by approximately fivefold (K H3K4-PHD1 Y310AD = 28.1 ± 1.3 μmol/L) compared with the binding affinity of the wild-type PHD1KDM5B to the H3 peptide. The PHD1KDM5B L326A mutant has an approximately two-fold weaker binding affinity (K H3K4-PHD1 L326AD = 14.1 ± 0.8 μmol/L) to the unmodified H3 peptide than the wild-type. These observations imply that the hydrophobic interactions between the side chains of L325 and Y310 in PHD1KDM5B and H3K4 contribute less to the binding than the hydrogen bond between H3K4 and D308 in PHD1KDM5B. The methylation of H3K4 may weaken these hydrophobic interactions because minimal space exists among residues L325 and Y310 in PHD1KDM5B and H3K4. This may explain the reduction in the binding affinities of PHD1KDM5B to H3K4me1 (by ~4-fold), H3K4me2 (by >10-fold) and H3K4me3 (by >15-fold) compared with the H3K4me0 peptide. Therefore, molecular recognition of the unmodified lysine 4 primarily occurs through hydrogen bonding to the unmodified epsilon amino group and steric elusion of the methyl groups on H3K4me3/2.
In addition, residues D308, Y310 and L326 are conserved among the members in the KDM5 PHD1 family (Fig. 3B) with the exception of residues E323 in KDM5C and F331 in KDM5D. Therefore, our structure may reveal that the PHD1 domain in other members of the KDM5 family functions as a specific reader of unmodified H3K4, although structural models of the PHD1 KDM5A, KDM5C and KDM5D in complex with unmodified H3K4 are not available.

Binding mode of PHD1 KDM5B to the H3K4me0 peptide. (A) The recognition modes of H3K4me0 by PHD1KDM5B, PHDBHC80, PHDARIE, PHD2DPF3b and PHDUHRF1 are displayed. For comparison, the recognition of H3K4me3 by PHDBPTF, PHDING2 and PHDTAF3 are also shown. Either H3K4me0 or H3K4me3 is depicted in yellow. (B) Sequence alignment of the PHD fingers (binding to H3K4me0) of KDM5 family members (BHC80, AIRE, DPF3b and UHRF1) and of the PHD fingers (binding methylated H3K4) of BPTF, ING2 and TAF3. The zinc-binding residues, H3A1-binding residues and H3R2-binding residues are highlighted in grey, blue and red, respectively. The H3K4-binding residues are highlighted in purple (in the KDM5 family), green (in all of the PHD fingers) and brown (in all of the PHD fingers except those of the KDM5 family)
Recognition of H3K4me0 by PHD1KDM5B affects KDM5B demethylase activity
The binding sites of unmodified H3K4 on PHD1KDM5B were first determined by an NMR titration binding assay (Fig. 2A and 2B). The majority of residues with chemical shifts larger than 0.3 ppm are involved in the interaction; this result is consistent with the results from the ITC assay (Table S1) and with the structural information of the complex. In the solution structure of PHD1KDM5B in complex with H3K4, residues D308, L325, D328 and W351 are involved in the interaction between PHD1KDM5B and H3K4 through hydrogen bonds, hydrophobic interactions, or salt bridges (Fig. 2). The single-site mutation of D308, L325, D328 or W351 to alanine disrupts the interaction between PHD1KDM5B and the H3K4me0 peptide. Based on these structural and biochemical analyses, we then designed three full-length KDM5B mutants (D308A, L325A/D328A and W351A) and tested whether the in vivo H3K4me2/3 demethylation by KDM5B is affected by disrupting the interaction between PHD1KDM5B and H3K4me0 through an immunofluorescence staining assay.
Compared with the wild-type (WT) KDM5B (99% of H3K4me3 was demethylated), the D308A mutant exhibited 79% demethylase activity on H3K4me3 (decreased by approximately 20%) (Fig. 4A). The mutants that completely lose their ability to binding to the unmodified H3K4 exhibited decreases in their demethylase activities to 72% (W351A) and 56% (L325A/D328A). For H3K4me2 demethylation (Fig. 4B), the wild-type KDM5B displays 93% demethylase activity, whereas the D308A, W351A and L325A/D328A mutants demonstrate 76%, 66% and 55% demethylase activities, respectively. In both cases, the L325A/D328A mutant has a higher effect on the demethylase activities of KDM5B. These data indicate that the KDM5B demethylase activity was partially affected by (but not dependent on) the interaction between the N-terminal PHD1 finger and the unmodified N-terminal H3K4 tail.

PHD1 KDM5B binding to H3K4me0 is important for KDM5B demethylase activity and KDM5B-mediated repression. Various mutants of KDM5B-myc proteins were expressed in 293T cells, and the histone modification levels were analyzed by immunofluorescent staining with (A) H3K4me3 and (B) H3K4me2 specific antibodies. The arrows indicate KDM5B-transfected cells. (C) KDM5B-mediated repression was affected by the disruption of the interaction between PHD1KDM5B and unmodified H3K4. The expression levels of BRCA1, CAV1, 14-3-3s and HOXA5 were analyzed in cells expressing wild-type and mutant KDM5B by real-time RT-PCR
Recognition H3K4me0 by PHD1KDM5B affects repression of tumor suppressor genes
KDM5B has been reported to function as a transcriptional repressor, and the knockdown of KDM5B increases the expression level of several tumor suppressor genes, including 14-3-3s, BRCA1, CAV1 and HOXA5 (Tan et al., 2003; Yamane et al., 2007). To illustrate the functional importance of the binding between PHD1KDM5B and the unmodified H3K4, we tested whether the mutants could affect the expression of tumor suppressor genes compared with wild-type KDM5B. As shown in Fig. 4C, the overexpression of WT KDM5B decreased the expression of tumor suppressor genes. Compared with WT KDM5B, the overexpression of the D308A, W351A and L325A/D328A mutants up-regulated the expression of the four genes to a lesser but still significant extent. Among the mutants, the L325A/D328A mutant increased the gene expression by more than twofold. Therefore, the gene expression results agree with the binding affinities of PHD1KDM5B and its mutants with the H3 peptide. The overexpression of the mutants is expected to produce fewer unmodified H3K4, thereby increasing the gene repression level. In combination, these findings support the model in which PHD1KDM5B specifically binds to the unmodified histone H3. Additionally, the H3 histone remains unmodified. This process is correlated with KDM5B demethylase activity and KDM5B-mediated gene repression.
Discussion
Comparison between PHD1KDM5B and other PHD fingers
The reported structure of PHDBHC80 in complex with unmodified H3K4 (PDB code: 2PUY) demonstrated that H3K4 residue forms hydrogen bonds with the side chains of D489 and E488 (Fig. 3A) (Lan et al., 2007). Moreover, the Cβ atom of H487 restricts the interaction of PHDBHC80 with methylated H3K4, which means that the H3K4me1 and H3K4me2 peptides cannot bind to PHDBHC80 (Lan et al., 2007). However, in the PHD1KDM5B-H3K4me0 complex, H3K4me0 recognition is enhanced by hydrophobic interactions of the side-chains of H3K4 with the aromatic ring of Y310 and the methyl group of L326. Therefore, the binding mode of PHD1KDM5B-H3K4me0 differs slightly from that of PHDBHC80. Recognition of H3K4me0 in PHD1KDM5B, PHDBHC80, PHDARIE (Chakravarty et al., 2009), the PHD finger in UHRF1 (i.e., PHDUHRF1, PDB code: 2LGG) (Hu et al., 2011; Wang et al., 2011; Lallous et al., 2011; Rajakumara et al., 2011) and the PHD2 finger in DPF3b (i.e., PHD2DPF3b, PDB code 2KWK) (Xie et al., 2012) occurs through a salt bridge between a conserved Asp (D) or Glu (E) residue in the PHD finger and the K4 side chain (Fig. 3). These differences result from the action of the conserved residues of glycine (G) in PHDBPTF and PHDING2 or cysteine (C) in PHDUHRF1 and PHD1KDM5B. In PHDBHC80, this space is occupied by residue M502. In comparison, methylated H3K4me3 recognition was found to occur primarily through the interaction between the positively charged H3K4me3 and the conserved aromatic residues, such as W32 in PHDBPTF, W238 in PHDING2 and W891 in PHDTAF3, as shown in Fig. 3. Other residues, including Y10 and Y17 in PHDBPTF, Y315 and M226 in PHDING2, and W868 and M882 in PHDING2, associate through hydrophobic interactions or aromatic Π-cation ion interactions to form the binding cage.
Thus, similar to PHDBHC80 and other PHD fingers (Fig. 3A), PHD1KDM5B does not use the aromatic cage to specifically identify H3K4, which is present in structurally characterized methylated lysine PHD fingers, such as the PHD fingers in BPTF (i.e., PHDBPTF, PDB code: 2F6J), ING2 (i.e., PHDING2, PDB code: 2G6Q) and TAF3 (i.e., PHDTAF3, PDB code: 2K17) (Fig. 3A) (Pena et al., 2006; Li et al., 2006). These PHD fingers adopt similar folds, engage the H3 peptide as an anti-parallel β-sheet on the surface and recognize the H3 N-amine and H3A1 side chain. H3R2 is buried in a pocket in PHDBPTF, PHDING2, PHDUHRF1 (Hu et al., 2011; Wang et al., 2011; Lallous et al., 2011; Rajakumara et al., 2011; Xie et al., 2012) and PHD2DPF3b (Zeng et al., 2010), bound in tiny surface grooves in PHDAIRE, PHDTAF3 and PHD1KDM5B, and not contacted in PHDBHC80.
The binding mode of PHD1KDM5B to H3K4me0 is also distinguished from the caging of the di- and tri-methyl lysine by aromatic residues, as identified in the polycomb (Pc) and heterochromatin protein I (HP1) chromodomain (Fischle et al., 2003; Nielsen et al., 2002; Jacobs and Khorasanizadeh, 2002; Min et al., 2003; Tan et al., 2003). The methylation of lysine 9 in histone H3 is recognized by HP1; this methylation directs the binding of other proteins to control chromatin structure and gene expression. The structures of the complex between the Drosophila HP1 chromodomain and the histone H3 tail with a di- or tri-methylated K9 display histone tail inserts as a β strand, completing the β-sandwich architecture of the chromodomain. The methylated lysine is caged by the side chains of the aromatic residues Y21, W42 and F45, whereas adjacent residues form discerning contacts with one face of the chromodomain. The structure of the Pc chromodomain in complex with a H3 peptide bearing trimethylated K27 (Fischle et al., 2003) demonstrates that the methylated H3K27 is caged by four aromatic residues (i.e., Y26, W47, W50 and Y54) preceding the ARKS motif.
Proposed biological function of PHD1KDM5B in demethylation by KDM5B
The binding of the PHD1KDM5B domain to the H3K4me0 peptide, which is a demethylation product of the full-length KDM5B, indicates that PHD1KDM5B may function downstream of KDM5B demethylase activity. This observation is similar to that found for BHC80 in LSD1-mediated H3K4me2 demethylation and repression (Lan et al., 2007). The regulation of histone methylation is highly dynamic and involves the actions of both a methyltransferase and demethylase on identical target promoters. Therefore, we suggest that PHD1KDM5B may be important in maintaining KDM5B at the target promoters and preventing the re-methylation of H3K4. The downstream effector of PHD1KDM5B is therefore required for the KDM5B-induced demethylation of H3K4me2/3. This was confirmed by our binding assay results, which showed that the removal of PHD1KDM5B resulted in decreased binding of KDM5B-N to the K4 unmethylated histone H3 peptide in vitro (Fig. 1C). Our findings indicate that PHD1KDM5B is important for KDM5B association with its reaction product H3K4me0 after demethylation. Thus, PHD1KDM5B operates both as a reader and a protector of unmethylated H3K4.
Similar to PHD1KDM5B, PHD1Lid has also been suggested to interact with H3K4me0 (Li et al., 2010). PHD1Lid was proposed to bind to non-DNA elements, such as local chromatin environments, during H3K4me2/3 demethylation by Lid. The C-terminal PHD3 region of Lid (i.e., PHD3Lid) was observed to specifically bind to H3K4me2/3 through the interaction of aromatic residues in the PHD finger with the positively charged methylated H3K4. By aligning the amino acid sequences of PHD3KDM5B, PHD3Lid and other PHD fingers that recognize the H3K4me2/3 peptide (Fig. 5), we found that the residues most likely interacting with the H3K4me2/3 site are highly conserved. Residues W1781 in PHD3Lid and W1512 in PHD3KDM5B are conserved corresponding to W32 in PHDBPTF, W238 in PHDING2, and W891 in PHDTAF3. We thus suggested that PHD3KDM5B might also specifically bind to methylated H3K4me2/3 and that the W1502 residue in PHD3KDM5B may have a biological function similar to that of residues W1771 in PHD3Lid, Y17 in PHDBPTF, M226 in PHDING2 and M882 in PHDTAF3. In mammalian cells, c-Myc prefers to bind to E-boxes located within a chromatin context that contain highly di- and tri-methylated nucleosomal histone H3K4 (Guccione et al., 2006). However, the mechanism through which Myc recognizes the chromatin landscape remains unclear. Here, we propose that KDM5B may utilize its H3K4me2/3-binding C-terminal PHD3 finger to tether Myc to its preferred chromatin context. This process may be enhanced by the interaction between PHD1KDM5B and the unmethylated H3K4me0 N-terminal tail, thereby permitting the selection of biologically important E boxes. Further experiments are required to more precisely define the roles of KDM5B PHD fingers in cell growth.

Sequence alignment of PHD fingers (binding to H3K4me3/2) of KDM5B (i.e., PHD3 KDM5B ), Lid (i.e., PHD3 Lid ), BPTF, ING2 and TAF3. The zinc-binding residues, H3A1-binding residues, H3R2-binding residues and H3K4me3/2-binding residues are highlighted in grey (and star on the top of KDM5B), blue, red and brown, respectively
In summary, we identified a specific interaction between PHD1KDM5B and the unmodified H3K4 peptide. We further provided structural insights into the binding. The specific recognition of unmodified H3K4 by the PHD1 domain of KDM5B is important for the KDM5B histone demethylase activity in cells and for the transcriptional repression of tumor suppressor genes.
Materials and methods
PHD1KDM5B preparation for crystallization and NMR experiments
The details of the preparation, purification, crystallization and determination of the PHDKDM5B (residues 306–360 aa) X-ray structure were previously described (Guo et al., 2011). The modified pGEX-6p-1 vectors expressed proteins with the N-terminal GST tag, which is removable by cleavage with a 3C protease, enabling the use of non-tagged proteins in our studies. Only the X-ray structure of free PHD1KDM5B was obtained.
A similar PHDKDM5B construct was used for the sample required in the NMR experiments. 13C- and 15N-labeled PHDKDM5B were prepared in M9 medium. Site-directed mutagenesis was performed using a QuikChange site-directed mutagenesis kit (Stratagene Inc. La Jolla, California, U.S.A). All of the DNA constructs were sequenced, and the molecular weights of the recombinant proteins were verified by mass spectrometry (MALDI).
H3 peptide synthesis
To study the binding affinities in vitro or to construct different NMR samples of the complex, H3K4 peptides without modifications (with the ARTKQTARKS sequence or ARTKQTARKSTGGKAPRKQLA sequence) or with some modifications (H3R2me1, R2 monomethylation; H3R2me2s, R2 symmetric dimethylation; H3R2me2a, R2 asymmetric dimethylation; H3K4me1/2/3, K4 mono-, di-, and tri-methylation; H3K9me1/2/3, K9 mono-, di- and tri-methylation; H3 peptide A1G, R2A, T3V, K4A, Q5E, T6V mutants without any modification) were purchased from GL Biochem Ltd. (Shanghai, China), and their purity was confirmed by HPLC and mass spectrometry.
GST pull-down and biotin pull down assays
For the biotin pull-down assay, biotin-labeled H3 peptides were used to pull down the PHD1KDM5B protein in order to determine the binding affinities. Briefly, 0.5 μg of the peptide was mixed with the protein at an identical molar ratio, and the mixture was incubated with streptavidin beads at 4°C overnight. After washing five times, the beads were boiled in SDS loading buffer and separated on a SDS-PAGE gel.
For the GST pull-down assay, histone peptides (0.5 μg) were incubated with 2–5 μg of purified recombinant GST-PHD1KDM5B for 2 h at 4°C in binding buffer (20 mmol/L Tris-HCl, pH 7.5, 150 mmol/L NaCl, 0.1% Triton X-100). The streptavidin beads (Upstate 16–126) were washed four times and stained with Coomassie blue.
NMR and isothermal titration calorimetry (ITC) binding assay
The 15N-labelled PHD1KDM5B and titrants (H3 peptide or its mutants) were mixed at a 1:6 molar ratio of PHDKDM5B:titrant in NMR buffer composed of 20 mmol/L Na2HPO3, 100 mmol/L NaCl, 0.01% NaN3, pH 7.4 and 10% D2O. The assignments of the cross peaks in the 2D 1H-15N HSQC spectrum were confirmed through NMR stepwise titration experiments using an increasing the molar ratio of PHDKDM5B:H3 as follows: 1:0.0, 1:0.6, 1:1.4, 1:2.0, 1:3.2, 1:6.2 and 1:7.2 (data not shown). The 1H-15N HSQC spectra were collected after each addition.
To investigate whether the PHD1KDM5B finger and its mutants interact with unmodified or methylated H3K4 or H3K4mutants, the binding affinities of PHD1KDM5B or its mutants to H3 peptides were studied. An ITC-200 microcalorimeter (GE Healthcare) was used with a buffer containing 20 mmol/L Tris, 150 mmol/L NaCl, and 1% Triton X-100, pH 7.5 at 25°C. The reference titration of small molecules in the buffer was subtracted from the experimental data, and the data were fitted using the Origin 7.0 (OriginLab Corporation) software. The results are summarized in Table S1.
NMR spectroscopy and analysis
The NMR samples contained 1.5 mmol/L uniformly 13C/15N-labelled PHDKDM5B and the unlabeled H3 peptide H3K4me0 in complex at a PHDKDM5B-to-H3-peptide molar ratio of 1:6 in NMR buffer (20 mmol/L Na2HPO3, 100 mmol/L NaCl, 0.01% NaN3, pH 7.4 and 10% D2O). All of the NMR experiments were performed at 20°C on a Varian Unity Inova 600 NMR spectrometer equipped with a triple resonances cryoprobe and pulsed field gradients. The standard suite of experiments for assigning the 1H, 13C and 15N backbone, determining the side-chain chemical shifts of PHDKDM5B in complex with the H3 peptide and collecting the NOE-based distance restraints were measured (Bax and Grzesiek, 1993; Clore and Gronenborn, 1998), and these included 2D 13C-edited HSQC and 15N-edited HSQC; 3D HNCA, HNCO, HN(CO)CA, HNCACB, CBCA(CO)NH, 15N-resolved HSQC-TOCSY and HCCH-TOCSY in both aliphatic and aromatic regions; 15N-resolved HSQC-NOESY; 13C-resolved HSQC-NOESY for both aliphatic and aromatic resonances; and 2D hbcbcgcdceheA and hbcbcgcdhdA spectra for the correlation of Cβ and Hδ or Hε in the aromatic ring that is used for aromatic proton assignment (Yamazaki et al., 1993). The proton NMR signals of the bound H3 peptides were assigned by analyzing the 2D 13C-filtered, 15N-filtered and J-resolved NOESY and TOCSY spectra recorded for the 13C- and 15N-labeled protein with the unlabeled H3 peptide H3K4me0 and the 2D 1H-1H COSY, NOESY and TOCSY spectra recorded for the unlabeled free H3 peptides in the NMR buffer mentioned above, respectively. The intermolecular NOEs between the labeled protein and the unlabeled H3 peptides were obtained by analyzing the 3D 13C-F1 edited and 13C/15N-F3 filtered NOESY spectra. The spectra were processed with the NMRPipe program (Delaglio et al., 1995) and analyzed using Sparky 3 (http://www.cgl.ucsf.edu/home/sparky/).
Determining the NMR structure
The calculations were performed using a standard simulated annealing protocol implemented in the XPLOR-2.19 program (NIH version)(Kuszewski and Clore, 2000). The inter-proton distance restraints derived from the NOE intensities were grouped into three distance ranges, namely 1.8–2.9 Å, 1.8–3.5 Å and 1.8–6.0 Å, which corresponds to strong, medium and weak NOEs, respectively. The dihedral angles phi and psi were derived from the backbone chemical shifts (HN, HA, CO and CA) using the program TALOS (Cornilescu et al., 1999). Slow-exchanging amide protons identified in the 2D 15N-1H HSQC spectra recorded after the H2O buffer was exchanged for a D2O buffer were used in the structure calculated with the NOE distance restraints to generate hydrogen bonds for the final structure calculation as previously described in the literature (Chakravarty et al., 2009). Constraints between the protein ligands and the zinc ion were added using a previously reported procedure (Neuhaus et al., 1992; Cao et al., 2003). A total of ten iterations were performed (50 structures in the initial eight iterations). In total, 100 structures were computed during the last two iterations, and the 20 conformers with the lowest energy were used to represent the 3D structures. The conformers of these two bundles (free PHD1KDM5B and PHD1KDM5B in complex with the H3K4me0 peptide) do not violate the following constraints: NOE > 0.3 Å and dihedral angle >3°. The entire structure statistics were evaluated with PROCHECK (Laskowski et al., 1993) and PROCHECK-NMR (Laskowski et al., 1996) and are summarized in Table 1. All of the structure figures were generated using the PyMOL (http://pymol.org/) and MOLMOL programs (Koradi et al., 1996).
Immunofluorescence staining
The 293T cells were transfected with pcDNA3-KDM5B-Myc-His WT and mutants using Lipofectamine 2000. After 48 h, the cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.5% Triton in PBS for 15 min. After blocking, the cells were incubated with a primary antibody (Millipore, anti-H3K4me2, 1:200 dilution), and washed three times with PBS, and incubated with fluorescence-labeled secondary antibody for 1 h (Molecular Probes, Alexa Fluor 555 goat anti-rabbit, 1:250 dilution). After extensive rinsing with PBS, cover slips were mounted with an antifade reagent and DAPI (Molecular Probes) and examined on an Olympus IX51 microscope.
Real-time qPCR assay
The pcDNA3-KDM5B-Myc-His WT and mutants were transfected into 293T cells with Lipofectamine 2000. Forty-eight hours after transfection, the total RNA was extracted using a standard protocol. Reverse transcription was conducted using the reverse transcriptase M-MLV from Promega. Real-time PCR was performed in triplicate using the SYBR Green PCR Mix (Promega) on an ABI 7500 sequence detection system (Applied Biosystems). Quantitative PCR reactions were performed under conditions that were standardized for each primer.
Abbreviations
- α-KG:
-
α-ketoglutarate
- JmjC:
-
Jumonji C
- LSD1:
-
lysine-specific demethylase 1
- WT:
-
wild-type
References
Bax A, Grzesiek S (1993) Methodological advances in protein NMR. Acc Chem Res 26(4):131–138
Cao C, Kwon K, Jiang YL, Drohat AC, Stivers JT (2003) Solution structure and base perturbation studies reveal a novel mode of alkylated base recognition by 3-methyladenine DNA glycosylase I. J Biol Chem 278(48):48012–48020
Chakravarty S, Zeng L, Zhou MM (2009) Structure and site-specific recognition of histone H3 by the PHD finger of human autoimmune regulator. Structure 17(5):670–679
Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, Hagman J, Hansen K, Shi Y, Zhang G (2006) Structural insights into histone demethylation by JMJD2 family members. Cell 125(4):691–702
Chen Z, Zang J, Kappler J, Hong X, Crawford F, Wang Q, Lan F, Jiang C, Whetstine J, Dai S, Hansen K, Shi Y, Zhang G (2007) Structural basis of the recognition of a methylated histone tail by JMJD2A. Proc Natl Acad Sci USA 104(26):10818–10823
Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128(6):1063–1076
Ciccone DN, Su H, Hevi S, Gay F, Lei H, Bajko J, Xu GL, Li E, Chen TP (2009) KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 461(7262):415–418
Cloos PA, Christensen J, Agger K, Maiolica A, Rappsilber J, Antal T, Hansen KH, Helin K (2006) The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 442(7100):307–311
Clore GM, Gronenborn A (1998) Determining the structures of large proteins and protein complexes by NMR. Trends Biotechnol 16(1):22–34
Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13(3):289–302
Couture JF, Collazo E, Ortiz-Tello PA, Brunzelle JS, Trievel RC (2007) Specificity and mechanism of JMJD2A, a trimethyllysine-specific histone demethylase. Nat Struct Mol Biol 14(8):689–695
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6(3):277–293
Drohat AC, Xiao G, Tordova M, Jagadeesh J, Pankiewicz KW, Watanabe KA, Gilliland GL, Stivers JT (1999) Heteronuclear NMR and crystallographic studies of wild-type and H187Q Escherichia coli uracil DNA glycosylase: electrophilic catalysis of uracil expulsion by a neutral histidine 187. Biochemistry 38(37):11876–11886
Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17(15):1870–1881
Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall’ Olio V, Zardo G, Nervi C, Bernard L, Amati B (2006) Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol 8(7):764–770
Guo X, Xu Y, Wang P, Li Z, Xu Y, Yang H (2011) Crystallization and preliminary crystallographic analysis of a PHD domain of human JARID1B. Acta Crystallogr Sect F 67(Pt 8):907–910
Horton JR, Upadhyay AK, Qi HH, Zhang X, Shi Y, Cheng X (2010) Enzymatic and structural insights for substrate specificity of a family of jumonji histone lysine demethylases. Nat Struct Mol Biol 17(1):38–43
Hu L, Li Z, Wang P, Lin Y, Xu Y (2011) Crystal structure of PHD domain of UHRF1 and insights into recognition of unmodified histone H3 arginine residue 2. Cell Res 21(9):1374–1378
Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y (2007) The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128(6):1077–1088
Jacobs SA, Khorasanizadeh S (2002) Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295(5562):2080–2083
Klose RJ, Yan Q, Tothova Z, Yamane K, Erdjument-Bromage H, Tempst P, Gilliland DG, Zhang Y, Kaelin WG Jr (2007) The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128(5):889–900
Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14(1):51–55, 29–32
Krishnakumar R, Kraus WL (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 39(5):736–749
Kuszewski J, Clore GM (2000) Sources of and solutions to problems in the refinement of protein NMR structures against torsion angle potentials of mean force. J Magn Reson 146(2):249–254
Lallous N, Legrand P, McEwen AG, Ramon-Maiques S, Samama JP, Birck C (2011) The PHD finger of human UHRF1 reveals a new subgroup of unmethylated histone H3 tail readers. PLoS One 6(11):e27599
Lan F, Collins RE, De Cegli R, Alpatov R, Horton JR, Shi X, Gozani O, Cheng X, Shi Y (2007) Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature 448(7154):718–722
Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 26:283–291
Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8(4):477–486
Lee MG, Norman J, Shilatifard A, Shiekhattar R (2007) Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell 128(5):877–887
Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442(7098):91–95
Li L, Greer C, Eisenman RN, Secombe J (2010) Essential functions of the histone demethylase lid. PLoS Genet 6:e1001221
Margueron R, Trojer P, Reinberg D (2005) The key to development: interpreting the histone code? Curr Opin Genet Dev 15(2):163–176
Martin C, Zhang Y (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6(11):838–849
Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O’Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448(7153):553–560
Min J, Zhang Y, Xu RM (2003) Structural basis for specific binding of polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17(15):1823–1828
Mosammaparast N, Shi Y (2010) Reversal of histone methylation: biochemical and molecular mechanisms of histone demethylases. Annu Rev Biochem 79:155–179
Neuhaus D, Nakaseko Y, Schwabe JW, Klug A (1992) Solution structures of two zinc-finger domains from SWI5 obtained using two-dimensional 1H nuclear magnetic resonance spectroscopy. A zinc-finger structure with a third strand of beta-sheet. J Mol Biol 228(2):637–651
Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, Rose NR, Offer J, Scheinost JC, Borowski T, Sundstrom M, Schofield CJ, Oppermann U (2007) Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 448(7149):87–91
Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED (2002) Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature 416(6876):103–107
Pelton JG, Torchia DA, Meadow ND, Roseman S (1993) Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci 2(4):543–558
Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG (2006) Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442(7098):100–103
Rajakumara E, Wang Z, Ma H, Hu L, Chen H, Lin Y, Guo R, Wu F, Li H, Lan F, Shi YG, Xu Y, Patel DJ, Shi Y (2011) PHD finger recognition of unmodified histone H3R2 links UHRF1 to regulation of euchromatic gene expression. Mol Cell 43(2):275–284
Shi Y, Whetstine JR (2007) Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 25(1):1–14
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119(7):941–953
Sims RJ 3rd, Nishioka K, Reinberg D (2003) Histone lysine methylation: a signature for chromatin function. Trends Genet 19(11):629–639
Szutorisz H, Canzonetta C, Georgiou A, Chow CM, Tora L, Dillon N (2005) Formation of an active tissue-specific chromatin domain initiated by epigenetic marking at the embryonic stem cell stage. Mol Cell Biol 25(5):1804–1820
Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y (2007) The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature 447(7144):601–605
Tan K, Shaw AL, Madsen B, Jensen K, Taylor-Papadimitriou J, Freemont PS (2003) Human PLU-1 has transcriptional repression properties and interacts with the developmental transcription factors BF-1 and PAX9. J Biol Chem 278(23):20507–20513
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439(7078):811–816
Wang C, Shen J, Yang Z, Chen P, Zhao B, Hu W, Lan W, Tong X, Wu H, Li G, Cao C (2011) Structural basis for site-specific reading of unmodified R2 of histone H3 tail by UHRF1 PHD finger. Cell Res 21(9):1379–1382
Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125(3):467–481
Xie S, Jakoncic J, Qian C (2012) UHRF1 double tudor domain and the adjacent PHD finger act together to recognize K9me3-containing histone H3 tail. J Mol Biol 415(2):318–328
Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y (2007) PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 25(6):801–812
Yamazaki T, Forman-Kay J, Kay LE (1993) Two-dimensional NMR experiments for correlating carbon-13. beta. and proton. delta./. epsilon. chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J Am Chem Soc 115(23):11054–11055
Yang Y, Hu L, Wang P, Hou H, Lin Y, Liu Y, Li Z, Gong R, Feng X, Zhou L, Zhang W, Dong Y, Yang H, Lin H, Wang Y, Chen CD, Xu Y (2010) Structural insights into a dual-specificity histone demethylase ceKDM7A from Caenorhabditis elegans. Cell Res 20(8):886–898
Zeng L, Zhang Q, Li S, Plotnikov AN, Walsh MJ, Zhou MM (2010) Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 466(7303):258–262
Zhang Y, Reinberg D (2001) Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev 15(18):2343–2360
Acknowledgements
This work was supported by funding from the National Basic Research Program of China under 2011CB966300, from National Science Foundation of China No. 21272261, 21275154 and 31100526, and Shanghai Pujiang Program No.13PJD006. We thank Prof. Degui Chen and the members in his lab (Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences) for help with in vitro biotin-H3 binding assay.
COMPLIANCE WITH ETHICS GUIDELINES
Yan Zhang, Huirong Yang, Xue Guo, Naiyan Rong, Yujiao Song, Youwei Xu, Wenxian Lan, Xu Zhang, Maili Liu, Yanhui Xu and Chunyang Cao declare that there is no conflict of interest.
This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Yan Zhang, Huirong Yang, and Xue Guo contributed equally to the paper.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, Y., Yang, H., Guo, X. et al. The PHD1 finger of KDM5B recognizes unmodified H3K4 during the demethylation of histone H3K4me2/3 by KDM5B. Protein Cell 5, 837–850 (2014). https://doi.org/10.1007/s13238-014-0078-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0078-4