
ABSTRACT
Transforming growth factor-β (TGF-β) members are key cytokines that control embryogenesis and tissue homeostasis via transmembrane TGF-β type II (TβR II) and type I (TβRI) and serine/threonine kinases receptors. Aberrant activation of TGF-β signaling leads to diseases, including cancer. In advanced cancer, the TGF-β/SMAD pathway can act as an oncogenic factor driving tumor cell invasion and metastasis, and thus is considered to be a therapeutic target. The activity of TGF-β/SMAD pathway is known to be regulated by ubiquitination at multiple levels. As ubiquitination is reversible, emerging studies have uncovered key roles for ubiquitin-removals on TGF-β signaling components by deubiquitinating enzymes (DUBs). In this paper, we summarize the latest findings on the DUBs that control the activity of the TGF-β signaling pathway. The regulatory roles of these DUBs as a driving force for cancer progression as well as their underlying working mechanisms are also discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Protein ubiquitination is a reversible process. Deubiquitinating enzymes (DUBs) function to remove covalently conjugated ubiquitins from their target proteins to regulate substrate activity and/or abundance (Nijman et al., 2005). DUBs have amongst others been implicated in cellular signaling pathways that control cell proliferation and differentiation. TGF-β/SMAD signaling can play a tumor promoting role in advanced cancer and certain essential components of this pathway, TGF-β receptors and SMADs are known to be downregulated via protein ubiquitination by E3 ligases (Massague, 2008a). Multiple DUBs have been shown to target ubiquitinated TGF-β/SMAD signaling components and to be associated with high risk for cancer metastasis, both in animal models and in clinical analysis (Eichhorn et al., 2012; Inui et al., 2011; Zhang et al., 2012a, b). As the DUBs are druggable proteins, these studies may provide possibilities for novel and effective therapeutic treatments (Cohen and Tcherpakov, 2010; Hoeller and Dikic, 2009). This paper revisits the signal transduction mechanisms and biological features of TGF-β/SMAD pathways, followed by an overview of the ubiquitination regulation of the TGF-β/SMAD pathways by ubiquitination and a brief introduction of the human DUB family. It finally highlights the newly identified DUB members acting on TGF-β/SMAD signaling as well as their emerging roles in the regulation of cancer invasion and metastasis.
TGF-β SIGNALING
Signaling pathways induced by the transforming growth factor-β superfamily
The TGF-β superfamily contains a number of structurally and functionally related secreted cytokines. Since TGF-β was discovered in 1983 (Frolik et al., 1983), more than 30 members of this family have been identified and verified. Members of the TGF-β family are characterized by the highly conserved cysteine residues, also known as the cystine knot (CK) motif (Galat 2011). According to the sequences similarities and their distinct downstream signaling pathways, the TGF-β superfamily can be divided into several subfamilies, including TGF-βs, bone morphogenetic proteins (BMPs), nodal, growth and differentiation factors (GDFs), Müllerian inhibitory factor (MIF), activins and inhibins (Massaous and Hata, 1997). Although different TGF-β members have distinct cellular functions, they all act on cells as dimers.
The TGF-β family members bind to the type I and type II serine/threonine kinase receptors on the cell surface. The serine/threonine kinase receptor family contains twelve members, that are seven type I receptors, also known as activin receptor-like kinases (ALKs), and five type II receptors (Huang et al., 2011; Massague, 2008b). Both type I and type II receptors are expressed ubiquitously in mammalian cells.
The canonical intracellular signaling induced by TGF-β ligands is mediated by SMAD family proteins. Based on their function differences, the SMAD family is divided into three groups, that are receptor-associated SMADs (R-SMADs), co-operating SMADs (Co-SMADs), and inhibitory SMADs (I-SMADs) (Ross and Hill, 2008). Only R-SMADs are targeted for activation via phosphorylation by the active type I receptor kinase. In general, diverse TGF-β ligand binds to and activates a characteristic combination or combinations of different type I and type II receptors on the plasma membrane, and targets specific R-SMADs for activation. Upon TGF-β-induced receptor complex formation, TβRII kinase phosphorylates TβRI, e.g. ALK5, on specific serine and threonine residues in its juxtamembrane. Subsequently, the activated ALK5 induces the phosphorylation of the R-SMADs SMAD2 and SMAD3, which can form heteromeric complexes with the Co-SMAD SMAD4. SMAD2/3 can be activated by TGF-βs, activins, and nodal upon complex formation between ALK4/5/7 and TGF-β type II receptor (TβRII) and activin receptor 2 (ACVR2). SMAD1/5/8, can be activated by BMP ligands through complex formation between the type I receptor ALK1/2/3/6 and BMP type II receptor (BMPRII) or ACVR2. The Co-SMAD SMAD4 functions as a central transducer in the TGF-β responses. The two I-SMADs, SMAD6 and SMAD7, enable tight control of TGF-β signaling through negative regulation: they can compete with Co-SMAD for the interaction with the phosphorylated R-SMADs and they can recruit SMURF E3 ubiquitin ligase to the type I receptors (Itoh and ten Dijke, 2007; Kavsak et al., 2000).
The heteromeric SMAD complexes formed by the activated R-SMADs and SMAD4 accumulate in the nucleus, where they regulate target gene expression (Fig. 1) (Heldin et al., 1997). In addition to this canonical SMAD-dependent TGF-β signaling pathway, there are other non-SMAD pathways that can be activated by the TGF-β receptors via either phosphorylation or direct interaction. These non-SMAD pathways include various branches, such as mitogen activated protein kinases (MAPKs) pathways, phophoinositde 3-kinase (PI3K)/Akt pathways, nuclear factor κB (NF-κB) pathways, and Rho-like GTPase pathways (Fig. 1) (Derynck and Zhang, 2003; Mu et al., 2012; Sanchez-Elsner et al., 2001; Zhang, 2009).

A schematic representation of the TGF-β signaling pathway. Upon ligand binding, the TGF-β type II receptor (TβRII) recruits and phosphorylates the type I receptor (TβRI) in the cell membrane, and then the activated type II-I heteromeric receptor complex can induce phosphorylation of R-SMADs. Activated R-SMADs can recruit Co-SMAD (SMAD4) for binding. The R-SMAD/Co-SMAD complexes translocate and accumulate in the nucleus and then initiate the expression of the target transcription factors with the help of other co-factors. TGF-β can in addition promote other intracellular (non-SMAD) signaling pathways, such as mitogen activated protein kinases (MAPKs) pathways, the phosphoinositide 3-kinase (PI3K)/AKT pathway, the nuclear factor κB (NF-κB) pathway, and the Rho-like GTPase pathway
Functions of TGF-β signaling
TGF-β/SMAD signaling is multifunctional in regulating cell growth, differentiation, apoptosis, migration and invasion/metastasis (Goumans and Mummery, 2000; Hogan, 1996; Massague et al., 2000; Proetzel et al., 1995; Sanford et al., 1997; Schier, 2003; Whitman, 1998). Disturbances of TGF-β/SMAD signaling are widely shown to be involved in human diseases, including hereditary hemorrhagic telangiectasia, fibrosis diseases, atherosclerosis, hereditary synostosis, hereditary chondrodysplasias, cleidocranial dysplasia and familial primary pulmonary hypertension (Blobe et al., 2000; Massague et al., 2000). In human cancer, TGF-β/SMAD signaling can have a dual role. In the early phase of tumor progression, TGF-β/SMAD plays a tumor suppressing role (Massague et al., 2000). On the contrary, TGF-β/SMAD can promote advanced tumor progression such as tumor cell invasion, dissemination/metastasis, and immune evasion (Massague, 2008a). Thus the functional outcome of the TGF-β response is context-dependent and determined both by cell, tissue, and cancer types.
TGF-β signaling inhibits cell proliferation in a multitude of cell types, including normal endothelial, epithelial, hematopoietic, and neural cells, certain types of mesenchymal cells, and especially many primary cancer cells (Massague et al., 2000). TGF-β can downregulate the c-Myc oncogene levels thereby counteracting Myc-induced cell proliferation via upregulation of cyclins and downregulation of p21 (also known as WAF1) (Dang, 1999; Warner et al., 1999). TGF-β can also induce growth arrest by its inhibitory role on cyclin-dependent kinases (CDK) via upregulation of p15 (also termed as INK4B) and p21 expressions and downregulation of CDC25A expression (Claassen and Hann, 2000; Iavarone and Massague, 1997). The tumor suppressing role of TGF-β/SMAD pathway seems particularly critical in the gastro-intestinal tract, since large subsets of pancreatic, gastric, and colon cancers carry mutations or deletions in TGF-β receptors or SMADs (Grady et al., 1999; Markowitz et al., 1995; Myeroff et al., 1995; Parsons et al., 1995; Schutte et al., 1996; Hahn et al., 1996; Schutte et al., 1996; Yakicier et al., 1999).
Advanced cancers such as gliomas, breast and prostate cancers usually do not acquire mutations in the core components of TGF-β/SMAD signaling, but can bypass the TGF-β/SMAD tumor-suppressive arms through other, more downstream (epi)genetic changes, allowing the tumor promoting arm of TGF-β/SMAD signaling to actively drive tumor cell progression (Jennings and Pietenpol, 1998; Jones et al., 2009; Takenoshita et al., 1998; Vincent et al., 1996; Xu et al., 2009). Tumors with such signatures are resistant to TGF-β/SMAD mediated growth arrest but can undergo epithelial-to-mesenchymal transition (EMT) and invasion/metastasis. EMT is a process required for embryonic development and wound healing, but is employed by tumor cells to invade normal tissue and/or spread to distant organs. During EMT, carcinoma cells lose cell polarity and cell-cell contacts, and acquire fibroblastic-like properties as evidenced by morphological observations at the invasive fronts of human tumors (Kalluri and Weinberg, 2009; Katsuno et al. 2012). The TGF-β/SMAD pathway is a critical regulator of EMT in development in vivo (Kaartinen et al., 1995). In tumor cells, SMAD3/SMAD4 mediates transcription of SNAIL and SLUG, two master regulators of the EMT process (Miyazono, 2009; Naber et al. 2013). TGF-β/SMAD signaling also strongly drives the appearance of various molecular hallmarks of cells undergoing EMT, such as the decreased expression of epithelial cell-cell junction proteins including E-cadherin and zona occludens 1 (ZO-1), and at the same time it can induce the expression of mesenchymal markers, such as N-cadherin, vimentin, α-smooth muscle actin (α-SMA), and fibronectin (Heldin et al., 2009; Huber et al., 2005; Moustakas and Heldin, 2007; Xu et al., 2009).
The TGF-β induced pathways also can enable advanced invasive tumor cells to disseminate to other organs and form metastatic lesions (Bos et al., 2010; Nguyen et al., 2009). TGF-β stimulated metastatic dissemination is typically studied in bone and lung metastases of breast and prostate tumors. For instance, the SMAD3/SMAD4 complex was found to mediate the induction of connective tissue growth factor (CTGF) and interleukin (IL-11), which are critical factors for bone metastasis (Deckers et al., 2006; Kang et al., 2005; Kang et al., 2003; Petersen et al., 2010). By inducing angiopoietin-like 4 (ANGPTL4), TGF-β primes dissemination towards the lung (Padua et al., 2008). An increasing amount of studies provide evidences that the TGF-β/SMAD pathway is widely involved in multiple processes of cancer metastasis, including early invasion, intravasation, and later extravasation and colony formation (Drabsch and ten Dijke, 2012).
UBIQUITINATION AND ITS ROLE IN TGF-β SIGNALING
Ubiquitin and ubiquitination
Ubiquitin is a small regulatory protein (76 amino acids) that exists in almost all kinds of eukaryotic cells. Ubiquitin has originally been characterized as a covalently attached signal for ATP-dependent proteasomal degradation of substrate proteins (Hershko and Ciechanover, 1998), although it also plays a role in both the lysosomal and autophagic degradation pathways (Clague and Urbe, 2010). In addition to the protein degradation pathways, ubiquitin attachment is also implicated in dynamic cellular events, such as the transduction of cellular signals, gene transcription as well as DNA damage and repair (Hunter, 2007; Jackson and Durocher, 2013). Ubiquitin contains seven lysine residues in its sequence and each of them allows polyubiquitin chain conjugation via a covalently linking to the carboxyl end of another ubiquitin (Pickart and Eddins, 2004).
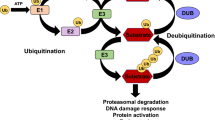
Ubiquitination is an enzymatic and post-translational modification process involving covalently linking of one ubiquitin (monoubiquitination) or more ubiquitins (polyubiquitination) to the substrate protein. The conjugation process of ubiquitin to the substrate normally requires three steps: a) the initial step is to activate the C-terminus of the ubiquitin protein by a ubiquitin-activating enzyme (E1), b) the intermediate step is to transfer and conjugate ubiquitin from the E1 enzyme and conjugate to an ubiquitin-conjugating enzyme (E2), c) the last step is to covalently conjugate the ubiquitin protein to the substrate protein which is normally facilitated by a substrate-specific ubiquitin ligase (E3) (Fig. 2) (Dikic, 2009; Pickart and Eddins, 2004; Schwartz and Ciechanover, 2009; Weissman, 2001). Two types of E3 ligases can facilitate this last step: one group of E3 ligases carries an E6-AP carboxyl terminus (HECT) domain, via which the E2 ligase can transfer the ubiquitin to the final substrate protein, the other group is characterized by a so-called really interesting new gene (RING) domain that may help to transfer E2-ubiquitin to the protein substrate (Dikic, 2009; Pickart and Eddins, 2004; Schwartz and Ciechanover, 2009; Weissman, 2001). Ubiquitination can alter the activity or localization of the substrate protein (mainly in case of monoubiquitination), target substrate proteins for degradation, or allow proteins to function as a scaffold (mainly via polyubiquitination) (Pickart and Eddins, 2004). In the case of polyubiquitination, there are at least 8 different types of poly ubiquitins linkages (Lysine-6, Lysine-11, Lysine-27, Lysine-29, Lysine-33, Lysine-48 and Lysine-63 polyubiquitination, and linear ubiquitination) can exist in the cells (Dikic, 2009; Weissman, 2001). Importantly different types of polyubiquitination linkages dictate distinct functions. For example, poly ubiquitins linked with Lysine-48 provide the main targeting signals for proteasomal degradation, whereas polyubiquitins linked with Lysine-63 enable the substrate protein to function as scaffolds to recruit other partners and thereby to participate in multiple cell processes, such as kinase activation, DNA repair, and protein synthesis (Schwartz and Ciechanover, 2009).

An overview of ubiquitination and deubiquitination processes. The conjugation process of ubiquitin to the substrate proteins normally requires three steps: a) the initial step is to activate the C-terminus of the ubiquitin protein by an E1 and this process requires ATP, b) the intermediate step is to transfer ubiquitin from the E1 and conjugate to an E2, c) the final step is to covalently conjugate the ubiquitin to the substrate protein which normally facilitated by an E3 enzyme. DUBs can catalyse the removal of ubiquitin from the conjugated substrates and also generate free ubiquitins from the newly translated polyubiquitins or recycle ubiquitins after the polyubiquitinated protein substrates are degraded (For further details see text)
Ubiquitination regulation in TGF-β signaling
Ubiquitination modifies a series of TGF-β pathway components, including receptors, R-SMADs, Co-SMAD, I-SMADs, and their regulators, via different E3 ubiquitin ligases (Inoue and Imamura, 2008). TβRI can be polyubiquitinated by SMAD-ubiquitination-related factor (SMURF) 1/2, WW domain-containing protein 1 (WWP1) and neural precursor cells-expressed developmentally down-regulated 4 (NEDD4)-2 with the help of the inhibitory SMAD7 (Ebisawa et al., 2001; Kavsak et al., 2000; Komuro et al., 2004; Kuratomi et al., 2005). This alters receptor stability on the membrane as well as the receptor internalization/endocytosis status and thus tightly restricts sensitivity of cells towards TGF-β stimulation. SMAD protein stability is also controlled by polyubiquitination. SMAD1 can be polyubiquitinated by SMURF1/2 and carboxyl terminus of Hsc70-interacting protein (CHIP) (Li et al., 2004; Zhang et al., 2001; Zhu et al., 1999). SMAD2 is reported to be polyubiquitinated by SMURF2, NEDD4L, or WWP1 (Kuratomi et al., 2005; Lin et al., 2000; Seo et al., 2004). SMAD3 is polyubiquitinated by CHIP (Xin et al., 2005). Phosphorylated SMAD2/3 can be polyubiquitinated by ARKADIA after the target gene transcription is initiated (Mavrakis et al., 2007). SMAD7 is shown to be targeted for polyubiquitination by ARKADIA and RNF12 (Koinuma et al., 2003; Liu et al., 2006; Zhang et al., 2012a; Zhang et al., 2012b). Similar to R-SMADs, SMAD4 could also be polyubiquitinated by the HECT domain ubiquitin E3 ligases SMURFs, WWP1, or NEDD-2 (Moren et al., 2005). Besides TGF-β receptors and SMADs, other key regulators of TGF-β signaling pathway can also be polyubiquitinated for degradation. As negative regulator of the TGF-β pathway, SNON is polyubiquitinated and targeted for degradation by SMURF2 or anaphase-promoting complex (APC) (Bonni et al., 2001; Stroschein et al., 2001).
In addition to activation of (canonical) signaling via the SMADs, the TGF-β receptor complex can also recruit TNF receptor-associated factor (TRAF) 4 and TRAF6, which then by K63-polyubiquitination activates the effector kinase TGF-β-activated kinase 1 (TAK1); TAK1 subsequently phosphorylates MAPK kinases, leading to activation of p38 or JNK (Sorrentino et al., 2008; Yamashita et al., 2008; Zhang et al., 2013a). Moreover, recent studies reveal a critical function for monoubiquitination on SMADs. The transcriptional activity of SMAD4 was shown to be antagonized upon monoubiquitination by Ectodermin/TRIM33/TIF1γ (Dupont et al., 2009). Similarly, monoubiquitination of R-SMADs triggered by SMURF has been shown to exert an inhibitory role (Inui et al., 2011; Tang et al., 2011). In addition, conjugation of a single ubiquitin molecule conjugation to SMAD6 by the E2 enzyme UBE2O appears already sufficient to attenuate the inhibitory function of SMAD6 on BMP signaling (Zhang et al., 2013b).
Deubiqutination and human dub family members
Deubiquitination
Ubiquitination is a reversible modification process and is counteracted by a process termed deubiquitination. Deubiquitination involves the removal of ubiquitin from its conjugates by deubiquitinating enzymes/deubiquitinases (DUBs) (Amerik and Hochstrasser, 2004; Nijman et al., 2005). DUBs are a large group of proteases that cleave ubiquitins from proteins (Nijman et al., 2005). DUBs also assist to generate free molecules from the newly translated polyubiquitins and recycle ubiquitins after the polyubiquitinated protein substrates are degraded (Fig. 2) (Komander et al., 2009). Therefore, DUBs play key roles in the regulation of signal transduction by controlling ubiquitin homeostasis thereby affecting the stability, activity and/of subcellular localization of proteins (Komander et al., 2009).
The human DUB family
The human genome encodes almost 90 DUBs of which 79 are predicted to be active. According to the sequence similarity and the possible functions, the DUBs family can be divided into 5 subfamilies, including ubiquitin-specific proteases (USPs), ubiquitin C-terminal hydrolases (UCHs), ovarian tumor proteases (OTUs), Machado-Joseph disease proteases (MJDs), and JAB1/MPN/Mov34 proteases (JAMMs) (Fig. 3) (Komander et al., 2009; Nijman et al., 2005; Reyes-Turcu et al., 2009).

A schematic summary of human DUB family. The DUB family can be divided into five subfamilies, including ubiquitin-specific proteases (USPs, 57 members), ubiquitin C-terminal hydrolases (UCHs, 4 members), ovarian tumor proteases (OTUs, 14 members), Machado-Joseph disease proteases (MJDs, 4 members), and JAB1/MPN/Mov34 proteases (JAMMs, 11 members)
Functions of DUBs
Since polyubiquitination can serve as a tag for protein destruction thus allows DUB mediated deubiquitination of such polyubiquitinated proteins will promote protein stability. USP1 for instance stabilizes inhibitors of DNA binding proteins (IDs) through deubiquitination (Williams et al., 2011). HAUSP (also termed as USP7) deubiquitinates p53, and is therefore considered to be an important positive regulator of p53 stabilization (Li et al., 2002). USP28 is overexpressed in colon and breast tumors, and by counteracting the ubiquitination activity of SCF-Fbxw7 ligase it causes the stabilization of cyclin E1 and c-Myc (Popov et al., 2007a; Popov et al., 2007b). Similarly, USP33 promotes centrosome biogenesis via specific and potent stabilization of the centriolar coiled coil protein CP110 (Li et al., 2013), and USP22 has been found to strengthen the NAD-dependent histone deacetylase Sirt1 to antagonize p53 activation (Lin et al., 2012).
Conceptually, removal of monoubiquitination by DUBs should also reverse for instance substrate localization or substrate-induced transcription activation in case monoubiqutination of the substrate has theses effects (see previous chapter). Indeed, such effects were reported for HAUSP-mediated ubiquitin removal of PTEN (phosphatase and tensin homologue deleted in chromosome 10) and FOXO (Forkhead box O) 4 (Song et al., 2008; van der Horst et al., 2006).
Another important function of DUBs is exemplified by their capability to reverse the non-degradative polyubiquitin chain conjugation on central signaling molecules. For instance, AMSH and AMSH-LP promote receptor trafficking by specifically cleaving Lysine-63 linked polyubiquitin chains from internalized receptors (McCullough et al., 2004; Sato et al., 2008) and the deubiquitinases CYLD, A20 and USP4 antagonize Lysine-63 polyubiquitin chain conjugation on TRAF6, thereby disrupting the docking sites for downstream innate immune signaling activation (Boone et al., 2004; Brummelkamp et al., 2003; Deng et al., 2000; Kovalenko et al., 2003; Trompouki et al., 2003; Turer et al., 2008; Xiao et al., 2012; Zhang et al., 2012a; Zhang et al., 2012b). Similarly, linear polyubiquitin chain formation on NF-κB essential modulator (NEMO) by the E3 ligase linear ubiquitin chain assembly complex (LUBAC) is cleaved by CYLD and more specifically by OTULIN (also termed as FAM105B) (Gerlach et al., 2011; Ikeda et al., 2011; Keusekotten et al., 2013; Niu et al., 2011; Rivkin et al.2013; Tokunaga et al., 2009). In the Wnt signal transduction pathway, CYLD inhibits β-catenin signaling by removing Lysine-63 linked ubiquitination from Dishevelled (Tauriello et al., 2010). Moreover, nuclear functions of DUBs in transcription and RNA processing have been uncovered (Clague et al., 2012). In this article, we will further focus on recent advances that help to understand the role of DUBs in TGF-β/SMAD signaling.
FUNCTIONAL DUBS IN TGF-β SIGNALING
Unlike the regulation of TGF-β signaling by ubiquitination, which has been intensely studied for the last decades, the role of DUB-mediated deubiquitination in the TGF-β signaling pathway is only recently emerging. It is until recently that a few reports just unveil this mystery in which several functional DUBs have now been identified and found to be potent TGF-β/SMAD modulators (Table 1) (Al-Salihi et al., 2012; Dikic, 2009; Eichhorn et al., 2012; Schwartz and Ciechanover, 2009; Wicks et al., 2005; Zhang et al., 2012a; Zhang et al., 2012b; Zhao et al., 2011).
UCH37 as the first identified DUB in TGF-β/SMAD pathway
UCH37, a member of the UCH enzymes subfamily, and also known as UCHL5 in mouse, has been identified as a SMAD3-binding partner (Wicks et al., 2005). Previously, it was shown to function as a component of the 26S proteasome and thus might play a role in the editing of polyubiquitinated protein substrates (Weissman, 2001). UCH37 also interacts with SMAD7 through the SMAD7 N-terminal domain (1–260 aa), and not via the PY motif, a region that mediates SMAD7’s binding to SMURF (Wicks et al., 2005). Via SMAD7, UCH37 can further be recruited to TβRI, where it removes polyubiquitin chains synthesized by SMURF (Wicks et al., 2005).
USP4 is a DUB for TGF-β type I receptor
USP4, a member of USP subfamily, is the first deubiquiting enzymes that have been identified in mammalian cells. USP4 is a very stable protein as it can deubiquitinate itself (Wada and Kamitani, 2006). In the past year, gathered observations by several groups have revealed that USP4 is widely involved in multiple signaling pathways including the Wnt/β-catenin pathway, the innate immune response pathway, p53 signaling pathway and in particularly the TGF-β/SMAD signaling pathway (Liu et al., 2002; Xiao et al., 2012; Zhang et al., 2012a; Zhang et al., 2012b; Zhao et al., 2009). In a genome wide gain-of-function screen that covered nearly 27,000 genes, USP4, as well as USP11/USP15 were found to play a strong activating role in TGF-β/SMAD signaling. It is not so surprising USP4/11/15 share the ability to potentiate TGF-β/SMAD signaling because they share highly conserved domains and similarity in their protein sequences (Fig. 4). As to underlying mechanism USP4 was demonstrated to deubiquitinate and stabilize TβRI in the plasma membrane through direct association (Zhang et al., 2012a; Zhang et al., 2012b) (Fig. 5). A series of in vitro and in vivo experiments showed that USP4 is a critical and selective regulator of TGF-β/SMAD signaling in mammalian cells and zebrafish embryos. The fact that USP4 is highly expressed in various cancers indicated a critical role for USP4 in the tumor-promoting arm of the TGF-β/SMAD pathway. Indeed, analysis in malignant breast cancer cells revealed that USP4 could regulate TGF-β-induced EMT, migration in vitro and stimulate TGF-β/SMAD signaling-dependent breast cancer invasion and metastasis in vivo (Zhang et al., 2012a; Zhang et al., 2012b). Importantly, USP4 could bind to itself and also interact with USP11 and USP15, and thus may be part of a DUB complex when exerting its function (Zhang et al., 2012a; Zhang et al., 2012b). Interestingly, USP4 was found also to associate with AKT and to be phosphorylated by AKT on it conserved Ser445 motif. This phosphorylation promotes USP4 localization in membrane and cytoplasm, where USP4 deubiquitylates TβRI. This study suggests that Akt activation in breast cancer cells induces USP4 to relocate and stabilize TβRI in the plasma membrane, and thereby enforces TGF-β-induced pro-tumorigenic responses (Zhang et al., 2012a; Zhang et al., 2012b). Moreover, aberrant over-activation of PI3K/AKT pathway is frequently observed in human cancers and this could blunt tumor suppressing pathways. PI3K/AKT activation may thus redirect TGF-β intracellular signaling and thereby contribute to its switch from tumor suppressor to tumor promoter.

Alignment of USP4 and its paralogs USP11 and USP15. The highly similar domain structure of USP4, USP11, and USP15 is schematically illustrated; the degree of identity is also shown. Overall, USP4 shares 46.7% identity with USP11, and 59.6% identity with USP15. USP11 shares 45.9% identity with USP15 (For further details see text)

Effects of USP4, USP15, and USP9X on TGF-β type I receptor and SMADs. As depicted schematically, USP4 associates with and deubiquitinates TβRI and recruits other DUBs. USP15 binds TβRI via SMAD7 and competes with SMURF2 to balance TβRI ubiquitination. The transcriptional activity of R-SMAD can be restricted by the SMURFs via monoubiquitination and reversed by USP15. USP9X reverses SMAD4 monoubiquitination that can be conjugated by Ectodermin/TIF1γ E3 ligase
USP11 is another DUB for TGF-β type I receptor
Like USP4, USP11 is involved in multiple signaling pathways. For instance, USP11 has been shown to associate with and stabilize RanGTP-associated protein RanBPM, BRCA2, HPV-16E7, nucleoprotein (Jennings and Pietenpol, 1998), and IκBα, depending on its DUB activity (Ideguchi et al., 2002; Liao et al., 2010; Lin et al., 2008; Schoenfeld et al., 2004; Sun et al. 2009). In a DUB activity independent manner, USP11 is involved in the IκB kinase α (IKKα)-p53 signaling pathway and also function in the regulation of DNA double-strand repair (Wiltshire et al., 2010; Yamaguchi et al., 2007). In addition to the USP4 study described above (Zhang et al., 2012a; Zhang et al., 2012b), an independent study identified USP11 as positive regulator of TGF-β signaling (Al-Salihi et al., 2012). In this study USP11 was identified by a proteomic approach in search for novel binding partners of TGF-β signaling components. USP11 was found to interact with SMAD7 and be recruited via SMAD7 to deubiquitinate TβRI and promote TGF-β signaling (Al-Salihi et al., 2012).
USP15 is a DUB for both TGF-β type I receptor and R-SMADs
A DUB RNAi library mediated loss-of-function screen also identified USP15 as a key regulator of TGF-β signaling (Eichhorn et al., 2012). Distinct from USP4, USP15 was not found to bind to activate TβRI directly; rather, it is recruited to the active receptor by SMAD7. In the model proposed by the authors, SMAD7 acts as a scaffold that brings both the ubiquitin E3 ligase SMURF2 and the deubiquitinase USP15 to the TβRI receptor (Fig. 5). When the level of (active) TGF-β is low, TβRI ubiquitinylation by SMURF2 is quickly removed by USP15 according to this model. However, when TGF-β signaling is increased, a higher level of SMAD7 expression is induced as a feedback response and this will make the amount of USP15 insufficient, thereby limiting the duration of TGF-β/SMAD signaling (Aggarwal and Massague, 2012; Eichhorn et al., 2012).
As described above, advanced human cancer cells that retain TGF-β/SMAD signaling but lack tumor suppressive responses can make use of the SMAD pathway to their advantages, and via SMAD3/SMAD4 stimulate pro-invasive and pro-metastatic target genes (for example, IL11, CTGF, CXCR4) and reprogram (EMT) phenotypes (Aggarwal and Massague, 2012). This happens frequently in aggressive breast carcinoma and glioblastoma. In this respect it is important to note that while functional linkage of USP4 to the TGF-β/SMAD pathway was shown by employing a breast cancer model, USP15 can enhance the tumorigenic effect of TGF-β in glioblastoma (Eichhorn et al., 2012).
Results from an independent screen using an RNAi library against human DUB family members also indicated the physiological relevance of USP15 in regulating TGF-β superfamily function. In this study USP15 was found to potentiate both the TGF-β pathway and the related BMP pathway by targeting mono-ubiquitinated R-SMADs for deubiquitination (Fig. 5) (Inui et al., 2011). Thus, USP15 is not only required for TGF-β signal transduction and biological functions, including TGF-β-induced cell arrest and cell migration, but also necessary for BMP-induced osteoblast differentiations. Moreover, Xenopus embryo analysis in this study also uncovered a role for USP15 in embryonal development in vivo, dependent on its effect on TGF-β superfamily signaling (Inui et al., 2011).
OTUB1 activates TGF-β signaling via activating (phospho-) SMAD2/3
Recently, OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) was found to act on R-SMAD as well (Herhaus et al., 2013). However, different from USP15, OTUB1 enhances TGF-β signaling by inhibiting the ubiquitination and degradation of active SMAD2/3 (and not the inactive un-phosphorylated form), because the association of OTUB1 to SMAD2/3 is phosphorylation dependent. Moreover, OTUB1 was found to antagonize SMAD2/3’s ubiquitination independent of its catalytic activity as it interacts with E2 enzymes and inhibits efficient ubiquitin transfer from E2 to E3. This mechanism is reminiscent to the mechanism described in an earlier study on OTUB1-mediated inhibition of ubiquitination (Wiener et al., 2012).
CYLD binds to Smad7
The deubiquitinase cylindromatosis (CYLD) was first identified as a tumor suppressor gene, mutations in patients with familial cylindromatosis (Bignell et al., 2000). As a member of USPs subfamily, CYLD can antagonize Lysine-63 polyubiquitin chain conjugation (Kovalenko et al., 2003; Trompouki et al., 2003b). As mentioned previously, CYLD is involved in NF-κB, Wnt/β-catenin and JNK signaling pathway (Reiley et al., 2004; Tauriello et al., 2010; Trompouki et al., 2003b). By using CYLD knock-out mice, a recent study shows that in TGF-β-treated T cells, CYLD deficiency causes enhanced TAK1 and p38 mitogen-activated protein kinase activities (Zhao et al., 2011). Accumulation of non-degraded polyubiquitin chains and enhanced activities of SMAD7 in the absence of CYLD led to a study on the putative role of CYLD in the TGF-β signaling (Zhao et al., 2011). This showed that CYLD can bind to SMAD7 and deubiquitinate SMAD7 at Lysine 360 and 374 residues, which are required for the activation of TAK1 and p38 signaling (Zhao et al., 2011).
USP9X associates with SMAD4
Although SMAD4 is not obligatory for TGF-β signaling, it is required to provide the highest response to signaling. SMAD4 stabilizes SMAD-DNA interaction complexes in the nucleus and also recruits transcriptional coactivators such as histone acetyltransferases to regulatory elements (Wrana, 2009; Yang and Yang, 2010). Compared with other components of the TGF-β/SMAD pathway, SMAD4 possesses a very long half-life and thus is a rather stable protein. Nevertheless, Ectodermin/TRIM33/TIF1γ, a member of TRIM protein family of RING domain E3 ubiquitin ligases, has been suggested to be a determinant of vertebrate gastrulation by targeting SMAD4 for polyubiquitination and degradation (Dupont et al., 2005). This hypothesis was adjusted in a later study by the same group, in which they showed that only the monoubiquitination of SMAD4 is mediated by Ectodermin (Dupont et al., 2009). Lysine 519 of SMAD4 was found to conjugate by Ectodermin with a single ubiquitin molecule in the nucleus, which impairs SMAD4’s binding affinity to R-SMADs. This monoubiquitinated SMAD4 stays in an inhibitory state and regains activity in the cytoplasm once it has been deubiquitinated by FAM/USP9X (Fig. 5) (Dupont et al., 2009). FAM was first discovered in the fly, where FAM stands for fat facets. In contrast to what has been shown for FAM/USP9X-mediated deubiquitinating of β-catenin, AF-6, AMPK-related kinase 5 (NUAK1), and microtubule-affinity-regulating kinase 4 (MARK4) (Al-Hakim et al., 2008; Taya et al., 1999; Taya et al., 1998), FAM/USP9X specifically removes the site directed monoubiquitin molecule but not the polyubiquitin chains from SMAD4 (Dupont et al., 2009).
Other possible DUBs involved in TGF-β signaling
Before the identification and characterization of human DUBs, certain deubiquitinating enzymes were already found to be involved in TGF-β/SMAD signaling, yet not known to act through deubiquitination. Associated molecule with the SH3 domain of STAM (AMSH), a member of JAMMs DUB subfamily, was first identified as a signal-transducing adaptor molecule (STAM) binding protein (Tanaka et al., 1999). AMSH was later found to antagonize the inhibitory effect of SMAD6 on BMP signaling through binding to SMAD6, and did not bind to R-SMAD or Co-SMAD (Itoh et al., 2001). Thus, it will be interesting to examine whether the stimulatory effect of AMSH on BMP signaling is dependent on its DUB activity. Another example is AMSH-2, also a member of the JAMMs subfamily, which has been demonstrated to enhance TGF-β/SMAD signaling when ectopic overexpressed (Ibarrola et al., 2004). Co-immunoprecipitation assays have indicated that AMSH-2 could associate with SMAD2 and SMAD7 (Ibarrola et al., 2004), but also in this case it is not yet known whether the DUB activity of AMSH-2 is required for the enhancement of TGF-β signaling.
DUBS AS THERAPEUTIC TARGETS
Because of their druggable enzymatic activity, DUBs can be considered as therapeutic targets. Although proteasome inhibitor has been approved for the therapy of multiple myeloma (Hoy, 2013), there are still no DUB inhibitors endorsed for clinical usage. However, multiple studies already revealed such possibilities. As an example, P1130-mediated inhibition of tumor-activated DUBs results in downregulation of antiapoptotic and upregulation of proapoptotic proteins, such as MCL-1 and p53, thereby causing tumor cell apoptosis (Kapuria et al., 2010). A selective inhibitor of the DUB USP14 could be effective against neurodegenerative diseases and myeloma (Lee et al., 2010). Using stereotaxis, direct incubation into brain tumors with PR-619, a broad-spectrum DUB inhibitor, could limit the concentrations of TβR-I and p-SMAD2, in which the effective target is considered to be USP15 (Eichhorn et al., 2012). With the availability of technologies for large scale screening, design and development specific small inhibitor molecules for specific DUBs is required and will be helpful for the generation of novel cancer therapeutics.
CONCLUSION
The increasing attention for the clinical importance of the TGF-β/SMAD pathway as a tumor promoter makes it more and more worthwhile to search for critical regulators of this pathway as putative therapeutic targets. Since deubiquitinating enzymes can be targeted with drugs, DUBs that control TGF-β/SMAD signaling are emerging as potential targets for cancer therapies (Cohen and Tcherpakov, 2010; Colland, 2010). Several studies utilizing DUB screening methods have provided detailed insights in and mapping of the dynamic functions of ubiquitination in TGF-β/SMAD signaling. Further understanding of the catalytic activity of DUBs, as well as of knowledge on their regulation and substrate specificity, will promote the development of DUB inhibitors as potential anti-cancer drugs. Several DUBs have been identified as driving forces that can trigger and/or enhance tumorigenic TGF-β/SMAD signaling. Among these, promising drug targets are apparently a group of highly-similar DUBs, including USP4, USP11, and USP15. For instance, it would be interesting to develop inhibitors for USP4/11/15 and examine their potentials for anti-invasive and anti-metastatic roles in aggressive human cancers such as breast cancer and glioblastoma.
References
Aggarwal K, Massague J (2012) Ubiquitin removal in the TGF-beta pathway. Nat Cell Biol 14:656–657
Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR (2008) Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J 411:249–260
Al-Salihi MA, Herhaus L, Macartney T, Sapkota GP (2012) USP11 augments TGFbeta signalling by deubiquitylating ALK5. Open Biol 2:120063
Amerik AY, Hochstrasser M (2004) Mechanism and function of deubiquitinating enzymes. Biochim Biophys Acta 1695:189–207
Bignell GR, Warren W, Seal S, Takahashi M, Rapley E, Barfoot R, Green H, Brown C, Biggs PJ, Lakhani SR et al (2000) Identification of the familial cylindromatosis tumour-suppressor gene. Nat Genet 25:160–165
Blobe GC, Schiemann WP, Lodish HF (2000) Role of transforming growth factor beta in human disease. N Engl J Med 342:1350–1358
Bonni S, Wang HR, Causing CG, Kavsak P, Stroschein SL, Luo KX, Wrana JL (2001) TGF-beta induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat Cell Biol 3:587–595
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O et al (2004) The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5:1052–1060
Bos PD, Nguyen DX, Massague J (2010) Modeling metastasis in the mouse. Curr Opin Pharmacol 10:571–577
Brummelkamp TR, Nijman SM, Dirac AM, Bernards R (2003) Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424:797–801
Claassen GF, Hann SR (2000) A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc Natl Acad Sci USA 97:9498–9503
Clague MJ, Urbe S (2010) Ubiquitin: same molecule, different degradation pathways. Cell 143:682–685
Clague MJ, Coulson JM, Urbe S (2012) Cellular functions of the DUBs. J Cell Sci 125:277–286
Cohen P, Tcherpakov M (2010) Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 143:686–693
Colland F (2010) The therapeutic potential of deubiquitinating enzyme inhibitors. Biochem Soc Trans 38:137–143
Dang CV (1999) c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol 19:1–11
Deckers M, van Dinther M, Buijs J, Que N, Lowik C, van der Pluijm G, ten Dijke P (2006) The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer Res 66:2202–2209
Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425:577–584
Dikic I (2009) Journal club. A new ubiquitin chain, a new signal. Nat Rev Mol Cell Biol 10:306
Drabsch Y, ten Dijke P (2012) TGF-beta signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev 31:553–568
Dupont S, Zacchigna L, Cordenonsi M, Soligo S, Adorno M, Rugge M, Piccolo S (2005) Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 121:87–99
Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L et al (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell 136:123–135
Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K (2001) Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem 276:12477–12480
Eichhorn PJ, Rodon L, Gonzalez-Junca A, Dirac A, Gili M, Martinez-Saez E, Aura C, Barba I, Peg V, Prat A et al (2012) USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat Med 18:429–435
Frolik CA, Dart LL, Meyers CA, Smith DM, Sporn MB (1983) Purification and initial characterization of a type beta transforming growth factor from human placenta. Proc Natl Acad Sci USA 80:3676–3680
Galat A (2011) Common structural traits for cystine knot domain of the TGFbeta superfamily of proteins and three-fingered ectodomain of their cellular receptors. Cell Mol Life Sci 68:3437–3451
Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW et al (2011) Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471:591–596
Goumans MJ, Mummery C (2000) Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44:253–265
Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim SJ et al (1999) Mutational inactivation of transforming growth factor beta receptor type II in microsatellite stable colon cancers. Cancer Res 59:320–324
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CJ, Hruban RH et al (1996) DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 271:350–353
Heldin CH, Miyazono K, ten Dijke P (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471
Heldin CH, Landstrom M, Moustakas A (2009) Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol 21:166–176
Herhaus L, Al-Salihi M, Macartney T, Weidlich S, Sapkota GP (2013) OTUB1 enhances TGF beta signalling by inhibiting the ubiquitylation and degradation of active SMAD2/3. Nat Commun 4:2519
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479
Hoeller D, Dikic I (2009) Targeting the ubiquitin system in cancer therapy. Nature 458:438–444
Hogan BL (1996) Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 10:1580–1594
Hoy SM (2013) Subcutaneous bortezomib: in multiple myeloma. Drugs 73:45–54
Huang T, David L, Mendoza V, Yang Y, Villarreal M, De K, Sun L, Fang X, Lopez-Casillas F, Wrana JL et al (2011) TGF-beta signalling is mediated by two autonomously functioning TbetaRI:TbetaRII pairs. EMBO J 30:1263–1276
Huber MA, Kraut N, Beug H (2005) Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 17:548–558
Hunter T (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 28:730–738
Iavarone A, Massague J (1997) Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-beta in cells lacking the CDK inhibitor p15. Nature 387:417–422
Ibarrola N, Kratchmarova I, Nakajima D, Schiemann WP, Moustakas A, Pandey A, Mann M (2004) Cloning of a novel signaling molecule, AMSH-2, that potentiates transforming growth factor beta signaling. BMC Cell Biol 5:2
Ideguchi H, Ueda A, Tanaka M, Yang J, Tsuji T, Ohno S, Hagiwara E, Aoki A, Ishigatsubo Y (2002) Structural and functional characterization of the USP11 deubiquitinating enzyme, which interacts with the RanGTP-associated protein RanBPM. Biochem J 367:87–95
Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz-Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J et al (2011) SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471:637–641
Inoue Y, Imamura T (2008) Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci 99:2107–2112
Inui M, Manfrin A, Mamidi A, Martello G, Morsut L, Soligo S, Enzo E, Moro S, Polo S, Dupont S et al (2011) USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol 13:1368–1375
Itoh S, ten Dijke P (2007) Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol 19:176–184
Itoh F, Asao H, Sugamura K, Heldin CH, ten Dijke P, Itoh S (2001) Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J 20:4132–4142
Jackson SP, Durocher D (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol Cell 49:795–807
Jennings MT, Pietenpol JA (1998) The role of transforming growth factor beta in glioma progression. J Neurooncol 36:123–140
Jones E, Pu H, Kyprianou N (2009) Targeting TGF-beta in prostate cancer: therapeutic possibilities during tumor progression. Expert Opin Ther Targets 13:227–234
Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J (1995) Abnormal lung development and cleft palate in mice lacking Tgf-Beta-3 indicates defects of epithelial–mesenchymal interaction. Nature Genetics 11:415–421
Kalluri R, Weinberg RA (2009) The basics of epithelial–mesenchymal transition. J Clin Invest 119:1420–1428
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J (2003) A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3:537–549
Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massague J (2005) Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci USA 102:13909–13914
Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ (2010) Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res 70:9265–9276
Katsuno Y, Lamouille S, Derynck R (2012) TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol 25:76–84
Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6:1365–1375
Keusekotten K, Elliott PR, Glockner L, Fiil BK, Damgaard RB, Kulathu Y, Wauer T, Hospenthal MK, Gyrd-Hansen M, Krappmann D et al (2013) OTULIN Antagonizes LUBAC Signaling by Specifically Hydrolyzing Met1-Linked Polyubiquitin. Cell 153:1312–1326
Koinuma D, Shinozaki M, Komuro A, Goto K, Saitoh M, Hanyu A, Ebina M, Nukiwa T, Miyazawa K, Imamura T et al (2003) Arkadia amplifies TGF-beta superfamily signalling through degradation of Smad7. EMBO J 22:6458–6470
Komander D, Clague MJ, Urbe S (2009) Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 10:550–563
Komuro A, Imamura T, Saitoh M, Yoshida Y, Yamori T, Miyazono K, Miyazawa K (2004) Negative regulation of transforming growth factor-beta (TGF-beta) signaling by WW domain-containing protein 1 (WWP1). Oncogene 23:6914–6923
Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G (2003) The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424:801–805
Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, Imamura T (2005) NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem J 386:461–470
Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP et al (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467:179–184
Li MY, Chen DL, Shiloh A, Luo JY, Nikolaev AY, Qin J, Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416:648–653
Li L, Xin H, Xu X, Huang M, Zhang X, Chen Y, Zhang S, Fu XY, Chang Z (2004) CHIP mediates degradation of Smad proteins and potentially regulates Smad-induced transcription. Mol Cell Biol 24:856–864
Li J, D’Angiolella V, Seeley ES, Kim S, Kobayashi T, Fu W, Campos EI, Pagano M, Dynlacht BD (2013) USP33 regulates centrosome biogenesis via deubiquitination of the centriolar protein CP110. Nature 495:255–259
Liao TL, Wu CY, Su WC, Jeng KS, Lai MM (2010) Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J 29:3879–3890
Lin X, Liang M, Feng XH (2000) Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem 275:36818–36822
Lin CH, Chang HS, Yu WC (2008) USP11 stabilizes HPV-16E7 and further modulates the E7 biological activity. J Biol Chem 283:15681–15688
Lin Z, Yang H, Kong Q, Li J, Lee SM, Gao B, Dong H, Wei J, Song J, Zhang DD et al (2012) USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol Cell 46:484–494
Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X (2002) Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108:837–847
Liu W, Rui H, Wang J, Lin S, He Y, Chen M, Li Q, Ye Z, Zhang S, Chan SC et al (2006) Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J 25:1646–1658
Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B et al (1995) Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268:1336–1338
Massague J (2008a) TGFbeta in cancer. Cell 134:215–230
Massague J (2008b) A very private TGF-beta receptor embrace. Mol Cell 29:149–150
Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103:295–309
Massaous J, Hata A (1997) TGF-beta signalling through the Smad pathway. Trends Cell Biol 7:187–192
Mavrakis KJ, Andrew RL, Lee KL, Petropoulou C, Dixon JE, Navaratnam N, Norris DP, Episkopou V (2007) Arkadia enhances Nodal/TGF-beta signaling by coupling phospho-Smad2/3 activity and turnover. PLoS Biol 5:e67
McCullough J, Clague MJ, Urbe S (2004) AMSH is an endosome-associated ubiquitin isopeptidase. J Cell Biol 166:487–492
Miyazono K (2009) Transforming growth factor-beta signaling in epithelial-mesenchymal transition and progression of cancer. Proc Jpn Acad B 85:314–323
Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A (2005) Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem 280:22115–22123
Moustakas A, Heldin CH (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 98:1512–1520
Mu Y, Gudey SK, Landstrom M (2012) Non-Smad signaling pathways. Cell Tissue Res 347:11–20
Myeroff LL, Parsons R, Kim SJ, Hedrick L, Cho KR, Orth K, Mathis M, Kinzler KW, Lutterbaugh J, Park K et al (1995) A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res 55:5545–5547
Naber HP, Drabsch Y, Snaar-Jagalska BE, Ten Dijke P, van Laar T (2013) Snail and Slug, key regulators of TGF-beta-induced EMT, are sufficient for the induction of single-cell invasion. Biochem Biophys Res Commun 435:58–63
Nguyen DX, Bos PD, Massague J (2009) Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer 9:274–284
Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123:773–786
Niu JX, Shi YL, Iwai K, Wu ZH (2011) LUBAC regulates NF-kappa B activation upon genotoxic stress by promoting linear ubiquitination of NEMO. Embo J 30:3741–3753
Padua D, Zhang XH, Wang Q, Nadal C, Gerald WL, Gomis RR, Massague J (2008) TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133:66–77
Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B (1995) Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res 55:5548–5550
Petersen M, Pardali E, van der Horst G, Cheung H, van den Hoogen C, van der Pluijm G, ten Dijke P (2010) Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene 29:1351–1361
Pickart CM, Eddins MJ (2004) Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 1695:55–72
Popov N, Herold S, Llamazares M, Schulein C, Eilers M (2007a) Fbw7 and Usp28 regulate myc protein stability in response to DNA damage. Cell Cycle 6:2327–2331
Popov N, Wanzel M, Madiredjo M, Zhang D, Beijersbergen R, Bernards R, Moll R, Elledge SJ, Eilers M (2007b) The ubiquitin-specific protease USP28 is required for MYC stability. Nat Cell Biol 9:U765–U771
Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T (1995) Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet 11:409–414
Reiley W, Zhang M, Sun SC (2004) Negative regulation of JNK signaling by the tumor suppressor CYLD. J Biol Chem 279:55161–55167
Reyes-Turcu FE, Ventii KH, Wilkinson KD (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem 78:363–397
Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, MacLean TA, Srikumar T, Huang H, Dunham WH, Fukumura R, Xie G et al (2013) The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 498:318–324
Ross S, Hill CS (2008) How the Smads regulate transcription. Int J Biochem Cell Biol 40:383–408
Sanchez-Elsner T, Botella LM, Velasco B, Corbi A, Attisano L, Bernabeu C (2001) Synergistic cooperation between hypoxia and transforming growth factor-beta pathways on human vascular endothelial growth factor gene expression. J Biol Chem 276:38527–38535
Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T (1997) TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 124:2659–2670
Sato Y, Yoshikawa A, Yamagata A, Mimura H, Yamashita M, Ookata K, Nureki O, Iwai K, Komada M, Fukai S (2008) Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature 455:358–362
Schier AF (2003) Nodal signaling in vertebrate development. Annu Rev Cell Dev Biol 19:589–621
Schoenfeld AR, Apgar S, Dolios G, Wang R, Aaronson SA (2004) BRCA2 is ubiquitinated in vivo and interacts with USP11, a deubiquitinating enzyme that exhibits prosurvival function in the cellular response to DNA damage. Mol Cell Biol 24:7444–7455
Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL, Bova GS, Isaacs WB, Cairns P, Nawroz H et al (1996) DPC4 gene in various tumor types. Cancer Res 56:2527–2530
Schwartz AL, Ciechanover A (2009) Targeting proteins for destruction by the ubiquitin system: implications for human pathobiology. Annu Rev Pharmacol Toxicol 49:73–96
Seo SR, Lallemand F, Ferrand N, Pessah M, L’Hoste S, Camonis J, Atfi A (2004) The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J 23:3780–3792
Song MS, Salmena L, Carracedo A, Egia A, Lo-Coco F, Teruya-Feldstein J, Pandolfi PP (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455:U811–U813
Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N, Zhang S, Heldin CH, Landstrom M (2008) The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 10:1199–1207
Stroschein SL, Bonni S, Wrana JL, Luo K (2001) Smad3 recruits the anaphase-promoting complex for ubiquitination and degradation of SnoN. Genes Dev 15:2822–2836
Sun W, Tan X, Shi Y, Xu G, Mao R, Gu X, Fan Y, Yu Y, Burlingame S, Zhang H et al (2009) USP11 negatively regulates TNFalpha-induced NF-kappaB activation by targeting on IkappaBalpha. Cell Signal 22:386–394
Takenoshita S, Mogi A, Tani M, Osawa H, Sunaga H, Kakegawa H, Yanagita Y, Koida T, Kimura M, Fujita KI et al (1998) Absence of mutations in the analysis of coding sequences of the entire transforming growth factor-beta type II receptor gene in sporadic human breast cancers. Oncol Rep 5:367–371
Tanaka N, Kaneko K, Asao H, Kasai H, Endo Y, Fujita T, Takeshita T, Sugamura K (1999) Possible involvement of a novel STAM-associated molecule “AMSH” in intracellular signal transduction mediated by cytokines. J Biol Chem 274:19129–19135
Tang LY, Yamashita M, Coussens NP, Tang Y, Wang X, Li C, Deng CX, Cheng SY, Zhang YE (2011) Ablation of Smurf2 reveals an inhibition in TGF-beta signalling through multiple mono-ubiquitination of Smad3. EMBO J 30:4777–4789
Tauriello DV, Haegebarth A, Kuper I, Edelmann MJ, Henraat M, Canninga-van Dijk MR, Kessler BM, Clevers H, Maurice MM (2010) Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol Cell 37:607–619
Taya S, Yamamoto T, Kano K, Kawano Y, Iwamatsu A, Tsuchiya T, Tanaka K, Kanai-Azuma M, Wood SA, Mattick JS et al (1998) The Ras target AF-6 is a substrate of the fam deubiquitinating enzyme. J Cell Biol 142:1053–1062
Taya S, Yamamoto T, Kanai-Azuma M, Wood SA, Kaibuchi K (1999) The deubiquitinating enzyme fam interacts with and stabilizes beta-catenin. Genes Cells 4:757–767
Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S et al (2009) Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol 11:123–132
Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G (2003) CYLD is a deubiquitinating enzyme that negatively regulates NF-kappa B activation by TNFR family members. Nature 424:793–796
Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, Shifrin N, Malynn BA, Ma A (2008) Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 205:451–464
van der Horst A, de Vries-Smits AMM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, Burgering BMT (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol 8:U1040–U1064
Vincent F, Hagiwara K, Ke Y, Stoner GD, Demetrick DJ, Bennett WP (1996) Mutation analysis of the transforming growth factor beta type II receptor in sporadic human cancers of the pancreas, liver, and breast. Biochem Biophys Res Commun 223:561–564
Wada K, Kamitani T (2006) UnpEL/Usp4 is ubiquitinated by Ro52 and deubiquitinated by itself. Biochem Biophys Res Commun 342:253–258
Warner BJ, Blain SW, Seoane J, Massague J (1999) Myc downregulation by transforming growth factor beta required for activation of the p15(Ink4b) G(1) arrest pathway. Mol Cell Biol 19:5913–5922
Weissman AM (2001) Themes and variations on ubiquitylation. Nat Rev Mol Cell Biol 2:169–178
Whitman M (1998) Smads and early developmental signaling by the TGFbeta superfamily. Genes Dev 12:2445–2462
Wicks SJ, Haros K, Maillard M, Song L, Cohen RE, Dijke PT, Chantry A (2005) The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-beta signalling. Oncogene 24:8080–8084
Wiener R, Zhang XB, Wang T, Wolberger C (2012) The mechanism of OTUB1-mediated inhibition of ubiquitination. Nature 483:U143–U618
Williams SA, Maecker HL, French DM, Liu J, Gregg A, Silverstein LB, Cao TC, Carano RA, Dixit VM (2011) USP1 deubiquitinates ID proteins to preserve a mesenchymal stem cell program in osteosarcoma. Cell 146:918–930
Wiltshire TD, Lovejoy CA, Wang T, Xia F, O’Connor MJ, Cortez D (2010) Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J Biol Chem 285:14565–14571
Wrana JL (2009) The secret life of Smad4. Cell 136:13–14
Xiao N, Li H, Luo J, Wang R, Chen H, Chen J, Wang P (2012) Ubiquitin-specific protease 4 (USP4) targets TRAF2 and TRAF6 for deubiquitination and inhibits TNFalpha-induced cancer cell migration. Biochem J 441:979–986
Xin H, Xu X, Li L, Ning H, Rong Y, Shang Y, Wang Y, Fu XY, Chang Z (2005) CHIP controls the sensitivity of transforming growth factor-beta signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J Biol Chem 280:20842–20850
Xu J, Lamouille S, Derynck R (2009) TGF-beta-induced epithelial to mesenchymal transition. Cell Res 19:156–172
Yakicier MC, Irmak MB, Romano A, Kew M, Ozturk M (1999) Smad2 and Smad4 gene mutations in hepatocellular carcinoma. Oncogene 18:4879–4883
Yamaguchi T, Kimura J, Miki Y, Yoshida K (2007) The deubiquitinating enzyme USP11 controls an IkappaB kinase alpha (IKKalpha)-p53 signaling pathway in response to tumor necrosis factor alpha (TNFalpha). J Biol Chem 282:33943–33948
Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE (2008) TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 31:918–924
Yang G, Yang X (2010) Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci 6:1–8
Zhang YE (2009) Non-Smad pathways in TGF-beta signaling. Cell Res 19:128–139
Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R (2001) Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA 98:974–979
Zhang L, Huang H, Zhou F, Schimmel J, Pardo CG, Zhang T, Barakat TS, Sheppard KA, Mickanin C, Porter JA et al (2012a) RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol Cell 46:650–661
Zhang L, Zhou F, Drabsch Y, Gao R, Snaar-Jagalska BE, Mickanin C, Huang H, Sheppard KA, Porter JA, Lu CX et al (2012b) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-beta type I receptor. Nat Cell Biol 14:717–726
Zhang L, Zhou F, Garcia de Vinuesa A, de Kruijf EM, Mesker WE, Hui L, Drabsch Y, Li Y, Bauer A, Rousseau A et al (2013a) TRAF4 Promotes TGF-beta receptor signaling and drives breast cancer metastasis. Mol Cell 51(5):559–572
Zhang X, Zhang J, Bauer A, Zhang L, Selinger DW, Lu CX, Ten Dijke P (2013b) Fine-tuning BMP7 signalling in adipogenesis by UBE2O/E2-230K-mediated monoubiquitination of SMAD6. EMBO J 32:996–1007
Zhao B, Schlesiger C, Masucci MG, Lindsten K (2009) The ubiquitin specific protease 4 (USP4) is a new player in the Wnt signalling pathway. J Cell Mol Med 13:1886–1895
Zhao Y, Thornton AM, Kinney MC, Ma CA, Spinner JJ, Fuss IJ, Shevach EM, Jain A (2011) The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor beta (TGF-beta) signaling and the development of regulatory T cells. J Biol Chem 286:40520–40530
Zhu H, Kavsak P, Abdollah S, Wrana JL, Thomsen GH (1999) A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 400:687–693
Acknowledgements
We are grateful to all members of laboratories for discussions. We thank Prof. Dr. Peter ten Dijke for careful reading and critical comments. We would like to apologize to the authors of those papers that we could not cite due to the space limitations. This project was supported by Zhejiang Provincial Natural Science Foundation of China (Grant No. R14C070002). This work was in part supported by Key Construction Program of the National ‘‘985’’ Project and Zhejiang University Special Fund for Fundamental Research, as well as the Fundamental Research Funds for the Central Universities.
Compliance with Ethics Guidelines
Juan Zhang, Xiaofei Zhang, Feng Xie, Zhengkui Zhang, Hans van Dam, Fangfang Zhou, Long Zhang declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhang, J., Zhang, X., Xie, F. et al. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 5, 503–517 (2014). https://doi.org/10.1007/s13238-014-0058-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0058-8