
Abstract
The myeloproliferative neoplasms (MPNs) are a group of clonal hematological malignancies characterized by a hypercellular bone marrow and a tendency to develop thrombotic complications and to evolve to myelofibrosis and acute leukemia. Unlike chronic myelogenous leukemia, where a single disease-initiating genetic event has been identified, a more complicated series of genetic mutations appear to be responsible for the BCR-ABL1-negative MPNs which include polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Recent studies have revealed a number of epigenetic alterations that also likely contribute to disease pathogenesis and determine clinical outcome. Increasing evidence indicates that alterations in DNA methylation, histone modification, and microRNA expression patterns can collectively influence gene expression and potentially contribute to MPN pathogenesis. Examples include mutations in genes encoding proteins that modify chromatin structure (EZH2, ASXL1, IDH1/2, JAK2V617F, and IKZF1) as well as epigenetic modification of genes critical for cell proliferation and survival (suppressors of cytokine signaling, polycythemia rubra vera-1, CXC chemokine receptor 4, and histone deacetylase (HDAC)). These epigenetic lesions serve as novel targets for experimental therapeutic interventions. Clinical trials are currently underway evaluating HDAC inhibitors and DNA methyltransferase inhibitors for the treatment of patients with MPNs.
Similar content being viewed by others

Introduction
Myeloproliferative neoplasms (MPNs) represent a diverse set of hematologic malignancies that have received intense scientific investigation in recent years with a goal of developing novel disease modifying strategies. In 2008, the World Health Organization (WHO) revised the classification of hematologic malignancies to reflect new molecular insights into the pathogenesis of these disorders. Currently, the MPNs include chronic myelogenous leukemia (CML), polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), systemic mastocytosis, chronic eosinophilic leukemia not otherwise specified, chronic neutrophilic leukemia and “MPN, unclassifiable”; CML, PMF, PV, and ET represent the four major clinical entities and will be the subject of this review (Tefferi et al. 2007b).
CML is distinct among the MPNs in that it is defined by a specific cytogenetic abnormality (Philadelphia chromosome) involving a balanced translocation between the long arms of chromosomes 9 and 22 [t(9;22)(q34;q11)]. The gene fusion product resulting from this translocation, BCR-ABL1, gives rise to a constitutively activated and unregulated cytoplasmic tyrosine kinase that causes uncontrolled proliferation and differentiation of hematopoietic cells (Deininger et al. 2000). The molecular understanding of this pathway led to development of imatinib mesylate (Gleevec)—an oral BCR-ABL1 inhibitor—which has revolutionized the treatment of this MPN. In the International Randomized Study of Interferon plus cytosine arabinoside and STI571, imatinib therapy was found to induce a complete cytogenetic remission in 76% of CML patients versus 15% of patients in the interferon arm leading to a 6-year overall survival of 88% (O’Brien et al. 2003; Hochhaus et al. 2009). In contrast to CML, pharmacologic interventions for the other common Philadelphia chromosome (Ph-) negative MPNs (PMF, ET, and PV) have not been shown to significantly alter disease progression and overall survival (Mascarenhas 2009).
In 2005, an activating point mutation (V617F) in the autoinhibitory region of the JAK2 tyrosine kinase was first documented in 96%, 50%, and 50% of patients with PV, ET, and MF, respectively. JAK2V617F has served as a target for the development of a number of tyrosine kinase inhibitors (Baxter et al. 2005; James et al. 2005; Kralovics et al. 2005; Levine et al. 2005). These novel agents have been tested in phases I, II, and III studies and as a class have been effective in palliating the constitutional symptoms and reducing symptomatic splenomegaly in the majority of patients (Pardanani et al. 2010a, b; Santos et al. 2010; Verstovsek et al. 2009, 2010; Hexner et al. 2009; Moliterno et al. 2009). However, these agents have to date not been shown to significantly improve cytopenias, restore normal bone marrow morphology, and induce cytogenetic remissions in MF patients. In fact, molecular responses, as demonstrated by significant reduction in the JAK2V617F allele burden, have not been achieved. Thus, newer therapies directed against epigenetic, immunological, and molecular alterations of these Ph-negative MPNs are necessary, and many are currently being evaluated in clinical trials. In this review, we discuss epigenetic alterations in the Ph-negative classic MPNs, specifically focusing on epigenetic therapies as they relate to the underlying pathophysiology of these blood cancers.
Philadelphia chromosome negative classic MPNs
The MPNs are collectively characterized by a hyperproliferative bone marrow and excessive myeloid cell production. An increased risk for venous and arterial thrombosis and transformation to acute leukemia exist and pose a serious threat of morbidity and mortality to patients. Cachexia, fatigue, global weakness, progressive splenomegaly, and constitutional symptoms (fever, night sweats, and weight loss) can plague patients with the various MPNs and are particularly troublesome in MF. Although elevated peripheral blood counts typify ET and PV, MF is most often characterized by anemia and thrombocytopenia. Standardized diagnostic criteria, validated risk stratification schema, and response criteria to therapeutic intervention exist for these related disorders which have recently been created to facilitate the evaluation of potential new therapeutic modalities (Dupriez et al. 1996; Barosi et al. 2005; Tefferi et al. 2006, 2007b; Passamonti et al. 2008; Cervantes et al. 2009).
Polycythemia vera
PV is defined by an increase in red cell mass in the absence of conditions that induce secondary erythropoiesis and specific diagnostic criteria exist to aid in confirming the diagnosis (Thiele and Kvasnicka 2009). The average age of onset is 60–65 years with an annual incidence ranging from 0.5 to 2.6 cases per 100,000 persons per year (Hoffman 2005). Patients with PV have been found to have a 1.6-fold higher mortality than control populations with the main causes of mortality and morbidity being arterial and venous thrombosis, hemorrhage, evolution to MF, and leukemic transformation (Hoffman 2005). PV is thought to arise from a multipotent hematopoietic progenitor cell or stem cell. Nearly 95% of patients express the JAK2V617F mutation (James et al. 2005; Verstovsek et al. 2006). Presently, treatment for low-risk individuals (<60 years of age and no history of thrombosis) is low-dose aspirin and phlebotomy to maintain the hematocrit below 45% for men and 42% for women. In higher-risk individuals, cytoreductive therapy such as hydroxyurea and interferon-alpha has been used with the goals to reduce the risk of thrombosis, normalize peripheral blood counts, decrease splenomegaly, and ameliorate hypercatabolic and constitutional symptoms (Scherber and Mesa 2011).
Essential thrombocythemia
ET, the most common MPN in the USA, is defined by a platelet count greater than 450,000 × 109/L (changed from 600,000 × 109/L in 2008 WHO classification update) in the absence of any other MPN or reason for reactive thrombocytosis (Tefferi et al. 2007b). Patients with ET generally have a similar age-matched median survival for the first decade after diagnosis that can shorten thereafter usually secondary to thrombosis or hemorrhage (Wolanskyj et al. 2006). In approximately 50% of patients with ET, JAK2V617F is expressed and in comparison to PV, the allele burden is lower (James et al. 2005). Like PV, treatment consists of aspirin and cytoreductive agents such as hydroxyurea or anagrelide for patients at high risk for thrombosis. Approximately 10% of ET patients over the first decade can transform to a myelofibrotic phase (post-ET-related myelofibrosis) with clinical features essentially indistinguishable from primary myelofibrosis (Wolanskyj et al. 2006).
Primary myelofibrosis
PMF is the least common of the classic Ph-negative MPNs and has the worst prognosis with a median survival of 3–5 years from the time of diagnosis (Rozman et al. 1991; Tefferi 2000). The annual incidence is 0.2–1.5 cases per 100,000 persons per year with predominance in males over 50 years of age. The JAK2V617F mutation is found in about half of patients with PMF (James et al. 2005). Myelofibrosis, arising from a background of polycythemia vera or essential thrombocythemia, is denoted post-PV/ET MF, and the treatment approach remains the same as PMF. Collectively these conditions are referred to simply as MF. Patients with MF can be risk stratified for risk of death from transformation to acute leukemia or catastrophic thrombosis and complications of portal hypertension by various risk stratification systems that are used mostly for research purposes in deciding appropriate treatment options (Passamonti et al. 2010; Dupriez et al. 1996; Dingli et al. 2006; Tefferi et al. 2007a; Cervantes et al. 2009; Tam et al. 2009). Therapeutic approaches for the treatment of MF include immunomodulatory agents such as thalidomide and lenalidomide in combination with prednisone, which have response rates of 20–40% (Mesa et al. 2003, 2010). Androgens have also been used to selectively manage the anemia associated with MF, with response rates close to 40% (Cervantes et al. 2005). A few prospective studies have used erythropoiesis-stimulating agents with conflicting results (Cervantes et al. 2006; Tsiara et al. 2007). Chemotherapeutic agents including hydroxyurea, melphalan, busulfan, and 2-chlorodeoxyadenosine have also been used to control the myeloproliferative aspects of the disease (Chang and Gross 1988; Tefferi et al. 1997; Petti et al. 2002). The only present approach capable of curing MF is allogeneic hematopoietic stem cell transplantation, which must be evaluated on a case-by-case basis and balanced against considerable transplant-related morbidity and mortality (Kroger et al. 2009; Rondelli et al. 2010).
The effects of Janus kinase 2 inhibitors on Ph-negative MPN patients
In 2005, with the discovery of the JAK2V617F mutation, a substantial breakthrough in the understanding of the pathogenesis of Ph-negative MPNs led to the rapid development of new class of agents. Within a year, preclinical studies demonstrated that a G to T point mutation in exon 14 of the JAK2 tyrosine kinase gene (valine-to-phenylalanine at codon 617) was associated with the development of an MPN-like phenotype—erythrocytosis, leukocytosis, splenomegaly, and eventually changes resembling transformation to myelofibrosis (Bumm et al. 2006; Lacout et al. 2006; Wernig et al. 2006; Zaleskas et al. 2006). In vivo murine studies rapidly spawned the development of new oral small molecule inhibitors designed to inhibit the JAK2V617F-induced constitutively active signaling pathway. For the first time in decades, a renewed sense of optimism for producing effective disease modifying agents for the treatment of MPNs brought laboratory investigators and clinician scientists to the same table.
One agent, INCB018424 (Incyte), a potent and selective JAK1/JAK2 inhibitor, which demonstrated preclinical benefits in a JAK2V617F expressing MPN mouse model, recently completed a phase 1/2 clinical trial (Quintas-Cardama et al. 2010; Verstovsek et al. 2010). At the 15-mg twice-daily dosing, 17 of 33 MF patients with or without the JAK2V617F mutation (52%) had an objective clinical response (>50% decrease in splenomegaly) for 12 months and significant reduction in symptoms such as weight loss, fatigue, night sweats, and pruritus. Grade 3 or 4 adverse events occurred in less than 10% of patients and are mainly due to myelosuppression. This agent is being investigated in a randomized, double-blind, placebo-controlled phase III study to assess overall clinical efficacy in spleen reduction and improvement in MF-related disease symptoms as measured by an MF-specific quality-of-life tool, and the results of this trial are expected to be revealed in the latter part of 2011 (Scherber and Mesa 2011; Mesa et al. 2009a).
Other JAK2 inhibitors undergoing clinical evaluation include the non-specific multi-tyrosine kinase inhibitor CEP-701 (Cephalon), the selective JAK2 inhibitor SB1518 (SBIO), TG101348 (TargeGen), and CYT387 (YM BioSciences; Pardanani et al. 2010a, b; Verstovsek et al. 2009). Studies with newer agents directed against the constitutive tyrosine kinase activity induced by the JAK2V617F mutation are promising but still limited. While offering significant improvement in clinical symptoms, these agents do not appear to alter disease progression which has led to the continued interest in investigating alternative targeted therapies such as mammalian target of rapamycin inhibitors, immunomodulatory drugs, and anti-fibrosing agents which act to exploit additional aberrant pathways and proposed mechanisms of disease pathobiology (Scherber and Mesa 2011; Hoffman and Rondelli 2007).
Other gene mutations influencing the MPN phenotype have also been identified. Mutations in exon 12 of JAK2 have been described in a fraction of JAK2V617F-negative PV patients (Scott et al. 2007). Somatic activating mutations in the codon of MPL (the receptor for thrombopoietin) W515L/K occur in 10% of patients with PMF and 8% of JAK2V617F-negative ET, and in some patients, the MPL and JAK2V617F mutation can coexist (Pardanani et al. 2006; Beer et al. 2008; Vannucchi et al. 2008). These mutations are of low frequency and unlikely to serve as worthy drug targets.
Epigenetic alterations involved in the pathogenesis of Ph-negative MPNs
The biologic events leading to the initiation and progression of MPNs are likely not only caused by the acquisition of genetic mutations, such as the JAK2V617F mutation, but may also due to epigenetic changes that do not affect the primary sequence of DNA but rather alter gene expression by remodeling chromatin. Chromatin remodeling is accomplished primarily through two main mechanisms: (a) post-translational modification of histones, such as methylation, acetylation, phosphorylation, ADP-ribosylation glycosylation, and ubiquitination. Among all the types of histone modifications, methylation and acetylation at specific lysine residues are considered crucial histone marks affecting chromatin structure and gene expression (Kouzarides 2007). (b) DNA methylation with the addition of a methyl group to cytosine–phosphate–guanine (CpG) dinucleotide repeats within gene regulatory DNA sequences modulates the transcription of various genes (Metivier et al. 2008).
The attachment of a methyl group to the five-carbon position of cytosine base located 5′ to a guanosine base in the CpG dinucleotide islands of gene promoter sites influences the access of transcriptional machinery to DNA (Mani and Herceg 2010). This enzymatic process is regulated by DNA methyltransferases (DNMT). DNMT3A and DNMT3B are involved in de novo DNA methylation and DNMT1 in the maintenance of DNA methylation. The methylation status of a particular gene is an important determinant of gene expression, and both DNA hypomethylation and hypermethylation patterns have been implicated in the pathogenesis of many cancer types (Feinberg and Tycko 2004; Esteller 2007).
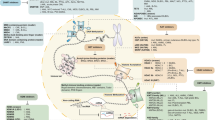
The addition of acetyl groups to lysine residue at the N terminus tail of histone proteins and the addition of methyl groups to lysine and arginine residues represent the best characterized histone modifications. The specific patterns of histone modifications define a code that dictates the dynamic recruitment of various transcription factors and the varied post-translational modification of histones by histone acetyl transferases (HATs), deacetylases (HDACs) or methyl transferases (HMTs), protein arginine methyltransferases (PRMTs), and DNA methyltransferases (DNMTs). These networks collectively play a critical role in modulating histone/histone and DNA/nucleosome interactions (Becker and Horz 2002). Dysregulation of these processes can result in silencing of tumor suppressor and cell differentiation genes, thereby promoting cell survival by blocking apoptosis and senescence and contributing to malignant transformation (Jenuwein and Allis 2001; Esteller 2008; Kondo et al. 2008). The complex interaction among these enzymes and the nucleosome result in a cumulative effect on chromatin structure. Figure 1 depicts the many varied and overlapping influences of HDACs, HMTs, DNMTs, and HATs directly on histone and DNA structure as well as the co-repression or activation of transcription factors. The shift in the balance of activity of one or more of these integral regulatory proteins will determine the transcriptional fate of numerous genes. Also indicated in the cartoon are two classes of therapeutic agents (histone deacetylase inhibitor (HDACi), DNMT1) which will be discussed in subsequent sections that can modify the epigenome in favor of overcoming transcriptional repression.

Active transcription is associated with hyperacetylation of histones, when an acetyl group (Ac) is added to specific lysines residing within the N terminal region of histones. The affinity between histone and DNA is reduced, leading to an open chromatin conformation that allows transcription factors (TF) and RNA polymerase to access the promoter of the target genes. Additionally, methylation of certain lysines on histones (H3K4, H3K36, and H3K79) located at promoter regions also leads to active transcription. In contrast, DNA methylation and hypoacetylation and/or methylation of certain other lysines on histones (H3K9, H3K27, and H4K20 residues) are associated with gene repression from formation of relatively condensed/inactive chromatin. These modifications are catalyzed by a number of chromatin-modifying enzymes including DNA methyltransferases (DNMTs), histone acetyltransferases (HATs)/histone deacetylases (HDACs), histone methyltransferases (HMTs), and histone demethylases (HDMs). The expression of transcriptionally inactive genes can be upregulated by exposure to DNMT-inhibitors and HDAC inhibitors
Other less well-characterized epigenetic modifications include post-transcriptional regulation of gene expression by a heterogeneous class of non-coding RNAs such as microRNAs (miRNAs) (Berdasco and Esteller 2010). MiRNAs bind to the 3′ untranslated region (3′UTR) of target mRNAs and either repress protein translation or cause mRNA degradation (Ambros 2004). MiRNAs (miR21, miR24, miR144, miR146a, miR150, miR155, miR221, miR222, miR223, miR451) play fundamental roles in the normal differentiation and activity of hematopoietic cells (Havelange and Garzon 2010). Data from both in vitro and in vivo studies indicate that miRNAs are important regulators of hematopoiesis and play a role in the pathogenesis of some acquired hematologic disorders, functioning either as tumor suppressors (miR-15/16) or as oncogenes (miR-17-92 cluster; Lawrie 2007). Microarray studies have defined miRNA signatures in hematopoietic cell lineages and related hematologic malignancies (Calin et al. 2004, 2005; Bruchova et al. 2007; Georgantas et al. 2007), and comparison of normal and patient samples has revealed aberrantly expressed miRNA that reflect a disease specific signature (Debernardi et al. 2007; Gramantieri et al. 2007; Meng et al. 2007; Venturini et al. 2007). Changes in miRNA expression can occur through different mechanisms including transcriptional deregulation, epigenetic alterations, gene mutations, DNA copy number abnormalities, and impaired miRNA processing (Deng et al. 2008). These disease specific-miRNA epigenetic signatures may provide a basis for new therapeutic interventions by specifically targeting miRNA expression.
MiRNA expression profiling of megakaryocytes in PMF but not ET has revealed that in the pre-fibrotic form of PMF, autonomous proliferation of the megakaryocytic lineage is associated with significant accumulation of miR-146b as compared to normal megakaryopoiesis (Hussein et al. 2009). These data reflect an active miRNA system in MPN megakaryocytes which appears to be one of the underlying defects associated with disease progression. Recently, Girardot et al. 2010 reported that in a fraction of MPN patient platelets, Mir 28 negatively regulates MPL expression. Mir 28 targets the 3′UTR region of MPL and inhibits its translation as well as other proteins potentially involved in megakaryocyte differentiation including E2F6, a transcription factor belonging to the E2F family and ERK2.
Two broad categories of epigenetic alterations in MPN pathophysiology have been observed. The first involves alterations in genes that encode proteins which influence chromatin structure. Alterations in TET2, ASXL1, EZH2, IDH1/2, JAK2V617F, and IKZF1 gene functions are examples of this first category and can lead to epigenetic dysregulation. TET2, ASXL1, IDH1/2, and EZH2 gene mutations are found alone or in combination with JAK2 or MPL mutations and influence the epigenetic regulation of transcription resulting in the possible silencing of putative tumor suppressor genes in MPNs. The second category includes the promoter site of genes critical for cell survival, differentiation, and proliferation. Examples of this group of genes in MPNs are provided in Table 1. We will now review the most current understanding of epigenetic dysregulation in Ph-negative MPNs.
Category I—gene alterations leading to epigenetic deregulation of Ph-negative MPNs
TET2
Mutations involving the ten to 11 translocation-2 (TET2) family gene located in the minimal loss of heterozygosity region at 4q24 have been identified in several myeloid malignancies (Delhommeau et al. 2009). The exact function of TET2 is not yet clear, but it appears to act as a tumor suppressor gene (Schaub et al. 2010; Delhommeau et al. 2009). Homozygosity for TET2 mutations as a result of uniparental disomy or deletion of the TET2 locus does not appear to confer a proliferative advantage to hematopoietic progenitor cell clones which would argue against a role as a tumor suppressor gene (Schaub et al. 2011). TET2 is a member of the α-ketoglutarate-dependent enzyme family that catalyzes the conversion of 5-methylcytosine of DNA to 5-hydroxymethylcytosine and induces subsequent DNA demethylation (Ito et al. 2010; Tahiliani et al. 2009). TET2 mutations have been reported in almost all coding regions including missense, nonsense, or frameshift mutations. Moreover, these mutations are not exclusively bi-allelic (both copies of TET2) and therefore considered TET2 loss of function mutations (Delhommeau et al. 2009). TET2 loss of function would be anticipated to result in DNA hypermethylation which has been recently reported in acute myeloid leukemia (AML) blast cells.
Overall, the frequency of TET2 mutations in Ph-negative MPNs has been reported to be 12–17% (Delhommeau et al. 2009; Tefferi et al. 2009b). A higher TET2 mutation frequency has been detected in older MPN patients (>60 years) and has been shown to be highly correlated with JAK2V617F allele burdening these patients (Tefferi et al. 2009b). In fact, studies support the role of TET2 in JAK2V617F-positive PV as not a disease-initiating event preceding the acquisition of the JAK2 mutation, but as a latter event that can confer a proliferative advantage to the JAK2V617F-bearing clone (Swierczek et al. 2010; Delhommeau et al. 2009). However, other studies utilizing colony-forming assays failed to demonstrate a consistent temporal relationship between the acquisition of the somatic mutations of TET2 and JAK2 (Schaub et al. 2010). TET2 mutations have largely not been uniformly shown to have prognostic significance; no influence was demonstrated on survival, rate of leukemic transformation, or thrombotic tendency in MPN patients (Hussein et al. 2010; Tefferi et al. 2009a, b). In contrast, TET2 mutations have been identified in blast phase MPN samples and not observed in the paired background MPN samples of both JAK2 wild-type and mutant MPNs obtained prior to blastic transformation (Abdel-Wahab et al. 2010a). This finding would suggest that the acquisition of TET2 mutations may be a possible step in MPN leukemic transformation.
ASXL1
ASXL1 is one of three mammalian homologs of the additional sex combs gene in Drosophila (69). The ASXL1 gene is located on chromosome 20q11 and encodes an enhancer of the trithorax group (trxG) and Polycomb group (PcG) proteins (ETP) chromatin modifier complex (Fisher et al. 2003). The PcG proteins (repressors) and trxG proteins (activators) serve to regulate gene expression of homeotic genes, such as Hox genes via histone methylation (Fisher et al. 2010a). PcG and trxG proteins function at the level of chromatin by forming multi-protein complexes: which are the three PRC1, Polycomb repressor complex 2 (PRC2), PhoRC and SET-1-like complex, BRM, and MLL supercomplex, respectively (Baskind et al. 2009). These complexes work together to establish and maintain methylation marks primarily on the tail of histones. Mammalian PcG and trx genes display hematopoietic lineage and differentiation stage-specific expression patterns and are required for normal hematopoiesis. Mammalian ASXL proteins are predicted to have dual activator/repressor functions depending on their cellular context. An ASXL1 knockout mouse model is characterized by a defect in frequency of differentiation of both myeloid and lymphoid cells, without an effect on hematopoietic stem cells and does not result in a myelodysplastic or leukemic phenotype (Fisher et al. 2010b). This would seem to indicate that ASXL1 mutations alone are not sufficient to induce malignant transformation.
ASXL1 mutations have been documented in myelodysplastic syndrome (MDS; 10%) and chronic myelomonocytic leukemia (40%) patients and most recently in 8% of MPN patients that were all negative for JAK2V617 (Carbuccia et al. 2009). The ASXL1 mutations were also found in the CD34+ cell population supporting the principle of a primitive hematopoietic stem as the origin of the MPN clone and further suggesting that acquisition of ASXL1 mutations can occur early in MPN pathogenesis. Although the ASXL1 mutations are associated with poor overall survival and increased risk for transformation to blast crisis in chronic myelomonocytic leukemia patients, it is not yet clear what influence it has on the behavior of Ph-negative MPNs.
EZH2
Mutations involving the enhancer of zeste homolog 2 (EZH2) gene located on chromosome 7q36.1, which encodes the catalytic component of the histone methyltransferase PRC2 have also been described in MPN patients (Stegelmann et al. 2010). PRC2 is a multi-protein enzyme complex responsible for the trimethylation of lysine 27 on histone H3 (H3K27me3; Cao and Zhang 2004; Simon and Lange 2008). The PRC2 complex contains multiple subunits: EZH2, SUz12, EED, and YY1. PRC2 can also recruit other Polycomb complexes, DNMTs, and HDACs to the gene site resulting in chromatin compaction and additional repressive activity (Chase and Cross 2011).
Activating and inactivating mutations of EZH2 have been reported in human malignancies. The EZH2 Y641 mutation (within the SET domain) which is found in lymphoma cells results in a gain of function with increased levels of H3K27me3 (Morin et al. 2010). The mutations associated with myeloid malignancies are thought to result in loss of histone methyltransferase activity. Forty-nine EZH2 mutations have been found in 42 individuals out of 614 patients with myeloid disorders (Ernst et al. 2010). Thirteen percent of MF patients in this cohort harbored an EZH2 mutation. A total of ten EZH2 mutations were identified in exons involving deletions, insertions, and missense mutations in patients with PMF, post-PV/ET MF, and MPN-associated acute myeloid leukemia. Microarray and SNP analysis did not show association with copy number alterations or uniparental disomy. Additionally, no association was seen with JAK2V617F allele burden. Degree of splenomegaly and leukocytosis was clinical findings found to be statistically associated in MPN patients expressing EZH2 mutations. Upregulation of EZH2 gene expression has been documented in MPNs, most frequently in PMF patients suggesting a potential role of tumor suppressor gene silencing as a mechanism in disease progression (Skov et al. 2010a). Additionally, EZH2 and ASXL1 mutations were not found to be mutually exclusive events in MPNs. Retrospective analysis of the presence of EZH2 mutations in archived MPN bone marrow samples has not been shown to have prognostic significance in PMF patients (Abdel-Wahab et al. 2010b).
Three-deazaneplanocin A (DZNep) is a carbocyclic adenosine analog that inhibits s-adenosylhomocysteine hydrolase and results in the accumulation of s-adenosylhomocysteine, disrupting methylation of targets by EZH2 (Chase and Cross 2011). Although the effects of DZNep are global and not specific to EZH2, this drug has been tested as a single agent in solid tumor cell lines and in combination with a HDACi in primary AML cells (Fiskus et al. 2009). The combination of this agent with the pan-HDACi, LBH589 (Panobinostat, Novartis), was shown in vitro to selectively induce apoptosis in AML primary cells and not normal CD34+ cells. This effect was correlated with reduction in EZH2 protein level and induction in p16, p21, and p27 gene expression. Combined therapy in a NOD/SCID mouse model with HL-60 AML led to an improvement in survival when compared to each agent alone (Fiskus et al. 2009). This compound is currently being investigated in early phase clinical trials.
Expression of miR-101-1 and 101-2 which negatively regulate EZH2 has been shown to be decreased in MPNs and displayed an inverse relationship with EZH2 mRNA expression (Swierczek et al. 2011). This may provide an additional mechanism for EZH2 gene dysregulation and contribute to MPN disease progression and disease severity.
IDH1/2
Isocitrate dehydrogenase 1 and 2, IDH1 and IDH2, located on chromosome 2q33.3 and 15q26.1, respectively, encode NADP+-dependent enzymes that catalyze oxidative decarboxylation of isocitrate to α-ketoglutarate (Tefferi et al. 2010). The IDH mutant has decreased affinity for isocitrate and instead converts α-ketoglutarate to hydroxyl-glutarate which has been implicated in malignant transformation (Dang et al. 2009). IDH gene mutations have been documented in solid tumors and de novo AML (Chou et al. 2010; Yan et al. 2009). A recent study in AML has revealed that the presence of IDH1/2 mutations result in production of 2-hydroxyglutarate and is associated with a specific global DNA hypermethylation signature (Figueroa et al. 2010). Both IDH1/2 mutations and TET2 mutations lead to similar hypermethylation signatures and patterns of impaired myeloid differentiation and increased expression of stem cell markers. Additionally, it has been shown that IDH1/2 mutations generate impaired enzymatic activity of α-ketoglutarate-dependent TET2 protein and result in increased stem cell/progenitor cell marker expression (Figueroa et al. 2010). Thus, the expression of IDH1/2 mutations can lead to a TET2-dependent epigenetic effect.
IDH1/2 mutational frequency in MPNs is approximately 0.8%, 1.9%, and 4.2% for ET, PV, and PMF, respectively (Tefferi et al. 2010). Thirty-eight IDH 1/2 mutations have been discovered in a large screening study of MPN patients and can coexist equally with mutations in JAK2, MPL, and TET2 (Tefferi et al. 2010). The types of IDH1/2 mutations seen in MPNs are distinctly different than the ones observed in brain tumors and overlapped with those documented in AML and include IDH1-R132, IDH2-R140, and IDH2-R172. Over 21% of patients with blast phase-related MPN carry an IDH1/2 mutation, and this was irrespective of JAK2V617F status (Green and Beer 2010; Pardanani et al. 2010c). This appears to indicate that IDH1/2 mutations can also influence the transformation of MPN to blast phase disease.
Interestingly, leukemic blasts and progenitor cells can possess both mutated IDH2 and JAK2V617F, and in other patients with MPN-transformed leukemia, the mutated IDH1/2 may be present in blasts with wild-type JAK2 and absent in the progenitor cells with JAK2V617F (Green and Beer 2010). These data raise the possibility of the presence of two subclones originating from a yet unidentified primary clone or two independent competing clones arising in the same individual. Further studies are needed to further clarify these findings and determine their possible significance.
IKZF1
Ikaros is a Kruppel-like zinc finger transcription factor that is integral to the development of normal hematopoiesis and is encoded by the Ikaros family zinc finger 1 (IKZF1) gene located at 7p.12 (Georgopoulos 2002; Westman et al. 2002). The exact mechanism by which this mutation influences chromatin remains unclear. IKZF1 influences maturation and differentiation of a variety of cell types at different stages of development including those of the hematopoietic system (Westman et al. 2002). IKZF1 interacts with the histone deacetylase repressor complexes NURD (HDAC1 and 2) and SIN3 which likely exerts a repressive influence on genes important in myelopoiesis.
IKZF1 mutations were first identified in cells from Ph-positive acute lymphocytic leukemia patients and are believed to play a role in leukemic transformation (Iacobucci et al. 2009). In a study of blast phase MPN patients, a recurrent loss of chromosomal region 7p.12 led investigators to the discovery of IKZF1 deletions in 21% of patients with blast phase MPN and only 0.2% of chronic phase MPN patients, providing a very compelling argument for a role of IKZF1 in leukemic transformation (Jager et al. 2010). IKZF1 mutants are associated with increased STAT5 expression and resultant activation of the JAK–STAT pathway (Jager et al. 2010). IKZF1 mutation appears to be a late event occurring after the acquisition of JAK2V617F, and its exact pathogenetic role in MPN leukemic transformation remains unclear (Jager et al. 2010).
JAK2V617F
Genome-wide methylation pattern studies on MPN patient samples demonstrate a distinct chromatin altered pattern in PMF when compared to PV/ET patient samples (Nischal et al. 2010). Both hyper- and hypomethylated loci were found in neutrophils of PMF patients. Hypomethylated promoter sites involved genes responsible for cytokine signaling and MAP kinases. The presence of JAK2V617F additionally was found to influence the degree of DNA hypomethylation and supports a proposed role for JAK/STAT pathway influence on the methylome and ultimately on gene transcription and disease phenotype (Nischal et al. 2010).
Recently, alternative pathways by which JAK2V617F might affect hematopoiesis have been identified, and Dawson and co-workers found that 35% of the JAK2 regulated genes did not contain a STAT5 binding site (Dawson et al. 2009). This group determined that JAK2 can be localized to not only the cytoplasm but also the nucleus where it phosphorylates histone H3 at tyrosine residue 41 (Y41). H3Y41 results in release of the transcriptional repression by heterochromatin protein 1α from chromatin. Furthermore, Liu and co-workers have shown that JAK2V617F phosphorylates and downregulates the activity of PRMT5, an arginine methyltransferase discussed below (Liu et al. 2011). This event leads to a gain of function that affects the gene expression pattern and the behavior of hematopoietic progenitor cells by downregulating histone arginine methylation.
PRMT5
The type II arginine methyltransferase PRMT5 catalyzes the symmetric dimethylation of arginine residues on histones H2a, H3, and H4 (Branscombe et al. 2001). PRMT5 is a target of JAK2-mediated phosphorylation and in JAK2V617F expressing cells leads to downregulation of PRMT5 activity and decreased global histone methylation. Forced PRMT5 gene over-expression in primary PV CD34+ cells results in a reduction in cell proliferation and differentiation and supports the role of downregulated PRMT5 activity via JAK2 mediated phosphorylation in the molecular pathogenesis of PV. This provides a very interesting pathogenetic view of JAK2-mediated chromatin modification as a downstream target of the activated tyrosine kinase pathway.
Category II—individual genes affected by epigenetic modification in MPN
SOCS
Suppressors of cytokine signaling (SOCS1, SOCS2, and SOCS3) are negative regulators of the JAK–STAT pathway and are both induced by and act in a negative feedback loop to downregulate JAK/STAT signaling (Greenhalgh and Hilton 2001). Epigenetic silencing of SOCS1/3 is an additional pathogenetic mechanism leading to cytokine signaling hypersensitivity. SOCS1 hypermethylation has been reported in a fraction of patients with Ph-negative MPNs and can be seen in both JAK2V617F-positive and JAK2 wild-type patients (Jost et al. 2007; Teofili et al. 2008). However, the methylation pattern that was observed in these studies was noted in SOCS1 exon 2 (also seen in normal control blood cells) but not the gene promoter site and thus the relevance of this observation to MPN pathogenesis is not evident. Hypermethylation of SOCS3 has been detected in PMF but not PV/ET patients. A trend for lower SOCS3 expression in JAK2V617F-negative PMF patients was noted in one study (Fernandez-Mercado et al. 2008). SOCS methylation status was not correlated with any identifiable clinical variables or outcome (Fourouclas et al. 2008). SOCS2 silencing by hypermethylation has also been shown in MPN-derived cell lines as well as primary MPN cells and can coexist in cells that carry the JAk2V617F mutation (Quentmeier et al. 2008).
SFRP1/2
Secreted Frizzled-related protein (SFRP) actively antagonizes the Wnt signaling pathway which is integral to the maintenance and proliferation of hematopoietic stem cells (Willert et al. 2003). Upregulation of the Wnt pathway and downregulation of SFRP has been shown in other hematologic malignancies. SFRP2 promoter hypermethylation was detected in 27%, 30%, and 26% of PV, ET, and PMF patient samples, respectively (Bennemann et al. 2010). Hypermethylation of SFRP-2 promoter site was not seen in any cases of CML.
PRV-1
Over-expression of polycythemia rubra vera-1 (PRV-1) mRNA, a GPI-linked protein expressed by neutrophils of patients with ET/PV, has been shown to be inversely related to the C30 promoter site methylation status (Jelinek et al. 2007). PRV-1 (CD177) is a hematopoietic cell surface receptor with yet unclear function normally expressed on neutrophils and can be upregulated in certain settings including, sepsis, pregnancy, and after granulocyte colony-stimulating factor administration. Although the mRNA transcript is over-expressed in all patients with PV, the protein product is not and is comparable to the level observed in normal control neutrophils (Klippel et al. 2002). The JAK2V617F allele burden was also inversely related to C30 methylation, and a trend toward significance was appreciated with an inverse relationship to JAK2V617F allele burden.
CXCR4
In MF, extramedullary hematopoiesis of CD34+ hematopoietic stem cells (HSC) in the spleen and other organs is responsible for many of the signs and symptoms attributed to this disease. This abnormal cell trafficking from the bone marrow niche is believed to be a result of reduced CD34+ cell expression of CXC chemokine receptor 4 (CXCR4), leading to constitutively mobilized peripheral blood HSC (Rosti et al. 2007). CXCR4 is a receptor for stromal-derived factor-1 (SDF-1; CXCL12) and induces normal HSC and progenitor cell chemotaxis to the marrow space. The CXCR4 receptor in PMF cells, compared to normal controls, is susceptible to epigenetic modification from hypermethylation of CpG islands within its promoter region (Bogani et al. 2008). Treatment with the demethylating agent 5-aza-2′-deoxycytidine (5-AzaD; DEC) in vitro increased membrane expression of CXCR4 and improved migration of CD34+ cells in the presence of SDF-1 (Shi et al. 2007). Moreover, sequential treatment of PMF CD34+ cells with 5-AzaD and trichostatin A (TSA), a HDAC inhibitor, increased preferential homing of PMF CD34+ cells to the bone marrow and not the spleen of NOD/SCID mice (Wang et al. 2009a, b). These agents also reduced the proportion of JAK2V617F-positive HPCs, homozygous HPCs, and cells that contained chromosomal abnormalities. Most recently, treatment with sequential 5-azacytidine (5-Aza)/TSA in a JAK2V617F+ PMF CD34+ cell transplant NOD/SCID mouse model led to dramatic reduction in number of these cells suggesting potential use of these agents in treating the mutated stem cell population in PMF (Wang et al. 2010).
HDAC
The HDAC family consists of 18 genes, subdivided into four classes based on their homology to yeast ortholog. The HDACs can be divided into two families, first is the Zn2+-dependent HDAC family composed of class I (HDACs 1, 2, 3 and 8), class II a/b (HDACs 4, 5, 6, 7, 9, and 10), and class IV (HDAC 11), and the second is the NAD-dependent class III SIRT enzymes (Biancotto et al. 2010). Histone acetylation is regulated by the dynamic and antagonistic action of two classes of enzymes, HDACs and HATs. HATs execute the transfer of an acetyl group from acetyl Co-A to the ε-amino group of lysine residues, whereas HDACs catalyze the removal of acetyl groups. Acetylation of histones and non-histone proteins can alter DNA–protein binding, protein–protein interaction, and/or subcellular localization. Considerable evidence indicates that the acetylation status of histones and non-histone proteins play a key role in the regulation of cellular signaling and disease progression (Haberland et al. 2009; Spange et al. 2009). For instance, global gene expression profiling of MPN patient blood cells revealed HDAC gene deregulation (Skov et al. 2010b). HDAC 9 and 11 gene over-expression was documented in various MPN subtypes, and an increase in HDAC 6 gene expression was observed during MPN disease progression. These data support the possible role of HDAC enzyme inhibition as a treatment approach to MPNs.
Epigenetic therapy for Ph-negative MPNs
HDAC inhibitors
HDACi are a novel class of structurally diverse natural and synthetic compounds that modulate a myriad of cellular functions by inhibiting HDAC activity. The ability of various HDACis such as LBH589 (Panobinostat, Novartis), ITF2357 (Givonostat, Italfarmaco SpA), and suberoylanilide hydroxamic acid (SAHA, Merck) to inhibit JAK2V617F-positive HPCs has been examined (Table 2 lists and describes these agents as well as the DNA methyltransferase inhibitors (DNMTi) currently being evaluated for the treatment of MPN patients). ITF2357 was shown in vitro to inhibit the proliferation of JAK2V617F cells by specifically decreasing the level of JAK2V617F protein, without associated changes in JAK2V617F mRNA levels, and inhibiting downstream signaling such as phosphorylation of STAT-5 (Guerini et al. 2008). Exposure of JAK2V617F-expressing cell lines to LBH589 has also been shown to result in the proteasomal degradation of JAK2V617F via disruption of the chaperone function of HSP90 and induced apoptosis in these cells (Wang et al. 2009b). Cotreatment with the JAK2 inhibitor TG101209 in both JAK2V617F-expressing cell lines and primary CD34+ MPN cells led to attenuated downstream JAK/STAT signaling and synergistic cytotoxicity that was selective to the malignant clone but was not observed in normal CD34+ hematopoietic stem cells. Preclinical studies have demonstrated the anti-proliferative activity of SAHA in JAK2V617F+-expressing cell lines (Akada et al. 2010). Selective reduction in the clonogenic growth of JAK2V617F+-expressing colonies suggested specificity for mutated JAK2-bearing cells. Using an established inducible JAK2V617F+ knock-in mouse model for PV, treatment with SAHA was shown to reduce splenomegaly, normalize hematocrit, and reduce the numbers of JAK2V617F-positive erythroid progenitor cells. These in vitro studies provided a foundation for the use of chromatin-modifying agents in clinical trials for MPNs (Table 3).
A phase II pilot study of ITF2357 at 50 mg orally twice daily for 24 weeks in patients with JAKV617F-positive PV, ET, and PMF showed three major responses among 16 MF patients treated (Rambaldi et al. 2010). This agent also improved pruritus and reduced splenomegaly in 75% of PV/ET and 38% of MF patients. A trend toward reduction in JAK2V617F allele burden was observed. No major grade III/IV adverse events were observed. LBH589 has been evaluated in two phase I/II studies. In the first study, the agent was used in both high-risk JAK2V617F-positive and JAK2V617F-negative PMF and post-ET/PV MF patients (Mascarenhas et al. 2009). This agent was associated with improvements in anemia in two patients and significant reduction in palpable splenomegaly. Thrombocytopenia was found to be the DLT and the recommended phase II dose was determined to be 25 mg given orally three times weekly. In the second study among 12 patients with PMF or post-ET/PV MF, four patients demonstrated reduction in spleen size greater than 50% (DeAngelo et al. 2010). One previously untreated JAK2V617F-positive patient demonstrated a partial response according to IWG response criteria. Three additional patients demonstrated clinical improvement lasting ≥8 weeks. Four patients had stable disease. Similar to the first study, the most common adverse event was thrombocytopenia. Phase II studies of LBH589 in this setting are currently underway and are anticipated to be reported next year. A single report exists of a patient with MF that achieved normalization of peripheral blood counts within 6 weeks of treatment with SAHA (Lee 2009).
LBH589 clinical activity has been correlated with biomarker response including reduction in downstream substrates of the JAK/STAT pathway (p-STAT3, p-STAT5, p-AKT, p-ERK1/2, and p-PIM protein levels), reduction in Bcl-Xl and MCL-1 as well as modest reduction in JAK2V617F allele burden in treated patients (DeAngelo et al. 2010). Additionally, Ac-α-tubulin and HSP70 levels were found to be elevated after treatment with LBH589 which supports the mechanism of both HDAC6 and HSP90 inhibition.
Thrombocytopenia is the major dose-limiting toxicity of HDACi treatment and can pose a serious clinical risk of bleeding in the treatment of patients with MF and low baseline platelet counts. Recently, in vitro studies on primary murine megakaryocytes and murine and human cell lines has revealed that the predictable clinical thrombocytopenia induced by LBH589 is not an effect on reduced platelet survival but instead a decrease in proplatelet formation (Bishton et al. 2011). Reductions in Rho-GTPase proteins Rac1, CDC42, and RhoA after exposure to LBH589 are correlated with an increase in phosphorylated myosin light chain 2 and postulated to reduce proplatelet production in megakaryocytes. Studies by Iancu-Rubin et al. (2010) show that LBH589-induced thrombocytopenia is also likely due to a reduction in proplatelet production mediated by hyperacetylation of tubulin which is an integral component of the microtubule cytoskeleton and necessary for proplatelet extension. This work highlights the importance of the impact of HDAC inhibition on non-histone protein function. The use of a thrombopoietin mimetic in combination with LBH589 to support the drug-induced toxicity of thrombocytopenia may offer an approach that will allow more effective clinical use in MPN patients that already have low platelet counts at baseline. Alternatively, LBH589 may prove to be very useful in the treatment of patients with ET, where reducing the platelet count would be advantageous.
DNA methyltransferase inhibitors
Two DNMTi, 5-Aza (Vidaza) and DEC (Decitabine) have been used extensively, and with success, for the treatment of MDS (Silverman et al. 2002, 2006; Kantarjian et al. 2007); these agents now also have rational application for the treatment of MPNs and have been examined in a number of phase I and II clinical trials. In two phase II studies, 5-Aza was tested as a 7- and 5-day course every 4 weeks in patients with refractory/relapsing PMF or post-ET/PV MF (Quintas-Cardama et al. 2008; Mesa et al. 2009b). No significant clinical response was seen although global hypomethylation was observed. Myelosuppression was the major adverse event in these two studies. DEC, however, has shown potential for clinical response in PMF as well as MF-BP (Mascarenhas et al. 2010; Danilov et al. 2009). In a phase II multi-center study of 20 patients with PMF treated with low-dose DEC as a subcutaneous injection, a 37% response rate was seen and two patients with MF-BP obtained a complete and partial response by WHO criteria (Odenike et al. 2008). In those patients with clinical response, a 61% mean reduction in circulating peripheral blood CD34+ cells was noted. However, no change in CXCR4 expression was seen. As is seen with 5-Aza, myelosuppression was the most common toxicity. A small retrospective study of the combined use of DEC with gemtuzumab ozogamicin in patients with MF and MF-BP supports a potential combination therapeutic approach (Al-Ameri et al. 2010).
Future studies will be designed combining both HDACi and DNMTi given either concomitantly or in sequence in MPN patients. Both scientific rationale as discussed above and clinical experience in the treatment of MDS provides a strong rationale for this treatment approach in MPNs. Additionally, as it has become clear that unlike CML which has a defined pathogenetic mechanism that can be exploited effectively, the Ph-negative MPNs are indeed more complex in their pathogenesis. These hematologic malignancies will likely require a therapeutic strategy including combinations of drugs that will affect both epigenetic events and intracellular signaling pathways in an attempt to target the MPN HSC clone. Studies combining the use of both HDACi and DNMTi together and with JAK2 inhibitors are being designed and will almost certainly herald a new era of combination therapy that will hopefully result in clinical advances.
Conclusion
The Ph-negative MPNs are a group of myeloid malignancies that have enjoyed a tremendous amount of attention in recent years due to successive laboratory based discoveries in molecular pathology stemming from the discovery of the JAK2V617F mutation in 2005. There is growing evidence of multiple mutational events that likely contribute to MPN pathogenesis and influence disease phenotype. Many of these gene mutations alter the MPN epigenome, and multiple genes have been identified as targets of epigenetic deregulation in MPN cells. Supported by sound scientific rationale, chromatin-modifying agents have been tested in early phase studies with a hint of efficacy and will continue to be tested more rigorously alone and in combination. An MPN-specific epigenetic signature is evolving and will likely soon play a vital role in MPN classification, prognostication, and treatment of these diverse hematopoietic stem cell neoplasms.
References
Abdel-Wahab O, Manshouri T et al (2010) Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 70(2):447–452
Abdel-Wahab O, Pardanani A et al (2010) Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. ASH Annu Meet Abstr 116(21):3070
Akada H, Hamada S et al (2010) Efficacy of vorinostat in a murine model of polycythemia vera. ASH Annu Meet Abstr 116(21):629
Al-Ameri AA-A, Kantarjian H et al (2010) Decitabine and gemtuzumab ozogamicin in patients with advanced myelofibrosis. ASH Annu Meet Abstr 116(21):5061
Ambros V (2004) The functions of animal microRNAs. Nature 431(7006):350–355
Aviram A, Witenberg B et al (2003) Detection of methylated ABL1 promoter in Philadelphia-negative myeloproliferative disorders. Blood Cells Mol Dis 30(1):100–106
Barosi G, Bordessoule D et al (2005) Response criteria for myelofibrosis with myeloid metaplasia: results of an initiative of the European Myelofibrosis Network (EUMNET). Blood 106(8):2849–2853
Baskind HA, Na L et al (2009) Functional conservation of Asxl2, a murine homolog for the Drosophila enhancer of trithorax and Polycomb group gene Asx. PloS One 4(3):e4750
Baxter EJ, Scott LM et al (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365(9464):1054–1061
Becker PB, Horz W (2002) ATP-dependent nucleosome remodeling. Annu Rev Biochem 71:247–273
Beer PA, Campbell PJ et al (2008) MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 112(1):141–149
Bennemann K, Schubert C et al (2010) Epigenetic downregulation of secreted Frizzled-related proteins in Philadelphia positive and Philadelphia negative myeloproliferative neoplasms. ASH Annu Meet Abstr 116(21):4647
Berdasco M, Esteller M (2010) Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev Cell 19(5):698–711
Biancotto C, Frige G et al (2010) Histone modification therapy of cancer. Adv Genet 70:341–386
Bishton MJ, Harrison SJ et al (2011) Deciphering the molecular and biologic processes that mediate histone deacetylase inhibitor-induced thrombocytopenia. Blood 117(13):3658–3668
Bogani C, Ponziani V et al (2008) Hypermethylation of CXCR4 promoter in CD34+ cells from patients with primary myelofibrosis. Stem Cells 26(8):1920–1930
Branscombe TL, Frankel A et al (2001) PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem 276(35):32971–32976
Bruchova H, Yoon D et al (2007) Regulated expression of microRNAs in normal and polycythemia vera erythropoiesis. Exp Hematol 35(11):1657–1667
Bumm TGP, Elsea C et al (2006) Characterization of murine JAK2V617F-positive myeloproliferative disease. Cancer Research 66(23):11156–11165
Calin GA, Liu CG et al (2004) MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci U S A 101(32):11755–11760
Calin GA, Ferracin M et al (2005) A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 353(17):1793–1801
Cao R, Zhang Y (2004) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 14(2):155–164
Capello D, Deambrogi C et al (2008) Epigenetic inactivation of suppressors of cytokine signalling in Philadelphia-negative chronic myeloproliferative disorders. Br J Haematol 141(4):504–511
Carbuccia N, Murati A et al (2009) Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 23(11):2183–2186
Cervantes F, Alvarez-Larrán A et al (2005) Efficacy and tolerability of danazol as a treatment for the anaemia of myelofibrosis with myeloid metaplasia: long-term results in 30 patients. Br J Haematol 129(6):771–775
Cervantes F, Alvarez-Larrán A et al (2006) Darbepoetin-alpha for the anaemia of myelofibrosis with myeloid metaplasia. Br J Haematol 134(2):184–186
Cervantes F, Dupriez B et al (2009) New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 113(13):2895–2901
Chang JC, Gross HM (1988) Remission of chronic idiopathic myelofibrosis to busulfan treatment. Am J Med Sci 295(5):472–476
Chase A, Cross NC (2011) Aberrations of EZH2 in cancer. Clin Cancer Res 17(9):2613–2618
Chou WC, Hou HA et al (2010) Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 115(14):2749–2754
Dang L, White DW et al (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462(7274):739–744
Danilov AV, Relias V et al (2009) Decitabine is an effective treatment of idiopathic myelofibrosis. Br J Haematol 145(1):131–132
Dawson MA, Bannister AJ et al (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461(7265):819–822
DeAngelo DJ, Tefferi A et al (2010) A phase II trial of Panobinostat, an orally available deacetylase inhibitor (DACi), in patients with primary myelofibrosis (PMF), post essential thrombocythemia (ET), and post polycythemia vera (PV) myelofibrosis. ASH Annu Meet Abstr 116(21):630
Debernardi S, Skoulakis S et al (2007) MicroRNA miR-181a correlates with morphological sub-class of acute myeloid leukaemia and the expression of its target genes in global genome-wide analysis. Leukemia 21(5):912–916
Deininger MW, Goldman JM et al (2000) The molecular biology of chronic myeloid leukemia. Blood 96(10):3343–3356
Delhommeau F, Dupont S et al (2009) Mutation in TET2 in myeloid cancers. N Engl J Med 360(22):2289–2301
Deng S, Calin GA et al (2008) Mechanisms of microRNA deregulation in human cancer. Cell Cycle 7(17):2643–2646
Dingli D, Schwager SM et al (2006) Prognosis in transplant-eligible patients with agnogenic myeloid metaplasia: a simple CBC-based scoring system. Cancer 106(3):623–630
Dupriez B, Morel P et al (1996) Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood 88(3):1013–1018
Ernst T, Chase AJ et al (2010) Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet 42(8):722–726
Esteller M (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat Rev Genet 8(4):286–298
Esteller M (2008) Epigenetics in cancer. The New England Journal of Medicine 358(11):1148–1159
Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4(2):143–153
Fernandez-Mercado M, Cebrian V et al (2008) Methylation status of SOCS1 and SOCS3 in BCR-ABL negative and JAK2V617F negative chronic myeloproliferative neoplasms. Leuk Res 32(10):1638–1640
Figueroa ME, Abdel-Wahab O et al (2010) Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18(6):553–567
Fisher CL, Berger J et al (2003) A human homolog of additional sex combs, ADDITIONAL SEX COMBS-LIKE 1, maps to chromosome 20q11. Gene 306:115–126
Fisher CL, Lee I et al (2010a) Additional sex combs-like 1 belongs to the enhancer of trithorax and Polycomb group and genetically interacts with Cbx2 in mice. Dev Biol 337(1):9–15
Fisher CL, Pineault N et al (2010b) Loss-of-function additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood 115(1):38–46
Fiskus W, Wang Y et al (2009) Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor Panobinostat against human AML cells. Blood 114(13):2733–2743
Fourouclas N, Li J et al (2008) Methylation of the suppressor of cytokine signaling 3 gene (SOCS3) in myeloproliferative disorders. Haematologica 93(11):1635–1644
Georgantas RW 3rd, Hildreth R et al (2007) CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A 104(8):2750–2755
Georgopoulos K (2002) Haematopoietic cell-fate decisions, chromatin regulation and Ikaros. Nat Rev Immunol 2(3):162–174
Girardot M, Pecquet C et al (2010) miR-28 is a thrombopoietin receptor targeting microRNA detected in a fraction of myeloproliferative neoplasm patient platelets. Blood 116(3):437–445
Gramantieri L, Ferracin M et al (2007) Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res 67(13):6092–6099
Green A, Beer P (2010) Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 362(4):369–370
Greenhalgh CJ, Hilton DJ (2001) Negative regulation of cytokine signaling. J Leukoc Biol 70(3):348–356
Guerini V, Barbui V et al (2008) The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia 22(4):740–747
Haberland M, Montgomery RL et al (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10(1):32–42
Havelange V, Garzon R (2010) MicroRNAs: emerging key regulators of hematopoiesis. Am J Hematol 85(12):935–942
Hexner E, Goldberg JD et al (2009) A multicenter, open label phase I/II study of CEP701 (Lestaurtinib) in adults with myelofibrosis; a report on phase I: a study of the myeloproliferative disorders research consortium (MPD-RC). ASH Annu Meet Abstr 114(22):754
Hochhaus A, O’Brien SG et al (2009) Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 23(6):1054–1061
Hoffman R (2005) Hematology: basic principles and practice. Elsevier Churchill Livingstone, Philadelphia
Hoffman R, Rondelli D (2007) Biology and treatment of primary myelofibrosis. Hematology Am Soc Hematol Educ Program 2007:346–354
Hussein K, Theophile K et al (2009) MicroRNA expression profiling of megakaryocytes in primary myelofibrosis and essential thrombocythemia. Platelets 20(6):391–400
Hussein K, Abdel-Wahab O et al (2010) Cytogenetic correlates of TET2 mutations in 199 patients with myeloproliferative neoplasms. Am J Hematol 85(1):81–83
Iacobucci I, Storlazzi CT et al (2009) Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell’Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 114(10):2159–2167
Iancu-Rubin C, Feller F et al (2010) Targeting non-histone protein acetylation impairs platelet production during normal megakaryocytopoiesis. ASH Annu Meet Abstr 116(21):2610
Ihalainen J, Juvonen E et al (1994) Calcitonin gene methylation in chronic myeloproliferative disorders. Leukemia 8(2):230–235
Ito S, D’Alessio AC et al (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466(7310):1129–1133
Jager R, Gisslinger H et al (2010) Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leukemia 24(7):1290–1298
James C, Ugo V et al (2005) A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 434(7037):1144–1148
Jelinek J, Li J et al (2007) Epigenetic control of PRV-1 expression on neutrophils. Exp Hematol 35(11):1677–1683
Jenuwein T, Allis CD (2001) Translating the histone code. Science 293(5532):1074–1080
Jones LC, Tefferi A et al (2004) RARbeta2 is a candidate tumor suppressor gene in myelofibrosis with myeloid metaplasia. Oncogene 23(47):7846–7853
Jost E, Do ON et al (2007) Epigenetic alterations complement mutation of JAK2 tyrosine kinase in patients with BCR/ABL-negative myeloproliferative disorders. Leukemia 21(3):505–510
Kantarjian HM, O’Brien S et al (2007) Survival advantage with decitabine versus intensive chemotherapy in patients with higher risk myelodysplastic syndrome: comparison with historical experience. Cancer 109(6):1133–1137
Klippel S, Strunck E et al (2002) Biochemical characterization of PRV-1, a novel hematopoietic cell surface receptor, which is overexpressed in polycythemia rubra vera. Blood 100(7):2441–2448
Kondo Y, Shen L et al (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40(6):741–750
Kouzarides T (2007) Chromatin modifications and their function. Cell 128(4):693–705
Kralovics R, Passamonti F et al (2005) A gain-of-function mutation of JAK2 in myeloproliferative disorders. The New England Journal of Medicine 352(17):1779–1790
Kroger N, Holler E et al (2009) Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood 114(26):5264–5270
Kumagai T, Tefferi A et al (2005) Methylation analysis of the cell cycle control genes in myelofibrosis with myeloid metaplasia. Leuk Res 29(5):511–515
Lacout C, Pisani DF et al (2006) JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 108(5):1652–1660
Lawrie CH (2007) MicroRNAs and haematology: small molecules, big function. Br J Haematol 137(6):503–512
Lee J (2009) Clinical efficacy of vorinostat in a patient with essential thrombocytosis and subsequent myelofibrosis. Ann Hematol 88(7):699–700
Levine RL, Wadleigh M et al (2005) Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 7(4):387–397
Liu F, Zhao X et al (2011) JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 19(2):283–294
Mani S, Herceg Z (2010) DNA demethylating agents and epigenetic therapy of cancer. Adv Genet 70:327–340
Mascarenhas J (2009) Risk adapted approach to the treatment of primary myelofibrosis. Hematology Education E H Association 14:192–199
Mascarenhas J, Wang X et al (2009) A phase I study of LBH589, a novel histone deacetylase inhibitor in patients with primary myelofibrosis (PMF) and post-polycythemia/essential thrombocythemia myelofibrosis (post-PV/ET MF). ASH Annu Meet Abstr 114(22):308
Mascarenhas J, Navada S et al (2010) Therapeutic options for patients with myelofibrosis in blast phase. Leuk Res 34(9):1246–1249
Meng F, Henson R et al (2007) MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 133(2):647–658
Mesa RA, Steensma DP et al (2003) A phase 2 trial of combination low-dose thalidomide and prednisone for the treatment of myelofibrosis with myeloid metaplasia. Blood 101(7):2534–2541
Mesa RA, Kantarjian H et al (2009a) Validation of the serial use of the myelofibrosis symptom assessment form (MF-SAF) for measuring symptomatic improvement: performance in 86 myelofibrosis patients on INCB018424 clinical trial. ASH Annu Meet Abstr 114(22):3917
Mesa RA, Verstovsek S et al (2009b) 5-Azacitidine has limited therapeutic activity in myelofibrosis. Leukemia 23(1):180–182
Mesa RA, Yao X et al (2010) Lenalidomide and prednisone for myelofibrosis: Eastern Cooperative Oncology Group (ECOG) phase 2 trial E4903. Blood 116(22):4436–4438
Metivier R, Gallais R et al (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452(7183):45–50
Moliterno AR, Hexner E et al (2009) An open-label study of CEP-701 in patients with JAK2 V617F-positive PV and ET: update of 39 enrolled patients. ASH Annu Meet Abstr 114(22):753
Morin RD, Johnson NA et al (2010) Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet 42(2):181–185
Nagy E, Beck Z et al (2003) Frequent methylation of p16INK4A and p14ARF genes implicated in the evolution of chronic myeloid leukaemia from its chronic to accelerated phase. Eur J Cancer 39(16):2298–2305
Nischal S, Zhou L et al (2010) Epigenomic profiling of myeloproliferative diseases reveal idiopathic myelofibrosis as an epigenetically distinct subgroup and highlights the epigenetic effects of Jak2V617F mutation. ASH Annu Meet Abstr 116(21):627
O’Brien SG, Guilhot F et al (2003) Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. The New England Journal of Medicine 348(11):994–1004
Odenike OM, Godwin JE et al (2008) Phase II trial of low dose, subcutaneous decitabine in myelofibrosis. ASH Annu Meet Abstr 112(11):2809
Pardanani AD, Levine RL et al (2006) MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood 108(10):3472–3476
Pardanani A, Gotlib JR et al (2010a) Longer-term follow up with TG101348 therapy in myelofibrosis confirms sustained improvement in splenomegaly, disease-related symptoms, and JAK2V617F allele burden. ASH Annu Meet Abstr 116(21):459
Pardanani A, George G et al (2010b) A phase I/II study of CYT387, an oral JAK-1/2 inhibitor, in myelofibrosis: significant response rates in anemia, splenomegaly, and constitutional symptoms. ASH Annu Meet Abstr 116(21):460
Pardanani A, Lasho TL et al (2010c) IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 24(6):1146–1151
Passamonti F, Rumi E et al (2008) A dynamic prognostic model to predict survival in post-polycythemia vera myelofibrosis. Blood 111(7):3383–3387
Passamonti F, Cervantes F et al (2010) A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 115(9):1703–1708
Pehlivan M, Sercan Z et al (2009) sFRP1 promoter methylation is associated with persistent Philadelphia chromosome in chronic myeloid leukemia. Leuk Res 33(8):1062–1067
Petti MC, Latagliata R et al (2002) Melphalan treatment in patients with myelofibrosis with myeloid metaplasia. Br J Haematol 116(3):576–581
Quentmeier H, Geffers R et al (2008) SOCS2: inhibitor of JAK2V617F-mediated signal transduction. Leukemia 22(12):2169–2175
Quintas-Cardama A, Tong W et al (2008) A phase II study of 5-azacitidine for patients with primary and post-essential thrombocythemia/polycythemia vera myelofibrosis. Leukemia 22(5):965–970
Quintas-Cardama A, Vaddi K et al (2010) Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood 115(15):3109–3117
Rambaldi A, Dellacasa CM et al (2010) A pilot study of the histone-deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol 150(4):446–455
Rondelli D, Boyer MW, Isola LM, Shore T, Bacigalupo A, Klisovic RB, Marchioli R, Goldberg JD, Hoffman R, Silverman LR (2010) First prospective study of allogeneic peripheral blood stem cell (PBSC) transplantation in patients with myelofibrosis in the United States: interim analysis of MPD-RC 101 protocol. J Clin Oncol 28:15s
Rosti V, Massa M et al (2007) The expression of CXCR4 is down-regulated on the CD34+ cells of patients with myelofibrosis with myeloid metaplasia. Blood Cells Mol Dis 38(3):280–286
Rozman C, Giralt M et al (1991) Life expectancy of patients with chronic nonleukemic myeloproliferative disorders. Cancer 67(10):2658–2663
Santos FPS, Kantarjian HM et al (2010) Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 115(6):1131–1136
Schaub FX, Looser R et al (2010) Clonal analysis of TET2 and JAK2 mutations suggests that TET2 can be a late event in the progression of myeloproliferative neoplasms. Blood 115(10):2003–2007
Schaub FX, Lehmann T et al (2011) Transition to homozygosity does not appear to provide a clonal advantage to hematopoietic progenitors carrying mutations in TET2. Blood 117(6):2075–2076
Scherber R, Mesa RA (2011) Future therapies for the myeloproliferative neoplasms. Curr Hematol Malig Rep 6(1):22–27
Scott LM, Tong W et al (2007) JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. The New England Journal of Medicine 356(5):459–468
Shi J, Zhao Y et al (2007) Effects of chromatin-modifying agents on CD34+ cells from patients with idiopathic myelofibrosis. Cancer Res 67(13):6417–6424
Silverman LR, Demakos EP et al (2002) Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol 20(10):2429–2440
Silverman LR, McKenzie DR et al (2006) Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol 24(24):3895–3903
Simon JA, Lange CA (2008) Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res 647(1–2):21–29
Skov V, Larsen TS et al (2010a) Enhanced gene expression of EZH2 in patients with primary myelofibrosis. ASH Annu Meet Abstr 116(21):4118
Skov V, Larsen TS et al (2010b) Increased gene expression of histone deacetylases in patients with Philadelphia-negative chronic myeloproliferative neoplasms. ASH Annu Meet Abstr 116(21):4119
Spange S, Wagner T et al (2009) Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41(1):185–198
Stegelmann F, Schlenk RF et al (2010) Mutations of EZH2 in myeloproliferative neoplasms with myelofibrosis: correlation with molecular and clinical data. ASH Annu Meet Abstr 116(21):4111
Suzuki R, Onizuka M et al (2007) Infrequent hypermethylation of WIF-1 promoter in BCR/ABL-negative myeloproliferative disorders. Tokai J Exp Clin Med 32(4):131–135
Swierczek S, Yoon D et al (2010) MicroRNA-101 is down-regulated in PV and ET granulocytes and its decrease is associated with over-expression of histone methyltransferase in EZH2 in MPN patients. ASH Annu Meet Abstr 116(21):1989
Swierczek SI, Yoon D et al (2011) Extent of hematopoietic involvement by TET2 mutations in JAK2V617F polycythemia vera. Haematologica 96:775–778
Tahiliani M, Koh KP et al (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324(5929):930–935
Tam CS, Kantarjian H et al (2009) Dynamic model for predicting death within 12 months in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. J Clin Oncol 27(33):5587–5593
Tefferi A (2000) Myelofibrosis with myeloid metaplasia. The New England Journal of Medicine 342(17):1255–1265
Tefferi A, Silverstein MN et al (1997) 2-Chlorodeoxyadenosine treatment after splenectomy in patients who have myelofibrosis with myeloid metaplasia. Br J Haematol 99(2):352–357
Tefferi A, Barosi G et al (2006) International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT). Blood 108(5):1497–1503
Tefferi A, Huang J et al (2007a) Validation and comparison of contemporary prognostic models in primary myelofibrosis: analysis based on 334 patients from a single institution. Cancer 109(10):2083–2088
Tefferi A, Thiele J et al (2007b) Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood 110(4):1092–1097
Tefferi A, Lim KH et al (2009a) Mutation in TET2 in myeloid cancers. N Engl J Med 361(11):1117–1118
Tefferi A, Pardanani A et al (2009b) TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 23(5):905–911
Tefferi A, Lasho TL et al (2010) IDH1 and IDH2 mutation studies in 1473 patients with chronic-, fibrotic- or blast-phase essential thrombocythemia, polycythemia vera or myelofibrosis. Leukemia 24(7):1302–1309
Teofili L, Martini M et al (2008) Epigenetic alteration of SOCS family members is a possible pathogenetic mechanism in JAK2 wild type myeloproliferative diseases. Int J Cancer 123(7):1586–1592
Thiele J, Kvasnicka HM (2009) The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Curr Hematol Malig Rep 4(1):33–40
Tsiara SN, Chaidos A et al (2007) Recombinant human erythropoietin for the treatment of anaemia in patients with chronic idiopathic myelofibrosis. Acta Haematologica 117(3):156–161
Vannucchi AM, Antonioli E et al (2008) Characteristics and clinical correlates of MPL 515 W > L/K mutation in essential thrombocythemia. Blood 112(3):844–847
Venturini L, Battmer K et al (2007) Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 109(10):4399–4405
Verstovsek S, Silver RT et al (2006) JAK2V617F mutational frequency in polycythemia vera: 100%, >90%, less? Leukemia 20(11):2067
Verstovsek S, Odenike O et al (2009) Phase I dose-escalation trial of SB1518, a novel JAK2/FLT3 inhibitor, in acute and chronic myeloid diseases, including primary or post-essential thrombocythemia/polycythemia vera myelofibrosis. ASH Annu Meet Abstr 114(22):3905
Verstovsek S, Kantarjian H et al (2010) Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 363(12):1117–1127
Wang JC, Chen W et al (2002) Hypermethylation of the P15INK4b and P16INK4a in agnogenic myeloid metaplasia (AMM) and AMM in leukaemic transformation. Br J Haematol 116(3):582–586
Wang X, Zhang W et al (2009a) Correction of the abnormal trafficking of primary myelofibrosis CD34+ cells by treatment with chromatin-modifying agents. Cancer Res 69(19):7612–7618
Wang Y, Fiskus W et al (2009b) Cotreatment with Panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 114(24):5024–5033
Wang X, Zhang W et al (2010) Sequential treatment of CD34+ cells from patients with primary myelofibrosis with chromatin-modifying agents eliminate JAK2V617F-positive NOD/SCID marrow repopulating cells. Blood 116(26):5972–5982
Wernig G, Mercher T et al (2006) Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 107(11):4274–4281
Westman BJ, Mackay JP et al (2002) Ikaros: a key regulator of haematopoiesis. Int J Biochem Cell Biol 34(10):1304–1307
Willert K, Brown JD et al (2003) Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 423(6938):448–452
Wolanskyj AP, Schwager SM et al (2006) Essential thrombocythemia beyond the first decade: life expectancy, long-term complication rates, and prognostic factors. Mayo Clinic Proc 81(2):159–166
Yan H, Parsons DW et al (2009) IDH1 and IDH2 mutations in gliomas. N Engl J Med 360(8):765–773
Zaleskas VM, Krause DS et al (2006) Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PloS One 1:e18
Acknowledgments
The authors would like to acknowledge Dr. Mingjiang Xu and Dr James Bieker for their help in reviewing sections of this manuscript.
Conflict of interest
Dr. Ronald Hoffman receives research support from Novartis and Celegene. Drs. Mascarenhas, Roper, and Chaurasia have no relevant conflict of interest to report.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mascarenhas, J., Roper, N., Chaurasia, P. et al. Epigenetic abnormalities in myeloproliferative neoplasms: a target for novel therapeutic strategies. Clin Epigenet 2, 197–212 (2011). https://doi.org/10.1007/s13148-011-0050-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-011-0050-6