
Abstract
Histone modifications have widely been implicated in cancer development and progression and are potentially reversible by drug treatments. The N-terminal tails of each histone extend outward through the DNA strand containing amino acid residues modified by posttranslational acetylation, methylation, and phosphorylation. These modifications change the secondary structure of the histone protein tails in relation to the DNA strands, increasing the distance between DNA and histones, and thus allowing accessibility of transcription factors to gene promoter regions. A large number of HDAC inhibitors have been synthesized in the last few years, most being effective in vitro, inducing cancer cells differentiation or cell death. The majority of the inhibitors are in clinical trials, unlike the suberoylanilide hydroxamic acid, a pan-HDACi, and Romidepsin (FK 228), a class I-selective HDACi, which are only approved in the second line treatment of refractory, persistent or relapsed cutaneous T-cell lymphoma, and active in approximately 150 clinical trials, in monotherapy or in association. Preclinical studies investigated the use of these drugs in clinical practice, as single agents and in combination with chemotherapy, hypomethylating agents, proteasome inhibitors, and MTOR inhibitors, showing a significant effect mostly in hematological malignancies. The aim of this review is to focus on the biological features of these drugs, analyzing the possible mechanism(s) of action and outline an overview on the current use in the clinical practice.
Similar content being viewed by others


Histones: structure and functions
Histones are alkaline proteins found in eukaryotic cell nuclei, which package and order the DNA into structural units called nucleosomes. They can be grouped into five major classes: H1/H5, H2A, H2B, H3, and H4. These are organized into two super-classes: core histones, H2A, H2B, H3, and H4 and linker histones, H1 and H5. Two core histones assemble to form one octameric nucleosome core particle by wrapping DNA around the protein spool in 1.65 Å left-handed super-helical coil. The linker histone H1 binds the nucleosome and the entry and exit sites of the DNA, thus locking the DNA into place and allowing the formation of higher order structure. A typical nucleosome is composed of an octamer of the four pairs of core histones H2A, H2B, H3, and H4 and ≈146 base pairs of DNA wrapped around. Histones undergo posttranslational modifications altering their interaction with DNA and nuclear proteins and which remodel the chromatin condensation status and gene expression.
Epigenetic modifications of the histone proteins
The H3 and H4 histones have long tails protruding from the nucleosome, which can be covalently modified. The core of H2A and H3 is susceptible as well. These modifications include methylation, phosphorylation, sumoylation, citrullination, acetylation, and ubiquitination. Combinations of modifications are thought to constitute the so-called “histone code.” Diverse biological processes such as gene regulation, DNA repair, and chromosome condensation seem to be regulated by this code.
Methylation
The addition of one, two, or three methyl groups is generally associated with transcriptional repression. However, methylation of some lysine and arginine by residues of histones, results in transcriptional activation. Methylation is operated by histone methyltransferases enzymes, histone-lysine N-methyltransferase and histone-arginine N-methyltransferase: both catalyze the transfer of one to three methyl groups from the co-factor S-adenosyl methionine to lysine and arginine residues of histone proteins. Methylation of histone 3 (H3) at lysine 4 (K4) is also associated with transcriptional activation, whereas methylation of H3 at K9 or 27 and of H4 at K20 is associated with transcriptional repression (Cameron et al. 1999; Esteller 2008; Kondo et al. 2008).
Phosphorylation
Relatively little is known about the enzymes that generate histone modifications. Phosphorylation of serine 10 in histone H3 has been shown to correlate with gene activation in mammalian cells (Nowak and Corces 2000). The mechanism by which phosphorylation contributes to transcriptional activation is not well understood. The addition of negatively charged phosphate groups to histone tails neutralizes their basic charge reducing their affinity for DNA. Furthermore, it has been found that several acetyltransferases have increased HAT activity on serine 10-phosphorylated substrates. Thus, phosphorylation may contribute to transcriptional activation through the stimulation of HAT activity on the same histone tail. Phosphorylation of histone H3 is also known to occur after activation of DNA damage signaling pathways. For example, a conserved motif found in the carboxyl terminus of yeast H2A and the mammalian H2A variant H2AX is rapidly phosphorylated upon exposure to DNA-damaging agents (Rogakou et al. 1999).
Serine 139 has been identified as the site for this modification, and its phosphorylation in response to damage is dependent on the phosphatidylinositol-3-OH kinase, and again serine 10 appears to play a key role (Cheung et al. 2000). Mec1, in yeast, Mec1-dependent serine 139 phosphorylation is apparently required for efficient non-homologous end-joining repair of DNA (Rogakou et al. 2000). This suggests that phosphorylation mediates an alteration of chromatin structure, which in turn facilitates repair.
Sumoylation
Histone H4 is modified by small ubiquitin-related modifier (SUMO) family proteins both in vivo and in vitro. H4 binds to the SUMO-conjugating enzyme and mediates gene silencing through recruitment of histone deacetylase and heterochromatin proteins (Shiio and Eisenman 2003).
Citrullination
Peptidylarginine deiminase (PAD) enzymes catalyze the conversion of arginine residues in proteins to citrulline residues. Citrulline is a non-standard amino acid that is not incorporated into proteins during translation, but can be generated posttranslationally by the PAD enzymes. One protein isozyme responsible for this modification, protein arginine deiminase 4 (PAD4), has also been proposed to “reverse” epigenetic histone modifications made by the protein arginine methyltransferases (Denis et al. 2009).
Acetylation
The core histone N-terminal domains are rich in positively charged basic amino acids, which can actively interact with DNA. Acetylation neutralizes the positive charges on histones and disrupts the electrostatic interactions between DNA and histone proteins, promoting chromatin unfolding which has been associated with gene expression (Gregory et al. 2001). Under physiological conditions, chromatin acetylation is regulated by the balanced action of histone acetyltransferases (HATs) and deacetylases (HDACs). The HATs transfer acetyl groups from acetyl coenzyme A onto the amino groups of conserved lysine residues within the core histones. Acetylation can neutralize the positive charge of histones, loosening their interactions with the negatively charged DNA backbone, and leading to a more “open” active chromatin structure favoring the binding of transcription factors for active gene transcription (Gregory et al. 2001).
In contrast, the re-establishment of the positive charge in the amino terminal tails of core histones catalyzed by HDACs is thought to tighten the interaction between histones and DNA, blocking the binding sites on promoter, thus inhibiting gene transcription. Obviously, a subtly orchestrated balance between the actions of HATs and HDACs is essential to the maintenance of normal cellular functions, shifting this balance in both senses might have dramatic consequences on the cell phenotypes such as carcinogenesis.
Ubiquitination
Ubiquitination represents another important histone modification (Jason et al. 2002). Histone H2A, the first protein identified to be ubiquitinated at the highly conserved residue, Lys 119 (Nickel and Davie 1989), can be monoubiquitinated, in the majority of the cases and polyubiquitinated, less frequently, in many tissues and cell types (Nickel and Davie 1989). In addition to H2A, H2B is also ubiquitinated (West and Bonner 1980). Only monoubiquitinated H2B has been reported, but like H2A, the ubiquitinated site has also been mapped to Lys 120 residue located at the C terminus of H2B in human. In addition to H2A and H2B, ubiquitination on H3 and H1 have also been reported (Chen et al. 1998; Pham and Sauer 2000).
Addition of a ubiquitin moiety to a protein involves the sequential action of E1, E2, and E3 enzymes. Removing the ubiquitin moiety, on the other hand, is achieved through the action of enzymes called isopeptidases (Wilkinson 2000).
Accumulating evidence indicates that ubiquitin plays an important role in regulating transcription either through proteasome-dependent destruction of transcription factors or proteasome-independent mechanisms (Conaway et al. 2002). Several studies suggest that not only histone ubiquitination (Krogan et al. 2003), but also deubiquitination (Henry et al. 2003), may participate in gene activation, linked to histone acetylation and methylation (Dover et al. 2002).
Histone deacetylases
In the 1970s, the Friend erythroleukemia cell line was found to differentiate in the presence of dimethyl sulfoxide or butyrate (Sato et al. 1971). Many compounds with the ability to promote differentiation of tumor cell lines, particularly those with a planar-polar configuration, induced accumulation of hyperacetylated histones.
This histone modification increased the spatial separation of DNA from histone and enhanced binding of transcription factor complexes to DNA (Lee et al. 1993). The first mammalian HDACs were cloned on the basis of their binding to known small molecule inhibitors of histone deacetylation. These genes were homologous to yeast transcriptional repressors, strengthening the evidence that histone deacetylation suppresses gene expression.
HDACs are a class of enzymes that remove acetyl groups from a ε-N-acetyl lysine amino acid on a histone. Contrarily to HAT, HDAC proteins are now also being referred to as lysine deacetylases (KDAC), as to more precisely describe their function rather than their target, which also includes numerous non-histone proteins. Eighteen human HDACs have been identified each with varying function, localization, and substrates (Table 2; Lane and Chabner 2009).
Classes of HDACs
HDACs can be divided in four different classes, based on the homologies between human and yeast reduced potassium dependency 3 (RPD3) enzymes.
Class I HDACs (HDACs 1, 2, 3, and 8) are related to the yeast RPD3 deacetylase and are primarily found in the nucleus with the exception of HDAC 3, which can be located both in the nucleus and the cytoplasm.
Class II HDACs are divided into two subclasses, class IIa (HDACs 4, 5, 7, and 9) and class IIb (HDACs 6 and 10), both homologous to the yeast Hda1 deacetylase. This class of HDACs is able to shuttle in and out of the nucleus responding to different signals.
Class III HDACs consists of seven HDACs (SIRT1 to SIRT7) and shows several homologies with the yeast silent information regulator 2. This class of HDACs has a unique catalytic mechanism requiring the co-factor NAD+ for activity.
Class IV of HDACs has only one member (Butler and Kozikowski 2008), HDAC 11, which shares similarities to both class I and class II HDACs.
Classes I, II, and IV require Zn2+ for activity.
Catalytic site and deacetylation mechanism
Histone proteins catalytically remove an acetyl group from a ε-N-acetyl lysine amino acid on a histone. The active site of HDACs consists of a cylindrical pocket, covered by hydrophobic and aromatic amino acids, where the lysine residue fits when deacetylation takes place; a zinc ion is located near the bottom of the cylindrical pocket, which is coordinated by amino acids and a single water molecule (Butler and Kozikowski 2008). During deacetylation, the water molecule acts as the nucleophile attacking the carbonyl, assisted by the zinc ion (Fig. 1). Prior to attacking the carbonyl group of the N-acetylated lysine, the water molecule is activated by an Asp–His charge relay system. The residues forming the cylindrical pocket and the adjacent cavity of classes I and II HDACs are highly conserved, although the residues lining the entrance of the pocket are not as conserved as the residues inside the pocket. Therefore, it is possible to design HDAC inhibitors (HDACis) that are isoform selective.

Water molecule acting as the nucleophile prior and during the attack of carbonyl, assisted by the zinc ion
HDAC expression in cancer tissues
Evidence has shown an implication of HDACs in cancer, as the high expression of HDAC isoenzymes correspond to hypoacetylation of histones in cancer cells compared to normal tissues (Nakata et al. 2004; Yoo and Jones 2006): HDAC 1 enzyme expression is higher in colon adenocarcinoma cells than in normal, while HDAC 2 expression is elevated in colon cancer, possibly as a result of adenomatosis poliposis coli gene deficiency.
A study conducted by Nakagawa showed a hyper expression of HDACs in a variety of cultured cancer lines and a broad panel of primary human lung, esophageal, gastric, colon, pancreas, breast, ovary, and thyroid cancers (Nakagawa et al. 2007).
High level class I HDAC expression was found in 75% esophageal, gastric, colon, and prostate cancers, as well as corresponding adjacent “normal” tissues. Esophageal and prostate cancers tended to exhibit more consistent overexpression of class I HDACs. Further studies suggest a correlation between advanced stage of lung carcinoma and high expression of class 1 HDAC 1 (Sasaki et al. 2004), as well as aggressive tumor histology, advanced stage of disease, and poor prognosis in patients with pancreatic carcinoma (Miyake et al. 2008).
Several retrospective studies, conducted on colon cancer primary cells, showed high level expression of HDAC 1 and 2 in gastric cancer; the higher expression correlated with nodes metastasis, undifferentiated histology, and poor prognosis (Weichert et al. 2008). High level expression of HDAC 1, 2, and 3 was observed in 70%, 74%, and 95% of 192 prostate cancers in a related study, where overexpression of HDAC 1 and/or HDAC 2 correlated with poorly differentiated tumors. Co-expression of elevated levels of the three HDACs was associated to an increased proliferation index, while overexpression of HDAC 2 alone was an independent index of poor prognosis.
Overexpression of HDAC 2 also correlates with advanced stage of disease and diminished survival of oropharyngeal carcinoma patients (Chang et al. 2009). In contrast, HDAC 1 expression in breast cancer is associated with estrogen receptor/progesterone (PR) expression at the earlier stage of disease (T as well as N classifications) and improved patient survival response to tamoxifen (Bicaku et al. 2008), and HDAC 6 expression is associated to improved patient survival (Zhang et al. 2004).
HDAC expression in hematological malignancies
In hematological malignancies, increased HDAC activity may lead to transcriptional repression of genes essential for hematopoietic differentiation, playing a critical role in the pathogenesis of several leukemias. In the case of acute promyelocytic leukemia (APL), the PML-RARα protein product of the t(15–17) translocation, and in core binding factor leukemia gene products AML1–ETO, product of the t(8–21) translocation and CBF–MYH 11, cause a transcriptional repression, aberrantly recruiting HDACs to genes promoter regions (Minucci and Pelicci 1999); (Minucci et al. 2001). CTCL cells undergo higher rates of apoptosis than normal lymphocytes in response to HDAC inhibitor treatment (Zhang et al. 2005). Sodium butyrate and tricostatin A induced apoptosis of acute myeloid leukemia (AML; HL60) and CML (K562) cell line, downregulating the expression of Daxx, without affecting the expression of Bcl-2 or Bcl-XL. A recent study reports anti-leukemic activity of valproic acid through induction of apoptosis, in CLL primary cells obtained from 14 patients (Stamatopoulos et al. 2009).
These findings supported drug development of HDACi that interacting with the catalytic region of HDACs, block HDAC activity, induce differentiation, cell growth arrest, or cell death in tumors (Mai et al. 2005; Nebbioso et al. 2005).
Histone deacetylase inhibitors
HDACis are compounds that are able to interact with the catalytic domain of histone deacetylases to block the substrate recognition ability of these enzymes, thus resulting in restoration of relevant gene expression (Finnin et al. 1999).
The vast majority of HDACis shares a common mechanism of action that consists with binding the catalytic domain of HDAC enzyme, thereby blocking substrate recognition and inducing gene expression. Most of the described HDACis primarily affect class I and class II HDACs, which are zinc dependent.
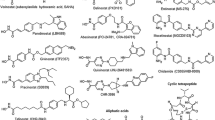
HDACis can be divided into four classes based on different chemical properties: short-chain fatty acids, hydroxamic acids, cyclic peptides, and benzamides (Table 1).
Short-chain fatty acids
This group includes Na butyrate, 4-phenylbutyrate, valproic acid, and phenyl acetate. The mechanism of action has not been well clarified yet, although a strong hypothesis exists in which the carboxylic function acts as zinc-binding group or competes with the acetate released in the deacetylation reaction by occupying the acetate escaping tunnel as described by Wang (Mai and Altucci 2009).
The butyrates (Na butyrate and 4 phenylbutyrate) inhibit the growth of several cancers, such as colon prostate and endometrial, but just at high concentration; although they both show effects on phosphorylation and methylation of histone and other nuclear proteins.
Sodium valproate (valproic acid, VPA), an “old” drug used in neurology as anticonvulsive and mood stabilizing, has been incidentally identified as HDACi and has shown anticancer effects in carcinoma cells, differentiation in hematopoietic cell lines, and also in clinical trials for hematological malignancies, such as leukemia, myelodysplastic syndrome (MDS), and lymphoma. VPA inhibits class I/IIa HDACs at very low concentrations compared to butyrates (Mai and Altucci 2009).
Hydroxamic acids
This class includes the majority of HDACis presently in use in clinical trials in hematological malignancies. They have a common structure characterized by a hydrophobic CAP group, able to interact with the rim of the catalytic tunnel of the enzyme, a polar connection unit (CU). Present in most of the HDACis, the CU can interact with the amino acids in the tunnel, and a 4- or 6-carbon unit hydrophobic spacer (linker), allowing the following zinc-binding group (ZBG) to reach and to complex the zinc ion and thus inhibiting the enzyme (Mai et al. 2005).
Past reports of the hydroxamic acid tricostatin A, first isolated as antifungal antibiotic (Tsuji et al. 1976), indicated the capacity of this drug to differentiate the Friend erythroleukemia cells. Further experiments have shown that the compound caused hyperacetylation due to hystone deacetylation inhibition.
The hybrid polar compounds (HPCs) are potent inducers of differentiation of murine erythroleukemia cells and a various other cancer cells (Andreeff et al. 1992; Mai et al. 2005). The progenitor of these compounds was hexamethylene bisacetamide, a drug that is able to induce remission in patients with myelodysplastic syndrome and acute myeloid leukemia, but cannot be used in clinical trials for the high dosage required and adverse side effects. The second generation of HPCs has shown a strong induction of apoptosis or cell differentiation at low doses (Mai and Altucci 2009).
The prototype of this class is the suberoylanilide hydroxamic acid (SAHA or Vorinostat). The chemical structure of SAHA is similar to VPA and the others, HPCs, with a CAP, a CU, and a ZBG. It has shown to induce acetylation in a vast variety of cell line and apoptosis, cell cycle arrest, and differentiation. SAHA is selective for HDAC 1, 2, 3, 4, 6, 7, and 9 and shows lower potency against HDAC 8.
In October 2006 the US FDA, approved SAHA in the therapy of refractory, relapsed cutaneous T-cell lymphoma (CTCL) and is at the present involved in a vast variety of clinical trials both in hematological malignancies, such as leukemia, MDS, lymphoma and myeloma, and solid tumors. The mechanism of action is still unclear because of the involvement of many pathways, including apoptosis, autophagy, and induction of ROS and DNA damage repair, all of them subsequent to re-expression of genes that become accessible to transcription factors, when hystone proteins are in an acetylated status.
The clinical efficacy of SAHA inspired the development of new analogues of the same class, as the indolyethylamino-methylcinnamyl hydro-amides LAQ-824 and LBH 589 (panobinostat; Arts et al. 2007).
Panobinostat, as SAHA, is in a vast variety of phase II/III clinical trials, both in solid tumors and hematological malignancies, such as lymphomas, multiple myeloma, MDS, acute myeloid leukemia, and CML. Its HDAC inhibition is strong against class I HDACs and less potent against class IIa.
Belinostat (PXD101) is a hydroxamic acid derivative, which has been administered as an infusion on days 1 to 5 of a 21-day cycle in a phase I study in patients with advanced B-cell malignancies refractory to standard therapy.
Cyclic peptides
The cyclic peptide Romidepsin, also known as FK-228, has been reported to induce cell cycle arrest and apoptosis in a variety of human cancer cells. In vitro it has shown a strong activity against HDAC 1 and 2, but also against HDAC 6 and HDAC 4, although it results to be weaker (Mai and Altucci 2009). The drug has been in clinical trials for CML and AML (Byrd et al. 2005; Piekarz et al. 2004) and has been approved in November 2009 for the treatment of refractory–relapsed CTCL (Kim et al. 2008).
Benzamides
This class of HDACis presents a different structure from the other classes because of the 2′-aminoanilide moiety, which can likely function as a weak zinc-chelating group into the tube-like active site of the deacetylase core of the enzyme as suggested by molecular modeling studies (Wang et al. 2005), or may contact key amino acids in the active site without Zn ion coordination. MS-275 (entinostat) inhibits preferentially HDAC 1, 2, and 3 and is inactive (IC50s > 10 μM) against HDAC 4, 6, 7, and 8 (Khan and La Thangue 2008). In clinical trials, it has been used in solid tumors, such as lung and breast cancer and metastatic melanoma, and in hematological malignancies, as CML, AML, CMML, and Hodgkin disease.
MGCD 0103 is a more recent benzamide that shows selectivity for HDAC I/II. It has been used in clinical trials mostly for hematological malignancies, such as AML, CML, non-Hodgkin lymphomas and refractory Hodgkin disease, where it has shown very encouraging results.
SIRT inhibitors
The compounds of this class of HDACis (class III) are much less validated as anticancer agents compared to HDAC I and II. This is also due to the fact that a low number of preclinical in vivo data exist presently and a small number of inhibitors are on the market.
Sirtuins play an important role in many cellular processes such as gene silencing and regulation of transcription factors. Members of the sirtuin family of NAD+-dependent protein deacetylases SIRT1 controls cell differentiation, metabolism, circadian rhythms, stress responses, and cellular survival. When tested in the human leukemia U937 cell line, SIRT inhibitors displayed high levels of apoptosis and cyto-differentiation. SIRT1 binds and deacetylates the histone acetyltransferase p300, which inhibits p300 enzymatic activity, which has the potential to promote hypoacetylation of nucleosomal histones and affect gene expression outcomes (Zhang et al. 2009). Cambinol is a SIRTi (against SIRT1 and -2, respectively) that induces apoptosis in BCL-6 expressing Burkit lymphoma cells, through acetylation of both BCL-6 and p53 (Heltweg et al. 2006). Finally, the anticancer action of SIRT inhibitors has been also reported to act via reactivation of methylated genes without evident DNA demethylation at the promoters level (Pruitt et al. 2006).
Mechanism of action
The clinical manifestation of aberrant HDAC expression is histology dependent. Each of the HDACs have different targets, not only interacting directly with chromatin through the acetylation of core histones (Ozdag et al. 2006) or “marking” chromatin for further recruitment of chromatin remodeling complexes (Choi and Howe 2009), such as hormone receptors, DNA repair enzymes, and signal transduction mediators chaperone proteins and cytoskeleton proteins, regulating cell proliferation and cell death (Dokmanovic and Marks 2005). Acetylation can either increase or decrease the function or stability of the proteins or protein–protein interaction (Glozak et al. 2005).
Gene expression
Acetylation generally results in an increase of the negative charge on histone core leading to an uncoiled chromatin structure, and accessibility to transcription factors of different genes, but the final gene expression is the result of the interaction of other alterations in chromatin structure mediated by DNA, such histone methylation, and the summation of activators and repressors recruited to the respective promoters (Ellis et al. 2008). It is important to stress that the number of genes detected with altered transcription is strongly influenced by the time of culture and concentration of HDACi. A short exposition and a low concentration of HDACi are associated with fewer changes, compared to consisting doses of the drugs and longer time points (Ellis et al. 2008).
The gene that seems to act on of the most induced by HDACis is a cyclin-dependent kinase, p21, which is regulated via p53 dependent as well as p53 independent; its expression correlates with an increase in the acetylation of histones associated with the p21 promoter region (Richon et al. 2000) and coincides with acetylation of histones H3 and H4.
HDACis are also able to increase the activity of 5-azacytidine and 5-aza-2′-deoxycytidine of the expression of suppressor gene promoters, silenced by aberrant methylation (Cameron et al. 1999). This mechanism of potentiation resulted to be more complex because recent studies suggest that TSA decreases the stability of DNMT3b mRNA, resulting in decrease of de novo methylation in endometrial cancer cells (Xiong et al. 2005).
Cell cycle regulation
HDACis have shown to impact the cell cycle by inducing, at low concentrations, predominantly G1 arrest (Marks et al. 1996) and G1 and G2/M arrest, at high concentration (Richon et al. 1997).
Cell cycle arrest is associated with decreased expression of cyclins A, B, and D, as well as their respective cyclin-dependent kinases, and induction of p21 and p27 (Emanuele et al. 2008). TSA, FK 228, deplete levels of kinethocore proteins and decreased phosphorylation of histone H3 in pericentrometric chromatin during G2 phase, while SAHA inhibits the transcription of aurora kinase A and B, resulting in apparent mitotic catastrophe (Dowling et al. 2005; Park et al. 2008; Robbins et al. 2005).
Regulation of the apoptotic pathways
Extrinsic pathway of apoptosis is initiated by the binding of death receptors to their ligands leading to activation of caspase-8 and caspase-10. HDACis upregulate the expression of the death receptors, Fas (Apo-1 or CD95), tumor necrosis factor (TNF) receptor-1 (TNFR-1), in transformed cells, but not in normal cells (Nakata et al. 2004). TRAIL and its receptor DR-5 were induced in the mouse model of APL by VPA (Insinga et al. 2005); TNF-related apoptosis-inducing ligand death receptors (DR-4 and -5) were induced in the mouse model of APL by VPA.
The intrinsic apoptosis pathway is mediated by mitochondria with the release of mitochondrial inter-membrane proteins, such as cytochrome c, apoptosis-inducing factor Smac, and the consequent activation of caspases. It is regulated, in part, by pro- and anti-apoptotic proteins of bcl-2 family (Insinga et al. 2005). Activation of the intrinsic apoptotic pathway is a major pathway for HDACis to induce cell death utilizing mechanisms that are still not well understood. HDACis facilitate the release of cytochrome c from mitochondrial inter-membrane space and activation of caspase-9 (Marks et al. 1996; Bolden et al. 2006), overexpression of Bcl-2 or Bcl-XL, while protecting the mitochondria, inhibit HDACi-induced apoptosis.
HDACis and autophagy
Autophagy is a mechanism of cellular proteins self-digestion by complexes called auto-phagosomes rich in lysosome, activated in cells during stress conditions, such as ATP deprivation, infections, and glucose deprivation. This mechanism of death is caspase independent and induced by nuclear, and p73 upregulation in response to cellular stress, and is p53 independent. HDACis regulate autophagy by inhibiting p73-mediated activation of a variety of genes including ATG5, ATG7, and p53 that can inhibit mTOR via activation of AMPK. Autophagy induced by HDACis seems related to inhibition of HDAC 1 (Rosenbluth et al. 2008).
HDACis and DNA damage repair
Induction of phosphorylation of the histone variant H2AX has been associated with the generation of double-strand DNA. A study conducted at M.D. Anderson Cancer Center showed that Vorinostat has the capacity to induce H2AX phosphorylation in two different cell lines (Molt4 and HL60). Induction of H2AX may have a role in the capacity to induce apoptosis of these drugs. In a model of irradiation, loss of H2AX resulted in clonogenic survival. This data is important, suggesting that HDACis may exert their cytotoxic effects by other mechanisms not directly linked to histone acetylation, but perhaps DNA damage (Sanchez-Gonzalez et al. 2006).
Induction of ROS by HDACis
Accumulation of ROS occurs in transformed cells cultured with HDACis, such as Vorinostat, TSA, butyrate, or MS-275 (Rosato et al. 2003; Ruefli et al. 2001; Ungerstedt et al. 2005; Xu et al. 2006). Accumulation of ROS might play an important role in HDACi-induced cell death. ROS accumulation occurs within 2 h of culture with HDACis, before disruption of mitochondria. Free radical scavengers such as N-acetylcysteine are able to decrease HDACi-induced apoptosis (Ruefli et al. 2001; Ungerstedt et al. 2005).
HDACis in clinical practice
Most promising results of the in vitro studies secured clinical trials based on the use of HDACis as single agents or in combination with other “epidrugs” and target therapies, or with the conventional chemotherapy either in solid tumors and hematological malignancies with the aim of improved response and reduced toxicity. Most of the pilot studies, to determine the safety and the efficacy of the compounds, have been conducted in hematological malignancies, nearly all of them in lymphoproliferative diseases first. The phase I clinical trials have shown that the drugs are safe, although with some side effects, for example cardiotoxicity that required the suspension of the study in some cases, and most of them are more effective in hematological malignancies than in solid tumors for different reasons that we will analyze later.
This review will attempt to summarize the most significant clinical trials conducted in the last few years in the hematological malignancies, attempting to group these diseases in three categories: lymphomas, myelodysplastic syndromes, and acute myelogenous leukemia and myeloma. These drugs have been experienced in myeloproliferative neoplasm, where encouraging results are coming out of studies performed with Givinostat alone in Polycythemya Vera and essential thrombocythemya (Rambaldi et al. 2008).
HDACis as single agents, the treatment of lymphoma
Lymphomas are heterogeneous diseases, with very different biologic features and clinical outcomes. Most of the phase 1 and 2 clinical trials that explore the safety and efficacy of HDAC inhibitors are conducted on relapse or refractory patients and tend to group the lymphomas as the same disease. Only subsequent studies divide the disease in morphological clinical categories, according to the WHO classifications. In this review, we aim to distinguish the results obtained in T-cell lymphomas from those obtained B-cell and Hodgkin lymphomas.
T-cell lymphomas
Lymphomas are very heterogeneous diseases, with different biologic features and clinical outcomes. Most of the phase 1 and 2 clinical trials that explore the safety and efficacy of HDAC inhibitors are conducted on relapsed or refractory patients and tend to group the lymphomas as the same disease. Only subsequent studies divide the disease in morphological clinical categories, according to the WHO classifications. One of the first studies in lymphomas was conducted with Vorinostat in 35 patients with advanced hematological malignancies (O’Connor et al. 2006). Five patients with Hodgkin lymphoma (HL) diffused large B-cell lymphoma (DLBCL) and cutaneous T-cell lymphoma and experienced tumor reduction, while collateral studies reported a transient (8 h with a peak at 2 h) increase in histone H3 acetylation in peripheral blood mononuclear cells. Due to the response shown in a CTCL patients treated with oral Vorinostat, a dose-finding study was initiated in 33 patients with relapsed/refractory CTCL exploring three different schedules of administration that showed 24% overall response (Mann et al. 2007). A second study (Olsen et al. 2007), which led to the Food and Drug Administration approval of Vorinostat in CTCL, was conducted in 74 patients with stage II B or higher CTCL. This study showed 33% ORR and assessed the 400 mg/day the optimum in terms of response/toxicity (Duvic et al. 2007). The side effects were almost the same in the two trials, predominantly nausea and diarrhea, while anemia and thrombocytopenia represented the main hematologic toxicity. Further studies, conducted with new HDACis, have investigated the efficacy of these drugs in CTCL and PTCL.
Panobinostat, administered orally at a MTD of 20 mg, 3 days a week on a 28-day cycle, showed to be safe and effective, achieving complete responses, partial response, stable disease with ongoing improvement, and progression on treatment, respectively, in 2, 4, 2, and 1 patient. Microarray data showed distinct gene expression response profiles over time following panobinostat treatment, with the majority of genes being repressed (Ellis et al. 2008). A recent clinical trial conducted on two cohorts of patients, previously treated with bexarotene or naïve, reported complete skin responses in 11 out of 62 pretreated patients, and 4 in 11 out of 33 naïve patients (Duvic et al. 2007).
Based on these results, a phase II study evaluating a MWF schedule of panobinostat in patients with relapsed CTCL is now ongoing (Ottmann et al. 2008).
Clinical efficacy has also been reported in a clinical trial conducted with Belinostat in patients with relapsed in CTCL and PTCL, showing in both diseases the same high rate of response (25%) and disease control (50% and 72%; Pohlman et al. 2009).
Interesting results have been reported in clinical trials with Romidepsin as a single agent. The first study conducted in 2001 at the National Cancer Institute reported responses in four patients with T-cell lymphoma (Piekarz et al. 2001). A recent analysis of a phase II study has reported 34% of response with four CR out of 71 patients treated with Romidepsin. The study presents a long duration of response with a median time to progression of 15.1 months (Bates). An international multicenter study confirmed these results in CTCL with 32% ORR and six CR (Kim et al. 2008). Promising results have been reported also in a Romidepsin single agent clinical trial with the treatment of relapsed–refractory PTCL, with 31% ORR, including four CR and 11 PR, signing the approval of the US Food and drug Administration in the therapy of refractory–relapsed CTCL (Piekarz et al. 2008).
B-cell and Hodgkin lymphomas
Initial encouraging results were obtained with Vorinostat studying its effect in B-cells neoplasm, particularly DLBCL. This study was not successful as expected from the phase I study, given that only one patient out of 18 showed complete response (CR), while 16 patients experienced progressive disease and one stable disease (SD; Crump et al. 2008a).
In HL, Vorinostat showed a partial response in one patient and a stable disease in nine out of 25 eligible patients tested. A phase I/II study conducted in non-Hodgkin lymphomas, with a schedule of 200 mg administered twice a day for 14 days on a 21-day cycle, showed CR in four patients, PR in two, and SD in four out of 17 patients in the study (Kirshbaum et al. 2008).
Panobinostat has been investigated in Hodgkin lymphomas in a phase IA/II multicenter study showing computed tomography partial response in 5 out 13 (38%) patients treated and a metabolic response by (18)F-fluoro-2-deoxy-d-glucose positron emission tomography scanning in 7/12 (58%) evaluable patients (Dickinson et al. 2009).
Mocetinostat has also been tested in B-cell lymphomas and HL. In the first case, a phase II clinical trial in patients with relapsed–refractory, DLBCL, and follicular lymphomas (FL) previously treated with a Rituximab-based regimen, showed one CR and three PR in DLBCL group and 13 SD; one patient with FL out of ten achieved a PR. Interestingly, DLBCL patients with SD presented 6 months to 1 year PFS (Bociek et al. 2008).
Very encouraging results have been showed in a phase II clinical trial in relapsed–refractory HL at a dose of 85 or 110 mg, three times a week. In the 110 mg cohort, two had CR and six PR, with a very long PFS (270–420 days); all patients in the 85 mg cohort showed tumor reduction, with one PR and one SD (Crump et al. 2008b). Finally, SD has been reported in 7 out of 13 patients evaluable in a phase II study conducted with Givinostat in HL, without severe toxicity (Carlo-Stella et al. 2008).
HDACis in the treatment of acute myeloid leukemias and myelodysplastic syndromes
The anti-leukemic activity showed in the preclinical studies opened the way to a large number of clinical trials in leukemia and myelodysplastic syndromes. Single agent clinical trials proved to be interesting, but produced limited results, possibly due to the fact that phase I and II clinical trials enrolled refractory–relapsed, heavily pretreated patients.
The first HDACi used in the treatment of AML and high risk MDS has been the valproic acid (VPA), a well-known drug, used in the therapy of epilepsy. Most of the pioneering studies reported clinical efficacy of VPA alone, but mostly in combination with all-trans retinoic acid (ATRA).
A pilot study of VPA/ATRA combination conducted in 11 elderly de novo AML patients (median age 82 years) showed complete marrow response in three patients, including one complete remission and two additional hematological improvements (Raffoux et al. 2005).
A German clinical trial, studying the combination VPA/ATRA, was also conducted in 26 poor-risk AML patients; one patient with de novo AML, out of 19 evaluable for response, achieved a minor response, two patients with AML secondary to myeloproliferative neoplasms reached partial response, while none achieved complete remission (Bug et al. 2005).
A subsequent study of the combination of VPA with two differentiating agents, 13-cis retinoic acid and 1.25 dihidroxyvitamin D3 in 19 previously untreated MDS or CMML patients, showed a response in three patients (16%), although eight patients had to discontinue the treatment for toxicity.
A phase II study has been conducted on 75 patients, 66 of which were treated with VPA monotherapy, in addition to ATRA in non-responders or relapsed, and nine patients were treated with ATRA + VPA from the beginning. Response rates were disease-depending: 52% response in MDS patients with normal blood count, 16% in AML, and 0% in CMML (Kuendgen et al. 2005).
Clinical evidences of the drug efficacy were shown in a study conducted in 58 patients with AML (32 AML secondary to MDS, 22 de novo AML, four AML secondary to myeloproliferative neoplasms) elderly or medically unfit to receive intense chemotherapy (Siitonen et al. 2007). Twenty-seven patients received VPA + ATRA from the start, and in 13 patients, ATRA was added later due to non-responders or relapse. The response rate was only 5%, but in 23 patients a decrease of blast count was observed (Kuendgen et al. 2006).
A study conducted combining VPA with ATRA and theophylline in 24 AML elderly patients showed that in 22 patients evaluable for response, nine responded with increased normal peripheral blood cell counts (Ryningen 2009).
The first evidence of the clinical efficacy of Vorinostat in AML has been shown in patients with advanced hematological disease, where the vast majority was represented by AML. Maximum tolerated dose was established as 200 mg twice a day. Clinical response was observed just in patients with AML, in terms of hematological improvement in 17% of the cases, whereas two patients presented CR and two CR with incomplete blood count recovery. None of the patients with ALL, CLL, and MDS experienced any response, despite hyperacetylation of histone H3 being observed in the blood and bone marrow cells of all the patients (Garcia-Manero et al. 2008a).
Panobinostat was also tested in the same category of patients (AM, ALL, and MDS) in the first clinical trial, administrated once a day intravenously, on day 1–7 on a 21-day cycle. The trial was discontinued because it caused asymptomatic QT elongation at the dose of 14 mg, but it showed anti-leukemic activity in terms of reduction of blasts number in the peripheral blood at all tested doses (Giles et al. 2006).
Preliminary results from an ongoing phase IA/II dose escalation study, with one arm receiving three times weekly dosing and the other three times alternate weekly dosing. Patients evaluable for response showed that anti-leukemic activity was dose- and schedule-dependent, with no response in patients treated alternative weekly, or those treated at doses <40 mg, while two CR, one CR without complete blood count reconstitution, and four patients with >50% reduction bone marrow or peripheral blood blasts (Ottmann et al. 2008).
Anti-leukemic activity has been reported in patients treated with Mocetinostat. The drug has been readmitted after suspension for episodes of pericarditis. A clinical trial conducted in patients with AML and high risk MDS reported three responses in AML patients, with two CR (Garcia-Manero et al. 2008b), despite another trial carried out on a less intensive regimen, which showed SD in four of 19 treated patients (Lancet et al. 2007).
Entinostat has not shown encouraging results in 12 refractory AML patients, with no CR or PR, but just a reduction in the blast count (Gojo et al. 2007). The study also showed that the drug is not tolerable on a daily schedule.
Finally, Romidepsin was tested at the dose of 13 mg/m2 once a week in a cycle of 4 weeks in a clinical trial that divided patients with chromosomal abnormalities (known to aberrantly recruit HDAC-repressive complexes) and patients lacking in these abnormalities. Romidepsin showed anti-leukemic activity in the cohort of patients carrying chromosomal abnormalities that may recruit HDACs: t(8;21), t(15;17) and inv16 (Odenike et al. 2008).
HDACis in the treatment of multiple myeloma
Several clinical trials have investigated the safety and efficacy of some HDACis used as single agents in the treatment of multiple myeloma (MM). The first study, carried out with Vorinostat, was nearly terminated by decision of the sponsor, and the MTD was not even found (Richardson et al. 2008). Modest results have been observed with panobinostat at the dose of 20 mg three times a week (Niesvizky et al. 2005), and complete absence of response in patients treated with Romidepsin was reported (Wolf et al. 2008), whereas clinical response in a clinical trial with Givinostat in 4 out of 14 patients was reported (Galli et al. 2010)
An association study of Romidepsin in combination with bortezomib in heavily pretreated myeloma patients has shown good tolerability in the 22 patients treated, with four CR, two VGPR, and six PR. The study is still ongoing (Harrison et al. 2008), and a combination study of bortezomib in combination with panobinostat is under evaluation (Siegel et al. 2008).
Toxicity profile of HDACis
The primary aim of the epigenetic drugs is to target the epigenetic HDAC-based alterations present in cancer, leading to a better outcome with possibly a minimal toxicity. The HDACis tested at the present in clinical trials have shown to be safe, although adverse events, sometimes severe, have been reported.
The most common toxicities have been related to gastrointestinal or constitutional symptoms, hematologic abnormalities (Pohlman et al. 2009), or taste disorders (Bates et al. 2010) and were mostly mild to moderate in severity. A dose escalating study of Vorinostat in 41 patients AML was recently reported. Grade 3/4 adverse events were predominantly fatigue, diarrhea, and thrombocytopenia.
One of the first clinical studies conducted with Panobinostat administered intravenously in patients with AML, ALL, and MDS was suspended for the asymptomatic grade 3 Fredericia correction factor (QTcF) prolongations reported (Giles et al. 2006); further trials of the drug conducted in lymphoma patients reported hyperglycemia, fatigue, diarrhea, and thrombocytemia (Ellis et al. 2008).
Also Romidepsin has shown cardiac toxicity (Kim et al. 2008), which requires intensive electrocardiogram and ejection fraction monitoring of patients receiving the drug, along with careful patient selection excluding those with a significant cardiac history, and the use of aggressive electrolyte replacement to reduce risks of QTcF prolongation. Although minor electrocardiogram changes are relatively frequent, the raised cardiac enzymes or altered left ventricular function due to drug administration have been reported thus far, even with prolonged treatment. Several episodes of pericarditis or pericardial effusion in MGCD0103 trials have been also reported (Bociek et al. 2008; Crump et al. 2008a; Garcia-Manero et al. 2008), decreasing the clinical interest for this drug. Patients with no signs or symptoms suggestive of pericardial disease are still in trials. Other reported toxicities of MGCD0103 include fatigue and gastrointestinal symptoms, with apparently lower hematologic toxicity than other HDACis reported thus far.
Conclusions
The analysis of the clinical trials based on HDACis confirmed the anticancer activity in hematological malignancies. HDACis showed very encouraging results in the treatment of CTCL and PTCL, although alternate results have been reached in leukemia, where the drugs did not dramatically change the outcome of the disease, when used as single agent. However, nearly all the completed trials have been conducted on refractory–relapsed heavily treated patients, where adjunctive molecular changes and mechanisms of drug resistance might have occurred, has to be considered.
Another reflection is that all HDACis have been tested without having a “rea” molecular biomarker of response. Note that hyperacetylation of histones has been suggested as one of the biomarkers of drug activity, despite the levels of hyperacetylation do not seem to correlate with the clinical response. In contradiction, constant hyperacetylation has been reported to correlate with a prolonged response in lymphomas (Bates et al. 2010).
On a different angle, all the studies analyzed in this review have been conducted using the HDACis as single agents even if with different schedules, but combination studies have just been completed or are ongoing (Tables 2 and 3), and might suggest interesting options for treatment.
What might be the next step?
Based on these evidences, the directions are clear. First of all, it will be necessary to identify a biomarker predictive of response to the therapy with HDACis thus helping in stratifying the patient to the correct treatment. This unfortunately has not yet been clearly identified (Stimson and La Thangue 2009; Prince et al. 2009).
Note that very recently HR23B has been suggested as a marker of response in CTCL (Khan et al. 2010). Moreover, the administration schedule allowing a persistent hyperacetylation might be an option in selected cases. Finally, in the combination studies, it will be significant to discriminate the “best” exact regimen of HDACis and chemotherapy in time frames, doses, and drugs.
A better understanding of the mechanism of action of HDACis will improve their use in clinical practice, identifying the “right” disease, the “correct” patient, and both the best combination and way of combination of drugs to be applied.
References
Andreeff M, Stone R, Michaeli J, Young CW, Tong WP, Sogoloff H, Ervin T, Kufe D, Rifkind RA, Marks PA (1992) Hexamethylene bisacetamide in myelodysplastic syndrome and acute myelogenous leukemia: a phase II clinical trial with a differentiation-inducing agent. Blood 80(10):2604–2609
Arts J, Angibaud P, Marien A, Floren W, Janssens B, King P, van Dun J, Janssen L, Geerts T, Tuman RW, Johnson DL, Andries L, Jung M, Janicot M, van Emelen K (2007) R306465 is a novel potent inhibitor of class I histone deacetylases with broad-spectrum antitumoral activity against solid and haematological malignancies. Br J Cancer 97:1344–1353
Bates SE, Zhan Z, Steadman K, Obrzut T, Luchenko V, Frye R, Robey RW, Turner M, Gardner ER, Figg WD, Steinberg SM, Ling A, Fojo T, To KW, Piekarz RL (2010) Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol 148:256–267
Bicaku E, Marchion DC, Schmitt ML, Munster PN (2008) Selective inhibition of histone deacetylase 2 silences progesterone receptor-mediated signaling. Cancer Res 68:1513–1519
Bociek RG, Kuruvilla J, Pro B et al (2008) Isotype-selective histone deacetylase (HDAC) inhibitor MGCD0103 demonstrates clinical activity and safety in patients with relapsed/refractory classical Hodgkin lymphoma (HL) [abstract]. J Clin Oncol (Meeting Abstracts) 26(Suppl 15):8507
Bolden JE, Peart MJ, Johnstone RW (2006) Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–784
Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG (2005) Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer 104:2717–2725
Butler KV, Kozikowski AP (2008) Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des 14:505–528
Byrd JC, Peterson BL, Gabrilove J, Odenike OM, Grever MR, Rai K, Larson RA (2005) Treatment of relapsed chronic lymphocytic leukemia by 72-hour continuous infusion or 1-hour bolus infusion of flavopiridol: results from Cancer and Leukemia Group B study 19805. Clin Cancer Res 11:4176–4181
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB (1999) Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 21:103–107
Carlo-Stella C, Guidetti A, Viviani S, Bonfante V, Valagussa P, Marchianò A, Crippa F, Zambelli S, Fasola C, Corradini P, Tarella C, Di Nicola M, Gianni AM (2008) Phase II Trial of Combination of the Histone Deacetylase Inhibitor ITF2357 and Meclorethamine Demonstrates Clinical Activity and Safety in Heavily Pretreated Patients with Relapsed/Refractory Hodgkin Lymphoma (HL) Blood (ASH Annual Meeting Abstracts) 112:2586
Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY (2009) Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol 45:610–614
Chen HY, Sun JM, Zhang Y, Davie JR, Meistrich ML (1998) Ubiquitination of histone H3 in elongating spermatids of rat testes. J Biol Chem 273:13165–13169
Cheung P, Allis CD, Sassone-Corsi P (2000) Signaling to chromatin through histone modifications. Cell 103:263–271
Choi JK, Howe LJ (2009) Histone acetylation: truth of consequences? Biochem Cell Biol 87:139–150
Conaway RC, Brower CS, Conaway JW (2002) Emerging roles of ubiquitin in transcription regulation. Science 296:1254–1258
Crump M, Coiffier B, Jacobsen ED, Sun L, Ricker JL, Xie H, Frankel SR, Randolph SS, Cheson BD (2008a) Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol 19:964–969
Crump M, Andreadis C, Assouline S et al (2008b) Treatmentof relapsed or refractory non-hodgkin lymphoma with the oral isotype-selective histone deacetylase inhibitor MGCD0103: interim results froma phase II study [abstract]. J Clin Oncol (Meeting Abstracts) 26(Suppl 15):8528
Denis H, Deplus R, Putmans P, Yamada M, Metivier R, Fuks F (2009) Functional connection between deimination and deacetylation of histones. Mol Cell Biol 29:4982–4993
Dickinson M, Ritchie D, DeAngelo DJ, Spencer A, Ottmann OG, Fischer T, Bhalla KN, Liu A, Parker K, Scott JW, Bishton M, Prince HM (2009) Preliminary evidence of disease response to the pan deacetylase inhibitor panobinostat (LBH589) in refractory Hodgkin Lymphoma. Br J Haematol 147:97–101
Dokmanovic M, Marks PA (2005) Prospects: histone deacetylase inhibitors. J Cell Biochem 96:293–304
Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A (2002) Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 277:28368–28371
Dowling M, Voong KR, Kim M, Keutmann MK, Harris E, Kao GD (2005) Mitotic spindle checkpoint inactivation by trichostatin a defines a mechanism for increasing cancer cell killing by microtubule-disrupting agents. Cancer Biol Ther 4:197–206
Duvic M, Talpur R, Ni X, Zhang C, Hazarika P, Kelly C, Chiao JH, Reilly JF, Ricker JL, Richon VM, Frankel SR (2007) Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 109:31–39
Ellis DJ, Lawman ZK, Bonham K (2008) Histone acetylation is not an accurate predictor of gene expression following treatment with histone deacetylase inhibitors. Biochem Biophys Res Commun 367:656–662
Emanuele S, Lauricella M, Tesoriere G (2008) Histone deacetylase inhibitors: apoptotic effects and clinical implications (Review). Int J Oncol 33:637–646
Esteller M (2008) Epigenetics in cancer. N Engl J Med 358:1148–1159
Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP (1999) Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401:188–193
Galli M, Salmoiraghi S, Golay J, Gozzini A, Crippa C, Pescosta N, Rambaldi A (2010) A phase II multiple dose clinical trial of histone deacetylase inhibitor ITF2357 in patients with relapsed or progressive multiple myeloma. Ann Hematol 89:185–190
Garcia-Manero G, Assouline S, Cortes J, et al (2008) Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 112:981–989
Garcia-Manero G, Assouline S, Cortes J, Estrov Z, Kantarjian H, Yang H, Newsome WM, Miller WH Jr, Rousseau C, Kalita A, Bonfils C, Dubay M, Patterson TA, Li Z, Besterman JM, Reid G, Laille E, Martell RE, Minden M (2008a) Phase 1 study of the oral isotype specific histone deacetylase inhibitor MGCD0103 in leukemia. Blood 112:981–989
Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, Faderl S, Koller C, Morris G, Rosner G, Loboda A, Fantin VR, Randolph SS, Hardwick JS, Reilly JF, Chen C, Ricker JL, Secrist JP, Richon VM, Frankel SR, Kantarjian HM (2008b) Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 111:1060–1066
Giles F, Fischer T, Cortes J, Garcia-Manero G, Beck J, Ravandi F, Masson E, Rae P, Laird G, Sharma S, Kantarjian H, Dugan M, Albitar M, Bhalla K (2006) A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin Cancer Res 12:4628–4635
Glozak MA, Sengupta N, Zhang X, Seto E (2005) Acetylation and deacetylation of non-histone proteins. Gene 363:15–23
Gojo I, Jiemjit A, Trepel JB, Sparreboom A, Figg WD, Rollins S, Tidwell ML, Greer J, Chung EJ, Lee MJ, Gore SD, Sausville EA, Zwiebel J, Karp JE (2007) Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 109:2781–2790
Gregory RI, Randall TE, Johnson CA, Khosla S, Hatada I, O'Neill LP, Turner BM, Feil R (2001) DNA methylation is linked to deacetylation of histone H3, but not H4, on the imprinted genes Snrpn and U2af1-rs1. Mol Cell Biol 21:5426–5436
Harrison SJ, Quach H, Yuen K et al (2008) High response rates with the combination of bortezomib, dexamethasone and the pan-histone deacetylase inhibitor romidepsin in patients with relapsed or refractory multiple myeloma in a phase I/II clinical trial [abstract]. ASH Annual Meeting Abstracts 112:3698
Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, Bedalov A (2006) Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 66(8):4368–4377
Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev 17:2648–2663
Insinga A, Minucci S, Pelicci PG (2005) Mechanisms of selective anticancer action of histone deacetylase inhibitors. Cell Cycle 4:741–743
Jason LJ, Moore SC, Lewis JD, Lindsey G, Ausio J (2002) Histone ubiquitination: a tagging tail unfolds? Bioessays 24:166–174
Khan O, La Thangue NB (2008) Drug insight: histone deacetylase inhibitor-based therapies for cutaneous T-cell lymphomas. Nat Clin Pract Oncol 5:714–726
Khan O, Fotheringham S, Wood V, Stimson L, Zhang C, Pezzella F, Duvic M, Kerr DJ, La Thangue NB (2010) HR23B is a biomarker for tumor sensitivity to HDAC inhibitor-based therapy. Proc Natl Acad Sci USA 107(14):6532–6537, Epub 2010 Mar 22
Kim Y, Whittaker S, Demierre MF et al (2008) Clinically significant responses achieved with romidepsin in treatment- refractory cutaneous T-cell lymphoma: final results from a phase 2B, international, multicenter, registration study [abstract]. ASH Annual Meeting Abstracts 112:263
Kirshbaum MH, Goldman BH, Cook JR, Zain JM, Forman SJ (ASH 2008) Vorinostat in relapsed or refractory indolent non Hodgkin Lymphoma. A California Center Consortium Study
Kondo Y, Shen L, Cheng AS, Ahmed S, Boumber Y, Charo C, Yamochi T, Urano T, Furukawa K, Kwabi-Addo B, Gold DL, Sekido Y, Huang TH, Issa JP (2008) Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet 40:741–750
Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11:721–729
Kuendgen A, Knipp S, Fox F, Strupp C, Hildebrandt B, Steidl C, Germing U, Haas R, Gattermann N (2005) Results of a phase 2 study of valproic acid alone or in combination with all-trans retinoic acid in 75 patients with myelodysplastic syndrome and relapsed or refractory acute myeloid leukemia. Ann Hematol 84(Suppl 1):61–66
Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N (2006) The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia. Cancer 106:112–119
Lancet JE, Nichols G, Assouline S et al (2007) A phase I study of MGCD0103 given as a twice weekly oral dose in patients with advanced leukemias or myelodysplastic syndromes (MDS) [abstract]. J Clin Oncol (Meeting Abstracts) 25(Suppl 18):2516
Lane AA, Chabner BA (2009) Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 27:5459–5468
Lee DY, Hayes JJ, Pruss D, Wolffe AP (1993) A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72:73–84
Mai A, Altucci L (2009) Epi-drugs to fight cancer: from chemistry to cancer treatment, the road ahead. Int J Biochem Cell Biol 41:199–213
Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G (2005) Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem 48:3344–3353
Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R (2007) FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 12:1247–1252
Marks PA, Richon VM, Rifkind RA (1996) Induced differentiation of cancer cells: second generation potent hybrid polar compounds target cell cycle regulators. Eur J Cancer Prev 5(Suppl 2):75–77
Minucci S, Pelicci PG (1999) Retinoid receptors in health and disease: co-regulators and the chromatin connection. Semin Cell Dev Biol 10:215–225
Minucci S, Nervi C, Lo Coco F, Pelicci PG (2001) Histone deacetylases: a common molecular target for differentiation treatment of acute myeloid leukemias? Oncogene 20:3110–3115
Miyake K, Yoshizumi T, Imura S, Sugimoto K, Batmunkh E, Kanemura H, Morine Y, Shimada M (2008) Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas 36:e1–e9
Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M (2007) Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep 18:769–774
Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T (2004) Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 23:6261–6271
Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, Weisz A, de Lera AR, Gronemeyer H, Altucci L (2005) Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med 11:77–84
Nickel BE, Davie JR (1989) Structure of polyubiquitinated histone H2A. Biochemistry 28:964–968
Niesvizky R, Ely S, DiLiberto M, Cho HJ, Gelbsthein UY, Jayabalan DS (ASH 2005) Multicenter Phase II Trial of the histone deacetylase Depsipeptide (FK 228) in the treatment of relapsed or refractory multiple myeloma (MM). Blood (Suppl) 106:2574
Nowak SJ, Corces VG (2000) Phosphorylation of histone H3 correlates with transcriptionally active loci. Genes Dev 14:3003–3013
O'Connor OA, Heaney ML, Schwartz L, Richardson S, Willim R, MacGregor-Cortelli B, Curly T, Moskowitz C, Portlock C, Horwitz S, Zelenetz AD, Frankel S, Richon V, Marks P, Kelly WK (2006) Clinical experience with intravenous and oral formulations of the novel histone deacetylase inhibitor suberoylanilide hydroxamic acid in patients with advanced hematologic malignancies. J Clin Oncol 24:166–173
Odenike OM, Alkan S, Sher D, Godwin JE, Huo D, Brandt SJ, Green M, Xie J, Zhang Y, Vesole DH, Stiff P, Wright J, Larson RA, Stock W (2008) Histone deacetylase inhibitor romidepsin has differential activity in core binding factor acute myeloid leukemia. Clin Cancer Res 14:7095–7101
Olsen EA, Kim YH, Kuzel TM, Pacheco TR, Foss FM, Parker S, Frankel SR, Chen C, Ricker JL, Arduino JM, Duvic M (2007) Phase IIb multicenter trial of vorinostat in patients with persistent, progressive, or treatment refractory cutaneous T-cell lymphoma. J Clin Oncol 25:3109–3115
Ottmann OG, Spencer A, Prince HM et al (2008) Phase IA/II study of oral panobinostat (LBH589), a novel pan-deacetylase inhibitor (DACi) demonstrating efficacy in patients with advanced hematologic malignancies [abstract]. ASH Annual Meeting Abstracts 112:958
Ozdag H, Teschendorff AE, Ahmed AA, Hyland SJ, Blenkiron C, Bobrow L, Veerakumarasivam A, Burtt G, Subkhankulova T, Arends MJ, Collins VP, Bowtell D, Kouzarides T, Brenton JD, Caldas C (2006) Differential expression of selected histone modifier genes in human solid cancers. BMC Genomics 7:90
Park JH, Jong HS, Kim SG, Jung Y, Lee KW, Lee JH, Kim DK, Bang YJ, Kim TY (2008) Inhibitors of histone deacetylases induce tumor-selective cytotoxicity through modulating Aurora-A kinase. J Mol Med 86:117–128
Pham AD, Sauer F (2000) Ubiquitin-activating/conjugating activity of TAFII250, a mediator of activation of gene expression in Drosophila. Science 289:2357–2360
Piekarz RL, Robey R, Sandor V, Bakke S, Wilson WH, Dahmoush L, Kingma DM, Turner ML, Altemus R, Bates SE (2001) Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood 98:2865–2868
Piekarz RL, Robey RW, Zhan Z, Kayastha G, Sayah A, Abdeldaim AH, Torrico S, Bates SE (2004) T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood 103:4636–4643
Piekarz R, Wright J, Frye R et al (2008) Results of a phase 2 NCI multicenter study of romidepsin in patients with relapsed peripheral T-cell lymphoma (PTCL) [abstract]. ASH Annual Meeting Abstracts 112:1567
Pohlman B, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A: Final results of a phase I study of oral Belinostat in patients with refractory, relapsed CTCL PTCL Blood (ASH 2009 abstract 920)
Prince HM, Bishton MJ, Harrison SJ (2009) Clinical studies of histone deacetylase inhibitors. Clin Cancer Res 15(12):3958–3969, Epub 2009 Jun 9
Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG, Baylin SB (2006) Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet 2:e40
Raffoux E, Chaibi P, Dombret H, Degos L (2005) Valproic acid and all-trans retinoic acid for the treatment of elderly patients with acute myeloid leukemia. Haematologica 90:986–988
Rambaldi A, Dellacasa CM, Salmoiraghi S, Spinelli O, Ferrari ML, Gattoni E, Guglielmelli P, Vannucchi AM, Barosi G, Tiziano Barbui (2008) A Phase 2A study of the Histone-Deacetylase Inhibitor ITF2357 in Patients with Jak2V617F Positive Chronic Myeloproliferative Neoplasms Blood (ASH Annual Meeting Abstracts) 112:100
Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, Sun L, Ricker J, Rizvi S, Oerth C, Atkins B, Fearen I, Anderson K, Siegel D (2008) Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma 49:502–507
Richon VM, Russo P, Venta-Perez G, Cordon-Cardo C, Rifkind RA, Marks PA (1997) Two cytodifferentiation agent-induced pathways, differentiation and apoptosis, are distinguished by the expression of human papillomavirus 16 E7 in human bladder carcinoma cells. Cancer Res 57(13):2789–2798
Richon VM, Sandhoff TW, Rifkind RA, Marks PA (2000) Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA 97:10014–10019
Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE, Sackett DL (2005) Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle 4:717–726
Rogakou EP, Boon C, Redon C, Bonner WM (1999) Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 146:905–916
Rogakou EP, Nieves-Neira W, Boon C, Pommier Y, Bonner WM (2000) Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J Biol Chem 275:9390–9395
Rosato RR, Almenara JA, Dai Y, Grant S (2003) Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2:1273–1284
Rosenbluth JM, Mays DJ, Pino MF, Tang LJ, Pietenpol JA (2008) A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol 28:5951–5964
Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW (2001) The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci USA 98:10833–10838
Ryningen A, Stapnes C, Lassalle P, Corbascio M, Bruserud O (2009) A subset of patients with high-risk acute myelogenous leukemia shows improved peripheral blood cell counts when treated with the combination of valproic acid, theophylline and all-trans retinoic acid. Leuk Res 33(6):779–787
Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, Garcia-Manero G (2006) Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood 108:1174–1182
Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Kiriyama M, Fukai I, Yamakawa Y, Fujii Y (2004) Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 46:171–178
Sato T, Friend C, De Harven E (1971) Ultrastructural changes in Friend erythroleukemia cells treated with dimethyl sulfoxide. Cancer Res 31:1402–1417
Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA 100:13225–13230
Siegel Dd, Sezer O, San Miguel JF et al (2008) A phase IB, Multicenter, open-label, dose-escalation study of oral panobinostat (LBH589) and I.V. bortezomib in patients with relapsed multiple myeloma [abstract]. ASH Annual Meeting Abstracts 112:2781
Siitonen T, Timonen T, Juvonen E, Terävä V, Kutila A, Honkanen T, Mikkola M, Hallman H, Kauppila M, Nyländen P, Poikonen E, Rauhala A, Sinisalo M, Suominen M, Savolainen ER, Koistinen P (2007) Valproic acid combined with 13-cis retinoic acid and 1, 25-dihydroxyvitamin D3 in the treatment of patients with myelodysplastic syndromes. Haematologica 92(8):1119–1122
Stamatopoulos B, Meuleman N, De Bruyn C, Mineur P, Martiat P, Bron D, Lagneaux L (2009) Antileukemic activity of valproic acid in chronic lymphocytic leukemia B cells defined by microarray analysis. Leukemia 23:2281–2289
Stimson L, La Thangue NB (2009) Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett 280:177–183
Tsuji N, Kobayashi M, Nagashima K, Yoshiharu W, Kenzo K (1976) A new antifungal antibiotic, Trichostatin. J Antibiot 29(1):1–6
Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, Ngo L, Holmgren A, Jiang X, Marks PA (2005) Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA 102:673–678
Wang DF, Helquist P, Wiech NL, Wiest O (2005) Toward selective histone deacetylase inhibitor design: homology modeling, docking studies, and molecular dynamics simulations of human class I histone deacetylases. J Med Chem 48(22):6936–6947
Weichert W, Roske A, Gekeler V, Beckers T, Ebert MP, Pross M, Dietel M, Denkert C, Rocken C (2008) Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol 9:139–148
West MH, Bonner WM (1980) Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res 8:4671–4680
Wilkinson KD (2000) Ubiquitination and deubiquitination: targeting of proteins for degradation by the proteasome. Semin Cell Dev Biol 11:141–148
Wolf JL, Siegel D, Matous J et al (2008) A phase II study of oral panobinostat (LBH589) in adult patients with advanced refractory multiple myeloma [abstract]. ASH Annual Meeting Abstracts 112:2774
Xiong Y, Dowdy SC, Podratz KC, Jin F, Attewell JR, Eberhardt NL, Jiang SW (2005) Histone deacetylase inhibitors decrease DNA methyltransferase-3B messenger RNA stability and down-regulate de novo DNA methyltransferase activity in human endometrial cells. Cancer Res 65:2684–2689
Xu Z, Meng X, Cai Y, Koury MJ, Brandt SJ (2006) Recruitment of the SWI/SNF protein Brg1 by a multiprotein complex effects transcriptional repression in murine erythroid progenitors. Biochem J 399:297–304
Yoo CB, Jones PA (2006) Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov 5:37–50
Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S, Iwase H (2004) HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res 10:6962–6968
Zhang C, Richon V, Ni X, Talpur R, Duvic M (2005) Selective induction of apoptosis by histone deacetylase inhibitor SAHA in cutaneous T-cell lymphoma cells: relevance to mechanism of therapeutic action. J Invest Dermatol 125:1045–1052
Zhang T, Berrocal JG, Gamble MJ, DuMond ME, Krishnakumar R, Yang T, Sauve AA, Kraus WL (2009) Enzymes in the NAD+ salvage pathway regulate SIRT1 activity at target gene promoters. J Biol Chem 284(30):20408–20417
Acknowledgments
This work was supported by: Associazione Italiana per la ricerca contro il cancro (AIRC to LA) European Union HEALTH-F4-2007-200767 “Apo-Sys.” The authors are not in conflict of interest. FPT is an Apo-Sys post-doctoral fellow.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tambaro, F.P., Dell’Aversana, C., Carafa, V. et al. Histone deacetylase inhibitors: clinical implications for hematological malignancies. Clin Epigenet 1, 25–44 (2010). https://doi.org/10.1007/s13148-010-0006-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13148-010-0006-2