
Abstract
As the fourth most diagnosed cancer, cervical cancer (CC) is one of the major causes of cancer-related mortality affecting females globally, particularly when diagnosed at advanced stage. Discoveries of CC biomarkers pave the road to precision medicine for better patient outcomes. High throughput omics technologies, characterized by big data production further accelerate the process. To date, various CC biomarkers have been discovered through the advancement in technologies. Despite, very few have successfully translated into clinical practice due to the paucity of validation through large scale clinical studies. While vast amounts of data are generated by the omics technologies, challenges arise in identifying the clinically relevant data for translational research as analyses of single-level omics approaches rarely provide causal relations. Integrative multi-omics approaches across different levels of cellular function enable better comprehension of the fundamental biology of CC by highlighting the interrelationships of the involved biomolecules and their function, aiding in identification of novel integrated biomarker profile for precision medicine. Establishment of a worldwide Early Detection Research Network (EDRN) system helps accelerating the pace of biomarker translation. To fill the research gap, we review the recent research progress on CC biomarker development from the application of high throughput omics technologies with sections covering genomics, transcriptomics, proteomics, and metabolomics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Despite being highly preventable, cervical cancer (CC) is the fourth most common gynecological malignancy threatening women health and lives due to the insufficient screening protocols, particularly in low‑ and middle‑income countries [1,2,3]. According to the World Health Organization (WHO), it is estimated that in 2018, approximately 570,000 cases of CC were diagnosed and about 311, 000 females died from the disease [4]. Persisting infection with high-risk subtypes of the human papilloma virus (hrHPV) is the main cause of cervical carcinogenesis [5].
Asymptomatic and non-specific nature in the early stages of CC often lead to late-stage diagnosis [6]. Cytology-based screening, known as Papanicolaou test (Pap smear) and HPV testing are the most frequently used methods for CC screening in the clinical practice [7]. However, the current screening programs have some limitations such as causing patient discomfort, the invasive and sensitive nature of the tests, as well as low levels of sensitivity and specificity. Early detection of disease is extremely important due to the availability of various treatment options which make CC curable [8]. The treatment options available for CC are surgery, radiation, chemotherapy, or in a combination, which may cause various side effects and no cure[9, 10]. The poorer prognosis and ineffective treatment in the advance stage of CC necessitate the development of new prognostic, diagnostic, and therapeutic strategies [11, 12].
A cancer biomarker is a substance or process indicative of the presence of cancer, which can be secreted by a malignancy itself, or as a specific body response to the presence of cancer [13]. The discovery of biomarkers including genes, DNA, RNA, proteins, enzymes, antigens, and other cellular and biological products paves the road to precision medicine for better patient outcomes through the classification of patients by probable disease risk, treatment and prognosis [14]. Thus, identification of CC biomarkers is expected to provide greater direction in strategizing the prevention and treatment of CC [15]. Various biomarkers concerning carcinogenesis, precancerous lesions, and CC have been described in many articles and reviews [15]. For instance, the well-known markers P16 and Ki-67 have demonstrated promising results as surrogate biomarkers of cervical neoplasia [16,17,18]. A recent meta-analysis confirmed that p16 and p16/Ki-67 immunocytochemistry has higher specificity for cervical intraepithelial neoplasia of grade 2 or worse (CIN2+) or cervical intraepithelial neoplasia of grade 3 or worse (CIN3+) than the hrHPV DNA testing [16]. Similar sensitivity was reported for dual staining and the hrHPV DNA testing. The application of p16/Ki-67 dual-stained cytology for detection of cervical precancer and cancers in various settings may limit the burden of over-detection such as unnecessary health care costs and potential adverse events due to overtreatment [16].
Omics technologies focused at the universal detection of genes (genomics), mRNA (transcriptomics), proteins (proteomics) and metabolites (metabolomics) in a biosample have revolutionized medical research [19]. It is possible to gather vast amounts of data of a particular type of molecules in a single experiment through these high throughput technologies [20]. A remarkable growth in the assay technologies which includes single nucleotide polymorphisms (SNP) arrays, gene expression microarrays and protein arrays continue to identify various novel biomarkers aimed for precision medicine [21]. Multi-omics approaches integrating omics data across different levels of cellular function enables better understanding of the molecular and clinical features of the disease, contributing to enhanced ability to address applications including disease subtyping and biomarker prediction [22].
To date, researchers have highlighted numerous biomarkers offering new prospects for translational CC research, however the focus on the contributions of high throughput omics technologies towards the process of CC biomarker development has not been extensively discussed [23,24,25,26]. Hence, the present review aims to summarize the various biomarkers associated with diagnosis, treatment and prognosis of CC discovered in the past five years through omics technologies at the aspects of genomics, transcriptomics, proteomics and metabolomics for precision medicine.
2 Methodology
This article is a general descriptive review summarizing various CC biomarkers discovered through high throughput omics technologies with most data cited ranging January 2016 to August 2021 for the most recent published study. A search was performed using online databases including Google Scholar, PubMed and Science Direct using search words and strings, mainly “cervical cancer”, “biomarker”, “omics”, “genomics”, “transcriptomics”, “proteomics” and “metabolomics”. Selection of articles was summarized in Fig. 1. Studies (original, review, systematic, meta-analysis) covering the following types of data were included and extracted: application of high throughput omics technologies with sections covering genomics, transcriptomics, proteomics and metabolomics as well as biomarkers associated with diagnosis, treatment and prognosis of CC. Studies were excluded if written in other languages than English due to language barrier.

Schematic representation of selection of articles for the review
3 Significance of biomarkers for CC
Accurate and predictable early screening of CC is crucial [27]. Although Pap smear can easily detect squamous lesions, it cannot detect glandular lesions as such lesions are only visible in histological examination via biopsy. On the other hand, although hrHPV DNA testing has become an important tool, the tests are limited by low specificity and inability to predict the infection outcome. Biomarkers may be implemented in various steps within the disease flowchart. The identification of biomarkers for CC will help to diagnose the conditions at early stage of disease development and help to control the condition from progressing to severe stage [8]. Utilization of biomarkers may help in making timely clinical management decisions such as further testing, treatment, colposcopy referral, increased surveillance or release to routine screening [27]. Biomarkers can also be applied to estimate the prognosis of patients, to determine the treatment impact, and to monitor the treatment progression. Biomarkers play a role in the development of precision medicine as the treatments to individual or subgroups of patients can be adjusted based on specific biomarkers for optimal patient outcomes [28].
4 Results and discussion
4.1 CC biomarkers discovered through genomics
Genomic markers causing genetic alterations have roles in the carcinogenesis and progression of CC. Genome-wide association studies (GWAS) and next generation sequencing (NGS) are the omics technologies widely used to investigate the genetic risk factors and mutation profiles in tumors, including CC [20, 29].
Numerous studies reported on the effect of SNPs on CC susceptibility [30, 31]. Heritability may be used to quantify the proportion of CC predisposition attributable to host genetic factors and it was estimated that shared genes account for 27% of CC heritability [32]. Human leukocyte antigen (HLA) genes exhibit statistically significant associations at the locus 6p21.3 (HLA class I and II genes) and two loci outside HLA at 4q12 (EXOC1), and 17q12 (GSDMB) [32,33,34]. With the lead SNP rs59661306 and rs7457728, novel-significant associations were identified at 5q14 and 7p11 respectively. Functional studies using cervical HeLa cell lines suggested the role of ARRDC3 gene in cell growth and susceptibility to HPV infection [35]. Disruption in apoptotic and immune function pathways at PAX8 and CLPTM1L and interaction between TP53 and XRCC1 increases the genetic susceptibility to CC [36]. The difficulty in interpreting GWAS associations limits the translation of the findings into clinical care [37, 38]. There have been concerns that the whole genome will be implicated in the disease predisposition and that the variants and genes reflected in association signals show no direct biological linkage to the disease [38]. Most of the disease-associated loci lie in the non-coding regions of the genome with regulatory role, questions regarding the genes regulated and cell types or physiological contexts the regulation occurs arise [37].
The persistent infection with hrHPV causes viral integration into the host genome up to 76.3% of CC cases with positive correlation to CIN grades [39], which can be detected with NGS. HPV integration, the key genetic mechanism reported at least 83% of HPV-associated CC commonly occur at particular fragile sites [40], significantly upregulate the gene expression and it has been associated with poorer rate of survival compared to those with episomal form of HPV. Therefore, the HPV integration status may consider as a promising biomarker for diagnosis, risk stratification, therapy, prediction of treatment responses and treatment monitoring [39, 40]. Analysis of blood samples with NGS technologies demonstrated the potential use of RNF213 mutation as a biomarker to monitor the treatment response to chemotherapy and radiotherapy [41]. Although NGS allows the whole sequence of cancer’s exome or genome to be obtained, not all information provided contribute substantially to the determination of the clinical decisions for cancer patients, for which smaller targeted sequencing panels are often more clinically practical [42]. NGS is also limited by the need for extensive analytic capabilities which may be costly. Other limitations include difficulties in identifying the driver mutations and confounding factor of tumor heterogeneity [43]. Table 1 shows the summary of studies on CC biomarkers discovered through genomics.
4.2 CC biomarkers discovered through transcriptomics
Microarrays and RNA sequencing (RNA-Seq) employs high throughput sequencing to capture the sequences of the whole transcriptome are the two key techniques used for transcriptome study [45]. Compared with microarrays, the identification of more differentially modulated transcripts, splice variants, and non-coding transcripts with higher fold-change by RNA-Seq technology provides additional data that may be informative for clinical prediction, mechanistic investigations or biomarker discovery [46, 47].
The noncoding RNAs (ncRNAs) are known as oncogenic drivers and tumor suppressors in CC [48, 49]. Epigenetic modifications including deregulated expression of ncRNAs and circular RNAs (circRNAs) involve in the initiation and promotion stages of CIN and cervical carcinoma [49]. MicroRNAs (miRNAs), long noncoding RNAs (lncRNAs) and circRNAs have also been associated with CC metastasis through the regulation of related genes, epithelial-mesenchymal transition, signaling pathways and interactions with microenvironment of tumors [50].
Small, single stranded miRNAs are the master modulators of genome which regulate up to 60% of protein-coding genes and they are involved in processes such as cell cycle regulation, differentiation, programmed cell death, angiogenesis, DNA repair or stress response [51]. Altered miRNAs can roughly be classified as oncogenic and oncosuppressor miRNAs, and both have been correlated with biological processes in CC progression [52]. Expression miR-29a and miR-21 are reported as the most frequently down- and up-regulated miRNAs respectively in the progression of invasive CC [53]. However, there was a small overlap between the results of microarray-based studies, with miR-10a, miR-20b, miR-9, miR-16 and miR-106a was found to be upregulated, whereas miR-99a, miR-203, and miR-195 were reported to be down-regulated [53]. Differences in study designs, populations, arrays used, convenience material-based studies and small sample size may be the plausible explanations for the variations. Improved performance has been reported with the combined use of miRNA markers [54]. A combination of six upregulated oncogenic miRNAs (miR-20a, miR-92a, miR-141, miR-183*, miR-210 and miR-944) showed enhanced accuracy for diagnosis of CC compared with individual use of any marker with an excellent AUC of 0.959, sensitivity of 91.4%, and specificity of 87.6% [55]. Cervical adenocarcinoma has been reported to be associated with higher rate of metastasis and treatment resistance than squamous cell carcinoma. Through transcriptome analysis, study reported the improved diagnostic performance for cervical adenocarcinoma from the combination of miR-192-5p, HNF1A-AS1, and VIL1 with an AUC of 0.911, which could be promising diagnostic biomarkers for cervical adenocarcinoma [56].
As miRNAs, the crucial roles of lncRNAs in cell growth, survival, cell cycle, differentiation and apoptosis have been demonstrated and their roles as molecular regulatory factors in CC may provide opportunities for early diagnosis and therapeutic targets to improve clinical outcomes [57, 58]. lncRNA microarray analysis revealed the oncogenic lncRNA‑AK001903 which promotes tumor progression in CC [59]. Transcriptomic and lncRNA-mRNA correlation analysis showed PCBP1-AS1 as a novel prognostic biomarker for CC. The elevated expression of PCBP1-AS1 is associated with tumor stage, TNM and invasion [60]. A recent study integrating the data of DNA methylation, copy number variation (CNV) and transcriptome to identify CNV-related lncRNAs for CC prognosis prediction have developed a 8-lncRNA (RUSC1-AS1, LINC01990, LINC01411, LINC02099, H19, LINC00452, ADPGK-AS1, C1QTNF1-AS1) signature with high AUC independent of clinical features, providing novel prognostic biomarkers for CC [61].
The differential expression of circRNAs in CC cells compared with normal cells suggests their potential roles and biological relevance in CC. CDR1as is one of the most well-identified circRNAs which sponges miRNA‐7, a tumor suppressor that has been associated with CC [62]. In vitro studies to investigate the roles of circRNAs in cervical carcinogenesis and progression reported upregulated circRNAs such as has_circ_0018289 (miR-497 sponge), has_circ_0018289 (miRNA-497 sponge), has_circ_0023404 (miRNA-136 sponge), has_circ_0000263 (miRNA-150-5p sponge), circRNA‐000284 (miRNA-506 sponge), has_circRNA_101996 (miRNA‐8075 sponge), circ‐ATP8A2 (miRNA‐433 sponge), circ_0067934 (miRNA‐545 sponge), circEIF4G2 (miRNA‐218 sponge) and circRNA8924, while the has_circ_0001445 (miRNA‐620 sponge) has been found to be downregulated [63]. The expression abundance, stability and specificity conferred by circRNAs make them as potential biomarker for cancers but further studies required as studies of circRNAs in CC, particularly their mechanisms of action are still at the nascent stage [63, 64].
Combined differential expression and differential co-expression analysis revealed, epidermis development-associated gene set around ZNF135 act as putative biomarker for the prevention and treatment of CC [65]. More recently, five out of the seven co-expressed gene modules identified by differential co-expression network analysis were reported to exhibit high capabilities for diagnosis and prognosis [11]. These gene modules were associated with biological processes including regulation of cell cycle, keratinization, degranulation of neutrophils as well as phospholipase D signaling pathway. AR, E2F4, ESR1, ETS1, FOXP3, GATA1, GATA2, GATA3, PRDM14, and YBX1 were the transcription factors regulating the module genes and ETS1 and GATA2 were found as the common regulatory elements in most modules. The incorporation of differential co-expression analyses in the search of molecular basis of complex diseases recommended to achieve systems-level understanding of the variation in disease phenotype in CC [11].
All low [e.g., in situ hybridization (ISH), subtractive hybridization (SH), Northern Blot (NB), ribonuclease protection assay (RPA), reverse transcription-polymerase chain reaction (RT-PCR)], medium- [e.g., expressed sequence tags (EST), Open Reading frame ESTs (ORESTES)] and high throughput [e.g. microarrays, serial analysis of gene expression (SAGE) and massively parallel signature sequencing (MPSS)] techniques have their pros and cons, with high throughput methods are characterized by big data production whereas low throughput methods offer higher specificity, sensitivity, and reproducibility [66, 67]. With that, there is a need for the high- and medium-performance techniques to be validated by low-performance techniques [66]. Combination of miRNA signatures with other different markers may help to improve risk stratification. Table 2 shows the summary of studies on CC biomarkers discovered through transcriptomics.
4.3 CC biomarkers discovered through proteomics
Protein microarrays, mass spectrometry (MS) and nuclear magnetic resonance (NMR) spectroscopy are some of high throughput techniques used in proteomics to determine protein expression levels which could not be achieved by conventional techniques such as one-dimensional SDS–polyacrylamide gel electrophoresis (1D SDS-PAGE) gels, Western Blot or enzyme-linked immunosorbent assay (ELISA) [69,70,71].
Membrane proteomics of one normal cervical (HCK1T) and there cervical cell lines, C33A (HPV-negative), SiHa (HPV16+), HeLa (HPV18+) have revealed the differentially expressed membrane proteins which are involved in cancer-associated biological pathways such as HIPPO, PI3K/Akt s and EIF2 signaling as well as cell cycle G2/M DNA damage checkpoint regulation which may be putative markers for diagnosis, prognosis and treatment [72]. Intracellular proteomics of the four cell revealed the upregulation of cofilin-1 [73]. Inhibition of matrix metalloproteases in cancer cell lines was found via secretome analysis of the cell lines, and this was further validated by zymography for MMP-2 and MMP-9, western blot analysis for ADAM10, CATD, FUCA1 and SΟD2, and multiple reaction monitoring (MRM) for CATD, CATB, SOD2, QPCT and NEU1 [74]. The biochemical similarities and differences among the four representative and informative cell lines reflect the aberrant pathways involved in cervical carcinogenesis, providing valuable information for the identification of biomarkers of cervical pathology [73].
Various protein markers have been identified through the proteomic analysis using biological samples including serum, cervical mucus, cervicovaginal fluid (CVF) and urine [8, 75,76,77,78,79,80,81]. Non-invasive measurement of tumor biomarkers in serum such as carcinoembryonic antigen (CEA), squamous cell carcinoma antigen (SCC-Ag) and carbohydrate antigen 19-9 (CA19-9) have been frequently employed in CC detection and monitoring but their specificity for CC detection and sensitivity for early stage detection are of unsatisfactory levels [8]. The significantly elevated levels of serum SCC-Ag, highly sensitive C-reactive protein (hs-CRP), and CA-125 in recurrence cervical patients indicates that these proteins could be potential biomarkers for the prediction of recurrence risk [75]. Vascular endothelial growth factor (VEGF) is the main mediator of angiogenesis which stimulates the formation of new blood vessels, contributing to tumorigenesis and cancer progression. It has been reported to be overexpressed in 63.07% of patients with cervical carcinoma compared to controls and it is associated with poor prognosis [76]. A recent meta-analysis concluded elevated expressions of VEGF and VEGF-C were significantly associated with poor survival outcome in patients with CC [77]. Angiopoietins also play important roles in angiogenesis. Serum angiopoietin 2 (sAng-2) and the ratio of sAng-1/sAng-2 reported as potential diagnostic and prognostic biomarkers in CC [78]. A non-targeted proteomic analysis of cervical mucus profiled the differently expressed proteins in cervical adenocarcinoma, including heme protein myeloperoxidase and apolipoprotein A–I (APOA1), which play roles in immune response and lipid metabolism respectively [79]. Self-sample collection of cervical tissue using brushes, tampons, swabs or lavages for subsequent DNA genotyping, cytology or immunohistochemistry is a good method to be considered for screening purpose. CVF which can simply be collected in a non-invasive manner offers new opportunities for the development of self-tests. Functional classification of CVF proteome using proteomics technologies shows various biological roles, particularly protein metabolism and modification as well as immunity and defense [80]. Alpha-actinin-4 (ACTN4) is one of the proteins in CVF found to be a promising biomarker for the development of a simple assay for self-screening of cervical (pre)cancer [80]. Urinary samples is another source of biomarkers that can be easily and non-invasively obtained. Study with urines reported a significant upregulation of leucine rich α 2 glycoprotein (LRG1) and isoform 1 of multimerin 1 (MMRN1), and downregulation of S100 calcium-binding protein A8 (S100A8), SERPINB3 and cluster of differentiation-44 antigen (CD44) in CC. Through the receiver operator characteristic curve (ROC) analysis, the combination of these proteins or individual use of LRG1 and SERPINB3 may be detection biomarkers for CC [81].
The high throughput technologies used in proteomics studies are still relatively old and the limitations in protein quantification, data collection, sensitivity and reproducibility restrict the discovery of clinically significant novel biomarkers [82]. Significant differences in type of biomarkers identified and concentration reported exist across the results reported, even with the use of same biological samples. Integration of information generated from proteomics and validation of proteins that have been identified as potential biomarkers may accelerate the development of individualized patient care through clinical proteomics [83]. Table 3 shows the summary of studies on CC biomarkers discovered through proteomics.
4.4 CC biomarkers discovered through metabolomics
Various studies have been conducted comparing the metabolomics profiles of blood, urine, cervicovaginal lavage and tissue samples in identifying diagnostic, predictive or prognostic biomarkers [84,85,86,87,88,89]. From the plasma metabolomics conducted using ultra-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry (UPLC-QTOF-MS) combined with multivariate statistical analysis, five differential metabolites including bilirubin, LysoPC(17:0), n-oleoyl threonine, 12-hydroxydodecanoic acid and tetracosahexaenoic acid were identified as the candidate biomarkers for CC with the area under curve (AUC) of 0.99 [84]. Phosphatidyl choline (15:0/16:0), phosphatidyl glycerol (12:0/13:0), actosylceramide (d18:1/16:0), D-Maltose, and phthalic acid with an AUC greater than 0.75, were pinpointed as potential prognostic biomarkers for cervical squamous cell carcinoma (SCC) by Zhou et al. through plasma metabolomics [85]. Another plasma metabolomics for diagnostic algorithm by Khan et al. [86] reported seven metabolites (adenosine monophosphate, aspartate, glutamate, hypoxanthine, lactate, proline, and pyroglutamate) which distinguished patients with CINs and CC from the healthy controls (AUC = 0.82 and 0.83 respectively). Metabolomics analysis of the urine samples using GC–MS to discriminate the HPV categories between patients revealed the closer metabolome of HPV + B (HPV positive with concomitant low and high-risk infections) with HPV − (HPV negative) than to HPV + H (HPV positive exclusively high-risk), suggesting the antagonism of HPV co-infections resulting from viral interference. Three urinary metabolites 5-oxoprolinate, erythronic acid (AUC = 0.92) and N-acetylaspartic acid (AUC = 0.91) identified differentiate those with HPV + H from the negative controls [87]. Metabolic analysis of cervicovaginal lavage revealed membrane lipids (3-hydroxybutyrate, eicosenoate, and oleate/vaccenate with excellent discrimination capacity AUC > 0.9) discriminated the invasive cervical carcinoma patients with the healthy controls and membrane lipids including sphingolipids, plasmalogens, and linoleate were positively correlated with genital inflammation. Non-Lactobacillus dominant communities resulted in perturbed metabolisms of amino acid and nucleotide, especially in high-grade dysplasia, connecting vaginal dysbiosis to cervical dysplasia, hence cervicovaginal metabolome may be a potential target for clinical interventions [88]. Tissue-based metabolomics to identify diagnostic biomarkers for HPV-associated cervical carcinoma showed decreased levels of α- and β-glucose, elevated levels of lactate and low-density lipoproteins as well as altered amino acid expression in HPV16-positive SCC or its precursor lesions compared with HPV-negative negative controls. The significantly upregulated expression of glycogen synthase kinase 3 beta (GSK3β) and glutamate decarboxylase 1 (GAD1) and decreased for pyruvate kinase muscle isozyme 2 (PKM2) and carnitine palmitoyltransferase 1A (CPT1A) in cervical lesions imply that increased aerobic glycolysis and disrupted lipid metabolism may confer advantages for tumor growth [89].
Although metabolomics has shown high potential in hypothesis generation and biomarker discovery, numerous challenges have to be addressed for the advancement of this relatively new omics field [90]. Difficulty in replicating the metabolomic biomarkers across various studies may be attributed to sample sources, population heterogeneity, experimental protocols, data parameter setting biological variations in metabolite turnover rates, thus limiting the application of novel cancer biomarkers in clinical settings [84]. Integrating metabolomics with other omics data may help to achieve improved translational outcomes [91]. Table 4 shows the summary of studies on CC biomarkers discovered through metabolomics.
4.5 Integrative multi-omics
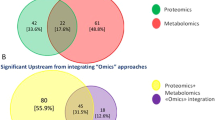
Integrative multi-omics approach (Fig. 2) involving the integration of gene expression profiles with genome-scale biomolecular networks on the CC transcriptomic datasets have revealed the reporter biomolecules at the levels of RNA, protein and metabolite. The potential biomarkers identified by the integrative multi-omics analysis were shown in Fig. 3. Other than the known biomarkers including BRCA1, ESR1, PCNA, FGFR2, CD86, EGFR, P2RX4, ETS1 and E2F4, novel biomolecules including receptors (EPHA4, EPHA5, EPHB2, EDNRA, EDNRB, NCOA3, NR2C1, and NR2C2), miRNAs (miR-192-5p, miR-193b-3p, and miR-215-5p), transcription factors (especially E2F4, ETS1, and CUTL1), other proteins (KAT2B, PARP1, CDK1, GSK3B, WNK1, and CRYAB), and metabolites (particularly arachidonic acids) have been identified as potential biomarkers for the purpose of screening or treatment of CC [12]. Six immune-related genes (chemokine receptor 7 (CCR7), CD3d molecule (CD3D), CD3e molecule (CD3E), and integrin subunit beta 2 (ITGB2), family with sequence similarity 133 member A (FAM133A), and tumor protein p53 (TP53)) identified as prognostic model to forecast the survival and response to immunotherapy to indicate immune status based on multi-omics data analyses [92]. Cervicovaginal microbiome plays a role in hrHPV susceptibility and clearance, and imbalanced cervicovaginal microbiome increases the risk of developing CC [93, 94]. Multi-omics combination of cervical microbiota data with urine metabolomics allows enhanced understanding of community functions in the disease and interactions with host by investigating the association between the host microbiome and circulating metabolites. Other than monitoring compositional changes of bacteria through urine metabolomics, identification of bacteria contributing to the circulating metabolites is also possible through functional characterization of cervicovaginal microbiota and urinary metabolome which may guide the development of diagnostic tools for self-testing [94].

Exploration of cervical cancer biomarkers using omics techniques

Potential biomarkers identified by the integrative multi-omics analysis
The limited resolving-power for the establishment of casual relationship between molecular signatures and the phenotypic manifestation of cancer hallmarks represents the limitation of single-level omics approaches [95]. On the contrary, investigation of cancer cells or tissues in multiple dimensions by multi-OMICS approaches which investigate cancer may potentially reveal the complicated molecular mechanisms underlying various phenotypes of cancer hallmark, analyze cellular response to treatment as well as contribute to the discovery of clinically relevant biomarkers. Conducting several omics may help to address the challenges arising from the individual use of omics approaches. Integration of omics data is vital for the interpretation of data but challenges arise as it involves computational and/or integration of data or concurrent analysis of multiple variables on multiple datasets [96]. Table 5 shows the summary of studies on integrative multi-omics approaches for CC biomarkers.
While biomarkers appear to be potential promising approach to decrease the CC disease burden, they may be too expensive to be applied as viable public health strategy [97]. Despite, in the cost-effectiveness study conducted by Termrungruanglert et al. [97], screening using HPV genotyping test combined with biomarker p16/Ki-67 dual stain cytology as the triage of HPV+ Thai patients aged 30–65 years old is expected to be more cost-effective (average quality-adjusted life years (QALYs) = 24.03, annual cost = $13,262,693) than the Pap cytology (average QALY = 23.98, annual cost = $7,713,251). The improved diagnostic accuracy for CIN2+ of HPV screening with p16/Ki-67 dual stain triage algorithm has enabled higher number of women with precancerous detected and treated in the earlier stages and resulted in lower prevalence and mortality rate [98]. However, the cost of screening, treatment and follow up might be increased due to increased number of patients who return at next screening. The much higher screening costs of the new algorithm had the greatest impact on the total cost [97]. Another study by Juan et al. [99] reported that co-testing (Pap plus HPV mRNA testing including genotyping for HPV 16/18) had greater effectiveness (lifetime QALYs per women screened = 23.01) compared with HPV primary (lifetime QALYs per women screened = 22.99) and lower total costs ($2326 for co-testing v s $2365 for HPV primary) despite the higher screening costs for co-testing.
This study has some limitations such as only online databases were used and there was limited access for some of the published articles. Reviewer and evidence selection bias may occur during screening of studies for the inclusion in this review, and bias may also arise in the primary studies included.
5 Conclusions
CC remains a global health issue which require more effective preventive and control strategies [100]. The limitations of current screening and diagnostic strategies for CC prompt the development of novel biomarkers to improve the clinical outcomes of CC patients [20]. In order to benefit the patients, the basic research achievements have to be applied to the clinics. Translational research is used to fill the gap between results of basic research in which biomarkers are discovered and their incorporation into clinical practice [101]. Relatively slow pace of cancer biomarkers being moved into clinical application, which could be attributed to the need of high-performance characteristics for a biomarker to be clinically useful, biology of tumors, inadequacy of the discovery design as well as cumbersome and costly validation process [13]. Regulatory requirements and the lack of reward for translational research also result in the biomarker research to remain stagnant at the discovery phase.
Large scale data provided by high throughput omics technologies has boosted the ability to identify molecular markers of disease processes. Improved patient care can be achieved with co-evolvement of high throughput analyses and biomarker-based precision medicine [20]. Despite, growing gap exists between the big data production and capacity to integrate, process and interpret data. The main challenge faced is to identify which data within the huge data obtained is of clinical relevance, which can be overcome by integrative multi-omics approaches [67]. Collaboration, data sharing, data integration and standards are essential in translating biomarker discovery into clinical use. A global Early Detection Research Network (EDRN) system should be formed to accelerate the pace of biomarker translation. For instance, the US National Cancer Institute (NCI)’s EDRN has been established with four main components, namely: (1) Biomarker Developmental Laboratories (BDLs) for the discovery, development and characterization of new biomarkers or refinement of existing biomarkers, (2) Biomarkers Reference Laboratories (BRLs) for analytical and clinical validation, (3) Clinical Validation Centers (CVCs) which carry out and support biomarker validation trials, and (4) Data Management and Coordinating Center (DMCC) that coordinates network, provides data management and protocol development supporting validation trials as well as conducts related theoretical and applied statistical researches [13]. EDRN aims to foster collaboration between investigators of various expertise and to encourage the rapid movement into clinical validation for successful translational research.
Data availability
Not applicable.
Code availability
Not applicable.
Abbreviations
- 1D SDS-PAGE:
-
One-dimensional SDS–polyacrylamide gel electrophoresis
- ACTN4:
-
Alpha-actinin-4
- AIS:
-
Adenocarcinoma in situ
- AMP:
-
Adenosine monophosphate
- APOA1:
-
Apolipoprotein A-I
- BDLs:
-
Biomarker Developmental Laboratories
- BRLs:
-
Biomarkers Reference Laboratories
- CA19-9:
-
Carbohydrate antigen 19–9
- CC:
-
Cervical cancer
- CCR7:
-
Chemokine receptor 7
- CD44:
-
Cluster of differentiation-44 antigen
- CEA:
-
Carcino-embryonic antigen
- CIN:
-
Cervical intraepithelial neoplasia
- circRNAs:
-
Circular RNAs
- CSCC:
-
Cervical squamous cell carcinoma
- CVCs:
-
Clinical Validation Centers
- CVF:
-
Cervicovaginal fluid
- DMCC:
-
Data Management and Coordinating Center
- EA:
-
Endocervical adenocarcinoma
- EDRN:
-
Early Detection Research Network
- ELISA:
-
Enzyme-linked immunosorbent assay
- EST:
-
Expressed sequence tags
- EVs:
-
Extracellular vesicles
- FAM133A:
-
Family with sequence similarity 133 member
- GC–MS:
-
Gas chromatography–mass spectrometry
- GO/KEGG:
-
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes
- GSEA:
-
Gene set enrichment analysis
- GSVA:
-
Gene set variation analysis
- GWAS:
-
Genome-wide association studies
- HLA:
-
Human leukocyte antigen
- HPV + H:
-
HPV positive exclusively high-risk
- HR-MAS NMR:
-
High-resolution magic angle spinning nuclear magnetic resonance
- HSIL:
-
High-grade squamous intraepithelial lesions
- ICC:
-
Invasive cervical cancer
- IGRPM:
-
Immune gene-related prognostic model
- IHC:
-
Immunohistochemistry
- ISH:
-
In situ hybridization
- ITGB2:
-
Integrin subunit beta 2
- LC–MS:
-
Liquid chromatography–mass spectrometry
- LC–MS/MS:
-
Liquid chromatography–tandem mass spectrometry
- LDL:
-
Low-density lipoprotein
- lncRNAs:
-
Long noncoding RNAs
- LSIL:
-
Low-grade squamous intraepithelial lesions
- MAMA-PCR:
-
Mutation analysis of mismatch amplification PCR
- miRNAs:
-
MicroRNAs
- MMRN1:
-
Multimerin 1
- MPSS:
-
Massively parallel signature sequencing
- MRM:
-
Multiple reaction monitoring
- MS:
-
Mass spectrometry
- NA:
-
Not available
- NB:
-
Northern Blot
- NCI:
-
National Cancer Institute
- ncRNAs:
-
Non-coding RNAs
- NGS:
-
Next generation sequencing
- NMR:
-
Nuclear magnetic resonance
- OR:
-
Odds ratio
- ORESTES:
-
Open reading frame ESTs
- PCR:
-
Polymerase chain reaction
- PPI:
-
Protein–protein interaction
- qPCR:
-
Quantitative polymerase chain reaction
- RNA-Seq:
-
RNA sequencing
- ROC:
-
Receiver operator characteristic curve
- RPA:
-
Ribonuclease protection assay
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- S100A8:
-
S100 calcium-binding protein A8
- SAGE:
-
Serial analysis of gene expression
- sAng-2:
-
Serum angiopoietin 2
- sAng:
-
Serum angiopoietin
- SCC-Ag:
-
Squamous cell carcinoma antigen
- SCC:
-
Squamous cell carcinoma
- SH:
-
Subtractive hybridization
- SNPs:
-
Single nucleotide polymorphisms
- TCT:
-
Thinprep cytologic test
- TP53:
-
Tumor protein p53
- QALYs:
-
Quality-adjusted life years
- UPLC-MS:
-
Ultra-performance liquid chromatography-mass spectrometry
- UPLC-QTOF-MS:
-
Ultra-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry
- WGCNA:
-
Weighted gene co-expression network analysis
References
Buskwofie A, David-West G, Clare CA. A review of cervical cancer: incidence and disparities. J Natl Med Assoc. 2020;112:229–32. https://doi.org/10.1016/j.jnma.2020.03.002.
Koh WJ, Abu-Rustum NR, Bean S, Bradley K, Campos SM, Cho KR, Chon HS, Chu C, Clark R, Cohn D, Crispens MA, Damast S, Dorigo O, Eifel PJ, Fisher CM, Frederick P, Gaffney DK, Han E, Huh WK, Lurain JR, Mariani A, Mutch D, Nagel C, Nekhlyudov L, Fader AN, Remmenga SW, Reynolds RK, Tillmanns T, Ueda S, Wyse E, Yashar CM, McMillian NR, Scavone JL. Cervical cancer, version 3.2019. JNCCN J Natl Compr Cancer Netw. 2019;17:64–84. https://doi.org/10.6004/jnccn.2019.0001.
Hull R, Mbele M, Makhafola T, Hicks C, Wang SM, Reis RM, Mehrotra R, Mkhize-Kwitshana Z, Kibiki G, Bates DO, Dlamini Z. Cervical cancer in low and middle income countrie s (review). Oncol Lett. 2020;20:2058–74. https://doi.org/10.3892/ol.2020.11754.
Mera SL. Cervical cancer. Med Lab Sci. 1991;48:155–60.
Li X, Hu SY, He Y, Hernandez Donoso L, Qu KQ, Van Kriekinge G, Zhao FH. Systematic literature review of risk factors for cervical cancer in the Chinese population. Women’s Health. 2018. https://doi.org/10.1177/1745506518816599.
Tekalign T, Teshome M. Prevalence and determinants of late-stage presentation among cervical cancer patients, a systematic review and meta-analysis. PLoS ONE. 2022;17: e0267571. https://doi.org/10.1371/JOURNAL.PONE.0267571.
Keeratichamroen S, Subhasitanont P, Chokchaichamnankit D, Weeraphan C, Saharat K, Sritana N, Kantathavorn N, Wiriyaukaradecha K, Sricharunrat T, Monique Paricharttanakul N, Auewarakul C, Svasti J, Srisomsap C. Identification of potential cervical cancer serum biomarkers in Thai patients. Oncol Lett. 2020;19:3815–26. https://doi.org/10.3892/ol.2020.11519.
Du S, Zhao Y, Lv C, Wei M, Gao Z, Meng X. Applying serum proteins and microRNA as novel biomarkers for early-stage cervical cancer detection. Sci Rep. 2020;10:1–8. https://doi.org/10.1038/s41598-020-65850-z.
Šarenac T, Mikov M. Cervical cancer, different treatments and importance of bile acids as therapeutic agents in this disease. Front Pharmacol. 2019. https://doi.org/10.3389/fphar.2019.00484.
Liu L, Wang M, Li X, Yin S, Wang B. An overview of novel agents for cervical cancer treatment by inducing apoptosis: emerging drugs ongoing clinical trials and preclinical studies. Front Med. 2021. https://doi.org/10.3389/fmed.2021.682366.
Kori M, Gov E, Arga KY. Novel genomic biomarker candidates for cervical cancer as identified by differential co-expression network analysis. Omi A J Integr Biol. 2019;23:261–73. https://doi.org/10.1089/omi.2019.0025.
Kori M, Arga KY. Potential biomarkers and therapeutic targets in cervical cancer: Insights from the meta-analysis of transcriptomics data within network biomedicine perspective. PLoS ONE. 2018;13: e0200717. https://doi.org/10.1371/journal.pone.0200717.
Wagner PD, Srivastava S. New paradigms in translational science research in cancer biomarkers. Transl Res. 2012. https://doi.org/10.1016/j.trsl.2012.01.015.
Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016. https://doi.org/10.1038/nrc.2016.56.
Volkova LV, Ilexander PA, Nadezhda ON. Cervical carcinoma: oncobiology and biomarkers. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms222212571.
Peeters E, Wentzensen N, Bergeron C, Arbyn M. Meta-analysis of the accuracy of p16 or p16/Ki-67 immunocytochemistry versus HPV testing for the detection of CIN2+/CIN3+ in triage of women with minor abnormal cytology. Cancer Cytopathol. 2019;127:169–80. https://doi.org/10.1002/cncy.22103.
Yu LL, Chen W, Lei XQ, Qin Y, Wu ZN, Pan QJ, Zhang X, Chang BF, Zhang SK, Guo HQ, Qiao YL. Evaluation of p16/Ki-67 dual staining in detection of cervical precancer and cancers: a multicenter study in China. Oncotarget. 2016;7:21181–9. https://doi.org/10.18632/oncotarget.8307.
Diouf D, Diop G, Fall C, Sarr S, Tidiane Diarra CA, Ngom AI, Ka S, Lo S, Faye O, Dem A. The association of molecular biomarkers in the diagnosis of cervical pre-cancer and cancer and risk factors in Senegalese. Asian Pacific J Cancer Prev. 2020;21:3221–7. https://doi.org/10.31557/APJCP.2020.21.11.3221.
Richard A, Louise PH. SAC review ‘Omic’ technologies: proteomics and metabolomics. Obstet Gynaecol. 2011;13:189–95.
Quezada H, Guzmán-Ortiz AL, Díaz-Sánchez H, Valle-Rios R, Aguirre-Hernández J. Omics-based biomarkers: current status and potential use in the clinic. Boletín Médico Del Hosp Infant México. 2017;74:219–26. https://doi.org/10.1016/j.bmhime.2017.11.030.
Matsui S. Genomic biomarkers for personalized medicine: development and validation in clinical studies. Comput Math Methods Med. 2013. https://doi.org/10.1155/2013/865980.
Subramanian I, Verma S, Kumar S, Jere A, Anamika K. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights. 2020. https://doi.org/10.1177/1177932219899051.
Yim E-K, Park J-S. Biomarkers in cervical cancer. Biomark Insights. 2006;1:215.
Sahasrabuddhe VV, Luhn P, Wentzensen N. Human papillomavirus and cervical cancer: biomarkers for improved prevention efforts. Future Microbiol. 2011. https://doi.org/10.2217/fmb.11.87.
Iida M, Banno K, Yanokura M, Nakamura K, Adachi M, Nogami Y, Umene K, Masuda K, Kisu I, Iwata T, Tanaka K, Aoki D. Candidate biomarkers for cervical cancer treatment: potential for clinical practice (review). Mol Clin Oncol. 2014;2:647–55. https://doi.org/10.3892/mco.2014.324.
Onyango CG, Ogonda L, Guyah B, Shiluli C, Ganda G, Orang’o OE, Patel K. Novel biomarkers with promising benefits for diagnosis of cervical neoplasia: a systematic review. Infect Agents Cancer. 2020. https://doi.org/10.1186/s13027-020-00335-2.
Molina MA, Carosi Diatricch L, Castany Quintana M, Melchers WJ, Andralojc KM. Cervical cancer risk profiling: molecular biomarkers predicting the outcome of hrHPV infection. Expert Rev Mol Diagn. 2020. https://doi.org/10.1080/14737159.2020.1835472.
Vargas AJ, Harris CC. Biomarker development in the precision medicine era: lung cancer as a case study. Nat Rev Cancer. 2016. https://doi.org/10.1038/nrc.2016.56.
Yang YC, Chang TY, Chen TC, Lin WS, Lin CL, Lee YJ. Replication of results from a cervical cancer genome-wide association study in Taiwanese women. Sci Rep. 2018;8:1–5. https://doi.org/10.1038/s41598-018-33430-x.
Das Ghosh D, Mukhopadhyay I, Bhattacharya A, Roy Chowdhury R, Mandal NR, Roy S, Sengupta S. Impact of genetic variations and transcriptional alterations of HLA class I genes on cervical cancer pathogenesis. Int J Cancer. 2017;140:2498–508. https://doi.org/10.1002/ijc.30681.
Alsbeih GA, Al-Harbi NM, Bin Judia SS, Khoja HA, Shoukri MM, Tulbah AM. Reduced rate of human papillomavirus infection and genetic overtransmission of TP53 72C polymorphic variant lower cervical cancer incidence. Cancer. 2017;123:2459–66. https://doi.org/10.1002/cncr.30635.
Leo PJ, Madeleine MM, Wang S, Schwartz SM, Newell F, Pettersson-Kymmer U, Hemminki K, Hallmans G, Tiews S, Steinberg W, Rader JS, Castro F, Safaeian M, Franco EL, Coutlée F, Ohlsson C, Cortes A, Marshall M, Mukhopadhyay P, Cremin K, Johnson LG, Garland S, Tabrizi SN, Wentzensen N, Sitas F, Little J, Cruickshank M, Frazer IH, Hildesheim A, Brown MA. Defining the genetic susceptibility to cervical neoplasia—a genome-wide association study. PLoS Genet. 2017;13: e1006866. https://doi.org/10.1371/journal.pgen.1006866.
Chen D, Juko-Pecirep I, Hammer J, Ivansson E, Enroth S, Gustavsson I, Feuk L, Magnusson PKE, McKay JD, Wilander E, Gyllensten U. Genome-wide association study of susceptibility loci for cervical cancer. J Natl Cancer Inst. 2013;105:624–33. https://doi.org/10.1093/jnci/djt051.
Shi Y, Li L, Hu Z, Li S, Wang S, Liu J, Wu C, He L, Zhou J, Li Z, Hu T, Chen Y, Jia Y, Wang S, Wu L, Cheng X, Yang Z, Yang R, Li X, Huang K, Zhang Q, Zhou H, Tang F, Chen Z, Shen J, Jiang J, Ding H, Xing H, Zhang S, Qu P, Song X, Lin Z, Deng D, Xi L, Lv W, Han X, Tao G, Yan L, Han Z, Li Z, Miao X, Pan S, Shen Y, Wang H, Liu D, Gong E, Li Z, Zhou L, Luan X, Wang C, Song Q, Wu S, Xu H, Shen J, Qiang F, Ma G, Liu L, Chen X, Liu J, Wu J, Shen Y, Wen Y, Chu M, Yu J, Hu X, Fan Y, He H, Jiang Y, Lei Z, Liu C, Chen J, Zhang Y, Yi C, Chen S, Li W, Wang D, Wang Z, Di W, Shen K, Lin D, Shen H, Feng Y, Xie X, Ma D. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nat Genet. 2013;45:918–22. https://doi.org/10.1038/ng.2687.
Takeuchi F, Kukimoto I, Li Z, Li S, Li N, Hu Z, Takahashi A, Inoue S, Yokoi S, Chen J, Hang D, Kuroda M, Matsuda F, Mizuno M, Mori S, Wu P, Tanaka N, Matsuo K, Kamatani Y, Kubo M, Ma D, Shi Y. Genome-wide association study of cervical cancer suggests a role for ARRDC3 gene in human papillomavirus infection. Hum Mol Genet. 2019;28:341–8. https://doi.org/10.1093/hmg/ddy390.
Bowden SJ, Bodinier B, Kalliala I, Zuber V, Vuckovic D, Doulgeraki T, Whitaker MD, Wielscher M, Cartwright R, Tsilidis KK, Bennett P, Jarvelin MR, Flanagan JM, Chadeau-Hyam M, Kyrgiou M. Genetic variation in cervical preinvasive and invasive disease: a genome-wide association study. Lancet Oncol. 2021;22:548–57. https://doi.org/10.1016/S1470-2045(21)00028-0.
Cano-Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet. 2020. https://doi.org/10.3389/fgene.2020.00424.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019. https://doi.org/10.1038/s41576-019-0127-1.
Hu Z, Zhu D, Wang W, Li W, Jia W, Zeng X, Ding W, Yu L, Wang X, Wang L, Shen H, Zhang C, Liu H, Liu X, Zhao Y, Fang X, Li S, Chen W, Tang T, Fu A, Wang Z, Chen G, Gao Q, Li S, Xi L, Wang C, Liao S, Ma X, Wu P, Li K, Wang S, Zhou J, Wang J, Xu X, Wang H, Ma D. Genome-wide profiling of HPV integration in cervical cancer identifies clustered genomic hot spots and a potential microhomology-mediated integration mechanism. Nat Genet. 2015;47:158–63. https://doi.org/10.1038/ng.3178.
Mühr LSA, Guerendiain D, Cuschieri K, Sundström K. Human papillomavirus detection by whole-genome next-generation sequencing: importance of validation and quality assurance procedures. Viruses. 2021;13:1323. https://doi.org/10.3390/v13071323.
Lee SY, Chae DK, Lee SH, Lim Y, An J, Chae CH, Kim BC, Bhak J, Bolser D, Cho DH. Efficient mutation screening for cervical cancers from circulating tumor DNA in blood. BMC Cancer. 2020;20:1–10. https://doi.org/10.1186/s12885-020-07161-0.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, Temple-Smolkin RL, Voelkerding KV, Nikiforova MN. Guidelines for validation of next-generation sequencing-based oncology panels: a joint consensus recommendation of the association for molecular pathology and college of american pathologists. J Mol Diagnostics. 2017. https://doi.org/10.1016/j.jmoldx.2017.01.011.
Basho RK, Eterovic AK, FundaMeric-Bernstam. Clinical applications and limitations of next-generation sequencing. Am J Hematol Oncol. 2015;11:17–22.
Liu GC, Zhou YF, Su XC, Zhang J. Interaction between TP53 and XRCC1 increases susceptibility to cervical cancer development: a case control study. BMC Cancer. 2019;19:1–9. https://doi.org/10.1186/s12885-018-5149-0.
Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T. Transcriptomics technologies. PLoS Comput Biol. 2017;13: e1005457. https://doi.org/10.1371/journal.pcbi.1005457.
Rao MS, Van Vleet TR, Ciurlionis R, Buck WR, Mittelstadt SW, Blomme EAG, Liguori MJ. Comparison of RNA-Seq and microarray gene expression platforms for the toxicogenomic evaluation of liver from short-term rat toxicity studies. Front Genet. 2019. https://doi.org/10.3389/fgene.2018.00636.
Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS ONE. 2014;9: e78644. https://doi.org/10.1371/journal.pone.0078644.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2017. https://doi.org/10.1038/nrc.2017.99.
Tornesello ML, Faraonio R, Buonaguro L, Annunziata C, Starita N, Cerasuolo A, Pezzuto F, Tornesello AL, Buonaguro FM. The role of microRNAs, long non-coding RNAs, and circular RNAs in cervical cancer. Front Oncol. 2020. https://doi.org/10.3389/fonc.2020.00150.
Cheng T, Huang S. Roles of non-coding RNAs in cervical cancer metastasis. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.646192.
Balacescu O, Balacescu L, Baldasici O, Tudoran O, Achimas-Cadariu P. The role of miRNAs in diagnosis, prognosis and treatment prediction in cervical cancer. Colposc Cerv Pathol. 2017. https://doi.org/10.5772/68011.
Wang J-y, Chen L-j. The role of miRNAs in the invasion and metastasis of cervical cancer. 2019. Biosci Rep. https://doi.org/10.1042/BSR20181377.
Pardini B, De Maria D, Francavilla A, Di Gaetano C, Ronco G, Naccarati A. MicroRNAs as markers of progression in cervical cancer: a systematic review. BMC Cancer. 2018;18:1–17. https://doi.org/10.1186/s12885-018-4590-4.
Jang JY, Kim YS, Kang KN, Kim KH, Park YJ, Kim CW. Multiple microRNAs as biomarkers for early breast cancer diagnosis. Mol Clin Oncol. 2021;14:1–9. https://doi.org/10.3892/mco.2020.2193.
Liu SS, Chan KKL, Chu DKH, Wei TN, Lau LSK, Ngu SF, Chu MMY, Tse KY, Ip PPC, Ng EKO, Cheung ANY, Ngan HYS. Oncogenic microRNA signature for early diagnosis of cervical intraepithelial neoplasia and cancer. Mol Oncol. 2018;12:2009–22. https://doi.org/10.1002/1878-0261.12383.
Xu J, Zou J, Wu L, Lu W. Transcriptome analysis uncovers the diagnostic value of miR-192-5p/HNF1A-AS1/VIL1 panel in cervical adenocarcinoma. Sci Rep. 2020;10:1–12. https://doi.org/10.1038/s41598-020-73523-0.
Dong JX, Su M, Chang W, Zhang K, Wu S, Xu T. Long non-coding RNAs on the stage of cervical cancer (review). Oncol Rep. 2017. https://doi.org/10.3892/or.2017.5905.
He J, Huang B, Zhang K, Liu M, Xu T. Long non-coding RNA in cervical cancer: from biology to therapeutic opportunity. Biomed Pharmacother. 2020. https://doi.org/10.1016/j.biopha.2020.110209.
Zhong G, Fang X, Xie Q, Wang Y, Lin Z, Lin R, Yao T. Long non-coding RNA AK001903 regulates tumor progression in cervical cancer. Oncol Lett. 2021;21:1–1. https://doi.org/10.3892/OL.2020.12338.
Li L, Peng Q, Gong M, Ling L, Xu Y, Liu Q. Using lncRNA sequencing to reveal a putative lncRNA-mRNA correlation network and the potential role of PCBP1-AS1 in the pathogenesis of cervical cancer. Front Oncol. 2021;11:769. https://doi.org/10.3389/fonc.2021.634732.
Zhong Q, Lu M, Yuan W, Cui Y, Ouyang H, Fan Y, Wang Z, Wu C, Qiao J, Hang J. Eight-lncRNA signature of cervical cancer were identified by integrating DNA methylation, copy number variation and transcriptome data. J Transl Med. 2021;19:1–16. https://doi.org/10.1186/s12967-021-02705-9.
Jiang C, Zeng X, Shan R, Wen W, Li J, Tan J, Li L, Wan R. The emerging picture of the roles of circRNA-CDR1as in cancer. Front Cell Dev Biol. 2020. https://doi.org/10.3389/fcell.2020.590478.
Chaichian S, Shafabakhsh R, Mirhashemi SM, Moazzami B, Asemi Z. Circular RNAs: a novel biomarker for cervical cancer. J Cell Physiol. 2020;235:718–24. https://doi.org/10.1002/jcp.29009.
Liu J, Zhu H, Fu L, Xu T. Investigating the underlying mechanisms of circular rnas and their application in clinical research of cervical cancer. Front Genet. 2021. https://doi.org/10.3389/fgene.2021.653051.
Fang SQ, Gao M, Xiong SL, Chen HY, Hu SS, Cai HB. Combining differential expression and differential coexpression analysis identifies optimal gene and gene set in cervical cancer. J Cancer Res Ther. 2018;14:201–7. https://doi.org/10.4103/0973-1482.199787.
Acevedo-Rocha CG, Munguía-Moreno JA, Ocádiz-Delgado R, Gariglio P. A transcriptome- and marker-based systemic analysis of cervical cancer. In: Rajamanickam R, editor. Topics on cervical cancer with an advocacy for prevention. London: InTech; 2012. https://doi.org/10.5772/30866.
D’Argenio V. The high-throughput analyses era: are we ready for the data struggle? High-Throughput. 2018. https://doi.org/10.3390/ht7010008.
Zhou Y, Shen L, Wang YZ, Zhou CC. The potential of CiRS-7 for predicting onset and prognosis of cervical cancer. Neoplasma. 2020;67:312–22. https://doi.org/10.4149/neo_2019_190415N334.
Neagu M, Bostan M, Constantin C. Protein microarray technology: assisting personalized medicine in oncology (review). World Acad Sci J. 2019. https://doi.org/10.3892/wasj.2019.15.
Aslam B, Basit M, Nisar MA, Khurshid M, Rasool MH. Proteomics: technologies and their applications. J Chromatogr Sci. 2017. https://doi.org/10.1093/chromsci/bmw167.
Martínez-Rodríguez F, Limones-González JE, Mendoza-Almanza B, Esparza-Ibarra EL, Gallegos-Flores PI, Ayala-Luján JL, Godina-González S, Salinas E, Mendoza-Almanza G. Understanding cervical cancer through proteomics. Cells. 2021;10:1854. https://doi.org/10.3390/cells10081854.
Pappa KI, Christou P, Xholi A, Mermelekas G, Kontostathi G, Lygirou V, Makridakis M, Zoidakis J, Anagnou NP. Membrane proteomics of cervical cancer cell lines reveal insights on the process of cervical carcinogenesis. Int J Oncol. 2018;53:2111–22. https://doi.org/10.3892/ijo.2018.4518.
Pappá KI, Lygirou V, Kontostathi G, Zoidakis J, Makridakis M, Vougas K, Daskalakis G, Polyzos A, Anagnou NP. Proteomic analysis of normal and cancer cervical cell lines reveals deregulation of cytoskeleton-associated proteins. Cancer Genom Proteom. 2017;14:253–66. https://doi.org/10.21873/cgp.20036.
Pappa KI, Kontostathi G, Makridakis M, Lygirou V, Zoidakis J, Daskalakis G, Anagnou NP. High resolution proteomic analysis of the cervical cancer cell lines secretome documents deregulation of multiple proteases. Cancer Genom Proteom. 2017. https://doi.org/10.21873/cgp.20060.
Guo S, Yang B, Liu H, Li Y, Li S, Ma L, Liu J, Guo W. Serum expression level of squamous cell carcinoma antigen, highly sensitive C-reactive protein, and CA-125 as potential biomarkers for recurrence of cervical cancer. J Cancer Res Ther. 2017;13:689–92. https://doi.org/10.4103/jcrt.JCRT_414_17.
Rahmani AH, Babiker AY, Alsahli MA, Almatroodi SA, Husain NEOS. Prognostic significance of vascular endothelial growth factor (VEGF) and Her-2 protein in the genesis of cervical carcinoma. Open Access Maced J Med Sci. 2018;6:263–8. https://doi.org/10.3889/OAMJMS.2018.089.
Zhang J, Liu J, Zhu C, He J, Chen J, Liang Y, Yang F, Wu X, Ma X. Prognostic role of vascular endothelial growth factor in cervical cancer: a meta-analysis. Oncotarget. 2017;8:24797–803. https://doi.org/10.18632/oncotarget.15044.
Yang P, Chen N, Yang D, Crane J, Yang S, Wang H, Dong R, Yi X, Xie L, Jing G, Cai J, Wang Z. The ratio of serum angiopoietin-1 to angiopoietin-2 in patients with cervical cancer is a valuable diagnostic and prognostic biomarker. PeerJ. 2017. https://doi.org/10.7717/peerj.3387.
Ma Z, Chen J, Luan T, Chu C, Wu W, Zhu Y, Gu Y. Proteomic analysis of human cervical adenocarcinoma mucus to identify potential protein biomarkers. PeerJ. 2020. https://doi.org/10.7717/peerj.9527.
Van Ostade X, Dom M, Tjalma W, Van Raemdonck G. Candidate biomarkers in the cervical vaginal fluid for the (self-)diagnosis of cervical precancer. Arch Gynecol Obstet. 2018. https://doi.org/10.1007/s00404-017-4587-2.
Chokchaichamnankit D, Watcharatanyatip K, Subhasitanont P, Weeraphan C, Keeratichamroen S, Sritana N, Kantathavorn N, Ayudthaya PDN, Saharat K, Chantaraamporn J, Verathamjamras C, Phoolcharoen N, Wiriyaukaradecha K, Paricharttanakul NM, Udomchaiprasertkul W, Sricharunrat T, Auewarakul C, Svasti J, Srisomsap C. Urinary biomarkers for the diagnosis of cervical cancer by quantitative label-free mass spectrometry analysis. Oncol Lett. 2019;17:5453–68. https://doi.org/10.3892/ol.2019.10227.
Zhang SQ, Pan SM, Liang SX, Han YS, Chen HB, Li JC. Research status and prospects of biomarkers for nasopharyngeal carcinoma in the era of high-throughput omics (review). Int J Oncol. 2021;58:1–12. https://doi.org/10.3892/ijo.2021.5188.
Mesri M. Advances in proteomic technologies and its contribution to the field of cancer. Adv Med. 2014;2014:1–25. https://doi.org/10.1155/2014/238045.
Yang K, Xia B, Wang W, Cheng J, Yin M, Xie H, Li J, Ma L, Yang C, Li A, Fan X, Dhillon HS, Hou Y, Lou G, Li K. A comprehensive analysis of metabolomics and transcriptomics in cervical cancer. Sci Rep. 2017. https://doi.org/10.1038/srep43353.
Zhou H, Li Q, Wang T, Liang H, Wang YY, Duan Y, Song M, Wang YY, Jin H. Prognostic biomarkers of cervical squamous cell carcinoma identified via plasma metabolomics. Med. 2019. https://doi.org/10.1097/MD.0000000000016192.
Khan I, Nam M, Kwon M, Seo SS, Jung S, Han JS, Hwang GS, Kim MK. Lc/ms-based polar metabolite profiling identified unique biomarker signatures for cervical cancer and cervical intraepithelial neoplasia using global and targeted metabolomics. Cancers. 2019;11:511. https://doi.org/10.3390/cancers11040511.
Godoy-Vitorino F, Ortiz-Morales G, Romaguera J, Sanchez MM, Martinez-Ferrer M, Chorna N. Discriminating high-risk cervical human papilloma virus infections with urinary biomarkers via non-targeted GC-MS-based metabolomics. PLoS ONE. 2018;13: e0209936. https://doi.org/10.1371/journal.pone.0209936.
Ilhan ZE, Łaniewski P, Thomas N, Roe DJ, Chase DM, Herbst-Kralovetz MM. Deciphering the complex interplay between microbiota, HPV, inflammation and cancer through cervicovaginal metabolic profiling. EBioMedicine. 2019;44:675–90. https://doi.org/10.1016/j.ebiom.2019.04.028.
Abudula A, Rouzi N, Xu L, Yang Y, Hasimu A. Tissue-based metabolomics reveals potential biomarkers for cervical carcinoma and HPV infection. Bosn J Basic Med Sci. 2020;20:78–87. https://doi.org/10.17305/bjbms.2019.4359.
Pinu FR, Goldansaz SA, Jaine J. Translational metabolomics: current challenges and future opportunities. Metabolites. 2019. https://doi.org/10.3390/metabo9060108.
Pinu FR, Beale DJ, Paten AM, Kouremenos K, Swarup S, Schirra HJ, Wishart D. Systems biology and multi-omics integration: viewpoints from the metabolomics research community. Metabolites. 2019. https://doi.org/10.3390/metabo9040076.
Xu F, Shen J, Xu S. Multi-omics data analyses construct a six immune-related genes prognostic model for cervical cancer in tumor microenvironment. Front Genet. 2021;12:734. https://doi.org/10.3389/fgene.2021.663617.
Dai W, Du H, Li S, Wu R. Cervicovaginal microbiome factors in clearance of human papillomavirus infection. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.722639.
Chorna N, Godoy-Vitorino F. A protocol for the multi-omic integration of cervical microbiota and urine metabolomics to understand human papillomavirus (HPV)-driven dysbiosis. Biomedicines. 2020;8:81. https://doi.org/10.3390/biomedicines8040081.
Chakraborty S, Hosen MI, Ahmed M, Shekhar HU. Onco-multi-OMICS approach: a new frontier in cancer research. Biomed Res Int. 2018. https://doi.org/10.1155/2018/9836256.
Karahalil B. Overview of systems biology and omics technologies. Curr Med Chem. 2016;23:4221–30. https://doi.org/10.2174/0929867323666160926150617.
Termrungruanglert W, Khemapech N, Tantitamit T, Havanond P. Cost effectiveness analysis of HPV primary screening and dual stain cytology triage compared with cervical cytology. J Gynecol Oncol. 2019. https://doi.org/10.3802/jgo.2019.30.e17.
Wentzensen N, Fetterman B, Castle PE, Schiffman M, Wood SN, Stiemerling E, Tokugawa D, Bodelon C, Poitras N, Lorey T, Kinney W. p16/Ki-67 dual stain cytology for detection of cervical precancer in HPV-positive women. J Natl Cancer Inst. 2015;107:djv257. https://doi.org/10.1093/jnci/djv257.
Felix JC, Lacey MJ, Miller JD, Lenhart GM, Spitzer M, Kulkarni R. The clinical and economic benefits of co-testing versus primary HPV testing for cervical cancer screening: a modeling analysis. J Womens Health. 2016;25:606–16. https://doi.org/10.1089/jwh.2015.5708.
Zhang X, Zeng Q, Cai W, Ruan W. Trends of cervical cancer at global, regional, and national level: data from the Global Burden of Disease study 2019. BMC Public Health. 2021;21:1–10. https://doi.org/10.1186/s12889-021-10907-5.
Guo H, Zhou X, Lu Y, Xie L, Chen Q, Keller ET, Liu Q, Zhou Q, Zhang J. Translational progress on tumor biomarkers. Thorac Cancer. 2015;6:665–71. https://doi.org/10.1111/1759-7714.12294.
Acknowledgements
We would like to thank the Director General of Health Malaysia for his permission to publish this article.
Funding
This work was supported by UCSI University Research Excellence & Innovation Grant (REIG) with project code REIG-FPS-2022/05.
Author information
Authors and Affiliations
Contributions
Conceptualization: LFT & MS; literature search and data analysis: LFT & MS; writing & editing the original draft preparation: LFT & MS; Review and editing: JR, MAR, MAB, MR funding acquisition: MAR; supervision: MR. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arip, M., Tan, L., Jayaraj, R. et al. Exploration of biomarkers for the diagnosis, treatment and prognosis of cervical cancer: a review. Discov Onc 13, 91 (2022). https://doi.org/10.1007/s12672-022-00551-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-022-00551-9