
Abstract
Kinases are the ideal druggable targets for diseases and especially were highlighted on cancer therapy. Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase and its aberrant signaling extensively implicates in the progression of most cancer types, involving in cancer cell growth, adhesion, migration, and tumor microenvironment (TME) remodeling. FAK is commonly overexpressed and activated in a variety of cancers and plays as a targetable kinase in cancer therapy. FAK inhibitors already exhibited promising performance in preclinical and early-stage clinical trials. Moreover, substantial evidence has implied that targeting FAK is more effective in combination strategy, thereby reversing the failure of chemotherapies or targeted therapies in solid tumors. In the current review, we summarized the drug development progress, chemotherapy strategy, and perspective view for FAK inhibitors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Focal adhesion kinase (FAK), encoded by the PTK2 gene (protein tyrosine kinase 2), is a non-receptor tyrosine kinase. It is well known that FAK is phosphorylated and activated by integrins or growth factors, which transduce extracellular signals into cells to response the dynamic changes in microenvironment [1, 2]. Extracellular matrix can support the tumorigenicity and the disease progression. Specifically, Integrin acts as the anchor of cells to its adjacent matrix components, and Integrin/FAK as a signaling bridge to connect the tumor microenvironment and the cancer cells, which helps the deteriorated cells to acclimate the cancer-associated contexts. FAK is ubiquitously overexpressed in a series of cancer types, consisting of breast, oral, colon, gastric, and ovarian cancers as well as hepatocellular carcinoma [3,4,5]. FAK exerts its functions through the phosphorylation on the corresponding downstream target proteins in the cytoplasm, thereby enhancing tumor cell adhesion and promoting tumor growth, cancer-stemness, invasion, and metastasis ability [6, 7], promoting cancer cell epithelial to mesenchymal transition (EMT), tumor angiogenesis, chemotherapeutic resistance, and fibrosis in the stroma. Besides, regulation of target protein function through FAK scaffolding activity also contributes to cancer progression [8]. Furthermore, FAK kinase quick response to cancer drug treatment predicate FAK kinase exerts a positive effect in cancers [8]. Therefore, inhibition of FAK kinase activity may effectively suppress tumor proliferation, metastasis, and chemo-resistance, and is expected to serve as a qualified strategy for cancers treatment. In this review, we briefly summarized the structure and functions of FAK and mainly introduced the research progress of FAK inhibitors regarding cancer treatment in the past decades.
2 FAK structural features and the function in cancer
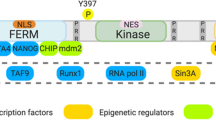
FAK is a 125 kDa protein consisting of three linearly arranged structural domains, including a trilobular structure N-terminus FERM structural domain located at the N-terminus. The FAT domain is located at the C-terminus. The catalytic kinase structural domain located between the FERM domain and FAT domain. Both terminus domains are separated from the kinase domain by linker regions that contain proline-rich regions (PRRs). (Fig. 1). It has been demonstrated that FAK contains multiple functional phosphorylation sites (Tyr397, Tyr407, Tyr576, Tyr577, Tyr861, Tyr925), of which Tyr397 is one of the most important phosphorylation site [9] and interacts directly with the Src family. Tyr576 and 577 are in the activation loop of the kinase domain, which are the main sites can be phosphorylated by Src family [9]. Tyr925 is phosphorylated and binds to the junction protein Grb2 to make FAK aggregate with integrins, while Tyr397 and Tyr 861 both could recruit other SH2 proteins [10].
2.1 N-terminus FERM domain
There are multiple protein binding sites for signal transduction proteins, cytoskeleton proteins, and integrin β subunits at the N-terminus. The main functional domain of this structural domain is FERM (four-point-one, ezrin, radixin, moesin) domain, which contains three closely related subdomains (F1, F2, F3), forming a cloverleaf shape [11]. Among them, the F1 and F2 subdomains interact with p53 to block apoptosis in tumor cells. The F2 subdomain regulates the kinase-independent activity and mediates cell survival. The F3 subdomain can arrest Mdm-2 to enhance ubiquitination of p53, which alleviates p53 independent cell apoptosis [11, 12]. In addition, the key tyrosine residue, Tyr397 is located at the N-terminus end of the FERM structural domain. Tyr397 autophosphorylation generates a high-affinity binding site for SH2 rich protein recruitment and subsequent activation of the FAK downstream pathway [13].
2.2 Central kinase domain
The central kinase domain has an activation loop that contains two important Tyr sites at576 and 577 which can be phosphorylated by Src and stimulate FAK kinase activity in turn [14]. The classic mechanism of FAK activation involves integrin receptor clustering upon the binding of cells to extracellular matrix (ECM) proteins, which leads to FAK autophosphorylation at Tyr397. FAK autophosphorylation at Tyr397 recruit Src-family kinases to phosphorylate FAK kinase activation loop atTyr576 and Tyr577, and finally formed a fully active FAK-SRC complex. Therefore, Tyr397 is one important phosphorylation located between FERM and the kinase region. Its autophosphorylation modulates the activity of FAK and consequently affects the biological functions and cell behaviors. Active FAK can phosphorylate various proteins such as the Src family, phospholipase C7, SHC adaptor protein, and growth factor connector protein 7 etc. [15].
2.3 C-terminus bomain
The C-terminus structural domain comprises two proline-rich regions (PRR2 and PRR3) and the Focal adhesion targeting (FAT) domain. Like the N-terminus FERM structural domain, the C-terminus region is also involved in various protein interactions. The C-terminus FAT is a functional domain for FAK adhesion to adhesive patches. The FAT domain contains binding sites for adhesion-associated proteins (such as paxillin and talin) that bind directly to integrins in the cytoplasmic region, thereby mediating the formation of the adhesion complex. In contrast, PRR2 and PRR3 at the C-terminus provide direct binding sites for proteins containing SH3 structures. Tyr861 and Tyr925 can be phosphorylated to form binding sites for proteins containing the SH2 structural domain. Thus, the C-terminus structural domain is also involved in regulating endogenous FAK function [14, 15].
2.4 The role of FAK in the cancer cell and tumor microenvironment remodeling
FAK regulation of cancer progression through “self-activation” and microenvironment remodeling for tumor seed preservation and niche cultivation (Fig. 2). Specifically, FAK activates FAK/PI3K, FAK/MAPK, FAK/p53, and other pathways related to cell growth, survival, and apoptosis [20]. Recent studies have demonstrated the role of FAK in promoting TME remodeling. In tumor-associated endothelial cells, FAK expression and phosphorylation levels of Tyr397 were elevated [21]. It was noted that stimulatory changes in EC migration are an essential component of angiogenesis, and that FAK activation downstream of growth factor, integrin, and cytokine receptors contributes to EC motility [22, 23]. Huang et al. showed that ECs acquire transformation into a mesenchymal stem cell (MSC)-like cells in glioblastoma (GBM), thus driving tumor resistance to cytotoxic therapy [24]. Furthermore, Jean et al. pointed out that FAK inhibition reduced tumor angiogenesis in the animal model of human ovarian cancer, indicating the positive role of FAK in angiogenesis [25].

structure of FAK protein and activation of FAK. 1.1 The protein structure of FAK which contains three major domains and three PPR small domains between the three major domains. 1.2 The activation of FAK. a Integrin binding to the relevant ligand on the extracellular matrix leads to Tyr397 autophosphorylation of FAK and FAK activation [16]. b Phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] binds to FERM mediated by integrins, the Tyr397 phosphorylation site is exposed and autophosphorylated [17]. Tyr397 phosphorylation recruits Src, which further phosphorylates Tyr576 and Tyr577 to release kinase domain from PERM domain, to make FAK reaches a fully activated state. c The receptor tyrosine kinases (RTKs) can directly activate the phosphorylation activation loop in FAK kinase region, thereby upregulating FAK kinase activity [18]. d Elevation pH reduces the stability of the FERM/kinase region interaction, resulting in phosphorylation of Y397 [19]. Activated FAK arrested p53 or convene Mdm-2 to enhance ubiquitination of p53 to block apoptosis in tumor cells. Talin and Paxillin bind to integrins in cytoplasmic regions, which can mediate the formation of adhesion complexes [1].

Multiple role of FAK in maintaining cancer malignancy. FAK kinase activation not only triggers ovarian cancer malignancy but also resulted in the recruitment of tumor-related cells including cancer-associated fibroblast, immune cells, endothelial cells as well as extracellular matrix remolding. FAK multiple roles in tumor progression evolution promise FAK inhibitor potential in mitigating tumor overgrowth, chemotherapy resistance, and immune escape
FAK, on the other hand, allows tumor cells to compromise with host immune cells in TME and evade the immune surveillance by recruiting immunosuppressive cells or secreting cytokines, cell immunity modulation [26]. Walsh et al. reported that the use of FAK inhibitors reduced leukocytes and macrophages infiltration and reduced tumor growth in a mammary carcinoma mouse model [27, 28]. Stokes et al. verified that pharmacological inhibition of FAK reduced TAMs within the tumor and reduced the size of the primary tumor in a pancreatic ductal adenocarcinoma mouse model [29].
Moreover, upon FAK activation, the ECM plays an important role in tumor progression by providing tumor cells with sustained proliferative signals, forming desmoplastic stroma, and evading growth inhibitory factors [30]. Finally, similar to TAMs, FAK inhibition in a bleomycin-induced fibrosis mouse model shows marked abrogation of lung fibrosis [31]. Overall, there is growing evidence for FAK, as a regulator of ECs, macrophage, and fibroblast signaling in TME, promotes the remodeling tumor microenvironment.
3 The development of FAK inhibitors
As reviewed above that FAK plays a vital role in many facets of tumors, and a consensus was widely reached in the science community that FAK is a promising target for the development of anti-cancer drugs. Many small-molecule FAK inhibitors have been developed and some even put forward to clinical trials ongoing or done. In this section, we summarized the progress in the development of FAK inhibitors.
3.1 ATP-competitive FAK inhibitors
Over the past decades, multiple preclinical and clinical-stage FAK inhibitors have been assessed for their effects in treating cancer diseases [9, 15, 32, 33]. Table 1 summarized five oral ATP-competitive FAK inhibitors that have been evaluated in clinical trials.
3.1.1 TAE226
TAE226, also known as NVP-226, inhibits FAK activity by blocking the linkage between FAK phosphorylation sites of Tyr397 and Tyr861 and ATP binding pocket. TAE226 has shown anti-tumor effects in preclinical in vivo and in vitro assays in non-small cell lung cancer (NSCLC) [34], Ewing’s Sarcoma (EWS) [35], Ph+Acute lymphoblastic leukemia (Ph+ALL) [36], oral squamous cell carcinoma (OSCC) [37], colorectal carcinoma [38] and pancreatic ductal adenocarcinoma (PDAC) [39]. Nevertheless, TAE226 had not been approved for clinical trials due to serious side effects on glucose metabolism [40].
3.1.2 VS-6062
VS-6062, also known as PF-562271 or PF00562271, is an orally administered biological agent that is a potent dual ATP-competitive inhibitor of FAK and FAK2. It blocks the phosphorylation of FAK Tyr397 and inhibits FAK overexpression in a dose-dependent manner, resulting in antitumor effects in rhabdomyosarcoma (RMS) [41], epithelial ovarian cancer (EOC) [42, 43] and PDAC [29]. Phase I clinical trials (NCT00666926) of VS-6062 have been completed as the first specific FAK inhibitor in clinical trials (head and neck cancer, prostate cancer and pancreatic cancer). This clinical trial has confirmed that VS-6062 has low toxicity and potent tumor-suppressive effects, toxicities included headache, nausea, vomiting, dehydration and edema [44].
3.1.3 PF-573228
PF-573228, also known as PF-228, which is current in preclinical studies, effectively blocks the phosphorylation of FAK Tyr397. PF-573228 can not only inhibit FAK signaling but also arrest tumor growth and invasion in bladder cancer [55], hemangioma [56], small cell lung cancer(SCLC) [57] and Neuroblastoma [58]. Meanwhile, tumor suppression and prolonged survival were also observed in animal models [55].
3.1.4 VS-6063
VS-6063, also known as PF-04554878 and defactinib, is a second-generation FAK inhibitor by suppressing of FAK Tyr397 phosphorylation. In detail, defactinib is now studied in nineteen clinical trials, five of which completed (NCT00787033, NCT01943292, NCT02913716, NCT01951690, NCT01778803), eight in recruiting status and two actives, but not yet recruiting. Ten are phases I studies, one of them in healthy patients, others eight are phase II studies. The phase I clinical trials in non-hematologic malignancies had been completed [46], which demonstrated a favorable and safe profile in these patients even including advanced solid tumors. The phase II clinical trials for Defactinib (VS-6063) have been completed in patients with KRAS mutant non-small cell lung cancer (NSCLC) and the drug was generally well tolerated and suitable for long-term dosing [59]. The results of these clinical trials suggest that the most common adverse events were nausea, vomiting, unconjugated hyperbilirubinemia, fatigue, headache, diarrhea [46, 47, 50, 60, 61].
Moreover, one study (NCT01951690) [61] on NSCLC patients harboring KRAS mutation was discontinued due the death of 76% of patients, in addition another study failed phase II multicenter clinical trial (NCT01870609) targeting malignant pleural mesothelioma stem cells [62]. There are still eight Phase I or II clinical trials under recruitment to evaluate the antineoplastic potential in ovarian cancer, non-small cell lung cancer and melanoma [63].
3.1.5 GSK2256098
GSK2256098 is an inhibitor targeting the FAK Tyr397 site In recent years, preclinical studies on GSK2256098 have found that it inhibits cell proliferation, migration and invasion of renal cell carcinoma [64], uterine cancer [65] and pancreatic ductal adenocarcinoma [66], and leads to lower tumor weight and less metastasis in mouse models. GSK2256098 is now studied in five clinical trials, three of which completed (NCT01938443, NCT00996671, NCT01138033). Preliminary results show that GSK2256098 was well tolerated with mild nausea, diarrhea, vomiting, decreased appetite and asthenia. A Phase II clinical trial (NCT02428270) in combination with trametinib for advanced pancreatic ductal carcinoma is in recruitment [67], The study is designed to evaluate the antitumor activity of GSK2256098 and Trametinib in patients with advanced pancreatic cancer, and commenced in April, 2016 and is expected to complete in December, 2022.
3.1.6 VS-4718
VS-4718, a potent reversible inhibitor of FAK, also known as PND-1186. An initial preclinical study by Buggio et al. indicated that VS-4718 monotherapy reduced the proliferation and increased the apoptosis of MM cells [68]. Jiang et al. reported that the single-dose FAK inhibition of VS-4718 greatly obstacles the tumor progression, increases two-folds survival rate in a human LSL-Kras (G12D) KPC mouse model [69]. Nevertheless, regarding the fact that all three of its clinical trials were terminated or withdrawn, the company did not announce the reasons [53].
3.1.7 IN10018
IN10018, also known as BI-853520, is a potent FAK inhibitor that inhibits the catalytic activity of FAK. IN10018 has been verified to inhibit Tyr397 phosphorylation in a series of human cancers and suppress tumor growth and progression in multiple mouse models of different cancer types [70]. In addition, IN10018 showed a manageable safety profile from a phase I study in Japanese and Taiwanese patients with solid tumors [54]. Six patients (29%) achieved a complete response (CR), suggesting that IN10018 exhibits good antitumor activity. IN10018 will soon enter into phase I clinical trials, including IN10018 in combination with cobimetinib for metastatic melanoma, with conventional chemotherapy for advanced plasma cancer, and with Docetaxel for gastric cancer [71, 72].
3.2 Specific FAK inhibitors
Specific FAK inhibitors that bind to distinct kinase domain sites and do not directly compete with ATP binding are being developed in recent years [73, 74]. These FAK inhibitors (C4, Y11, Y15, and R2) have the potential for high FAK specificity, they show anti-tumor activity in cells and xenograft mouse models. Moreover, they have been reported can enhance the anti-tumor activity of other chemotherapeutics but have not been rigorously tested in clinical trials.
3.2.1 Y11
Y11 is a small molecule inhibitor of FAK designed by computer modeling combined with a functional assay approach, which directly bound to the N-terminus domain of FAK to preventTyr397 autophosphorylation. The in vitro tests showed that Y11 significantly decreased Tyr397 phosphorylation producing cell growth inhibition of colon cancer cell line SW620 and breast cancer cell line BT474 and also showed tumor growth inhibition for a colon cancer xenograft model [75].
3.2.2 Y15
Y15 was directly bound to FAK phosphorylated site Tyr397 in the FERM structural domain and inhibited Tyr397 phosphorylation in a time and dose-dependent manner. Y15 did not target homologous Pyk-2, c-Src, c-RAF, EGFR, IGFR, PDGFR, PI3K, VEGFR-3, and c-Met [76]. From in vitro studies, Y15 significantly inhibited cancer cell viability in six cancer cell lines, including breast cancer, thyroid cancer, colon cancer [77] glioblastoma tumor [78], Lung cancer [79] and Ewing’s sarcoma [80]. Further evidence indicated that Y15 promoted the pancreatic cancer cells apoptosis and inhibited the cell adhesion in a dose-dependent manner [81]. From in vivo assessment, Y15 was effective in causing regression of pancreatic cancer, inducing synergistic effects when combined with Gemcitabine [81].
3.2.3 C4
C4 inhibits FAK activity by hampering the interaction of the C-terminus region of FAK and is currently developed with preclinical experiments [82] C4 treatment resulted in FAK inactivation, reduced cell viability and proliferation, cell cycle arrest and apoptosis in pancreatic cancer cells. Mechanismly, C4 highly specific disrupt FAK-VEGFR3 interactions resulted in cell cycle arrest [83]. C4 increased the sensitivity of cancer cells to Gemcitabine in vitro and inhibited tumor growth in vivo [84].
3.2.4 R2
R2 Compound can specifically disrupt the FAK-p53 interaction by specifically blocking their binding site, increase the transcriptional activity of p53, which is currently assessed in clinical trials. R2 reduced the tumor volume of HCT116 colon cancer model. Notably, the efficacy of R2 in treating colon cancer was even better than that of standardized treatments. It also showed synergistic anticancer effects when combined with 5-fluorouracil or doxorubicin [85].
3.3 FAK inhibitors in combination with anti-cancer drugs improves efficacy
Over the past few decades, treatment modalities for metastatic cancer cells have evolved from cytotoxic chemotherapy to targeted therapies. Therapeutic interventions combined with multiple target anti-cancer agents against different but interrelated tumorigenic mechanisms are more likely to eliminate cancer cells and reduce the likelihood of drug resistance development. For example, VEGF inhibitor and carboplatin separately target tumor angiogenesis and cell DNA replication, but their combination showed a synergic effect. Ongoing clinical trials, supported by in vitro and in vivo experimental studies, suggests that cytotoxic drugs are more effective in combination with some specific targeted therapies.
This section described studies of ATP-competitive FAK inhibitors (Table 2) and specific FAK inhibitors (Table 3) in combination with other anti-cancer drugs in vivo and in vitro over the past 5 years.
3.3.1 Respiratory system tumors
PF573228 in combination with erlotinib reduced cell viability and tumor growth in EGFR TKI-resistant non-small cell lung cancer (NSCLC) more effective than treatment with erlotinib alone in the A549 mouse xenograft model57. VS-6063 in combination with Gefitinib inhibited NSCLC tumor growth both in vivo and in vitro [48]. In addition, ABT263 enhanced the efficacy of Y15, showing synergistic effects in a series of lung cancer cell lines [79].
In clinical trials, VS-6063 in combination with RO5126766 for NSCLC is in Phase I clinical recruitment, the study is designed to determine the maximum tolerated dose (MTD) and recommended Phase II dose (RP2D) of VS-6063 combined with VS-6766 in NSCLC, and commenced in December, 2017 and is expected to complete in July, 2022 [86].
3.3.2 Digestive system tumors
In pancreatic cancer, Y15 exhibited synergic anti-cancer effects with Gemcitabine [81], GSK2256098 reversed Gemcitabine-related chemoresistance [87]. C4 increased the sensitivity of tumor cells to Gemcitabine chemotherapy in vitro [83].
In pancreatic ductal adenocarcinoma (PDAC), TAE-226 in combination with Nab-paclitaxel inhibited PDAC progression and prolonged survival in hormonal mice by inhibiting cancer cell growth, invasion, and induction of apoptosis [39]. VS-6063 in combination with Nab-paclitaxel also showed synergistic effects in the treatment of PDAC [39]. Besides, PF573228 can restore cell sensitivity to lexatumumab-induced apoptosis in PDAC and showed significant inhibition of pancreatic tumor growth in xenograft mice [88]. VS-6063 synergistically performs with the mTOR inhibitor everolimus by blocking feedback AKT activation in pancreatic neuroendocrine tumors (PanNETs) [89]. Of note, VS-6063 in combination with pembrolizumab for advanced pancreatic cancer is in Phase II clinical trials (NCT03727880), the purpose of this study is to evaluate if reprograming the tumor microenvironment by targeting FAK following chemotherapy can potentiate anti-programmed death-1 (PD-1) antibody, and is expected to complete in August, 2022 [90].
In colon cancer, the combination of FAK inhibitor Y15 and the Src inhibitor PP2 reduced colon cancer cell viability more effectively than each single treatment. The combination inhibited cell growth and enhanced the efficacy of chemotherapy both in vitro and in vivo [91]. Interestingly, Y15 and the HAS inhibitor 4-methylumbelliferone (4-MU) reduced the viability of colon cancer cells in a dose-dependent manner [77]. Furthermore, R2 is able to sensitize colon cancer cells to adriamycin and 5-fluorouracil [85].
3.3.3 Bone tumors
In Ewing's sarcoma, TAE226 enhanced the efficacy of conventional chemotherapy [35]. PF-562,271 and Aurora kinase inhibitors synergistically inhibited the proliferation of Ewing’s sarcoma cells and significantly suppressed tumor progression [92].
In multiple myeloma (MM), both in vivo and in vitro, VS-4718 resensitized MM cells to the proteasome inhibitors bortezomib and carfilzomib [68].
3.3.4 Reproductive system tumors
In ovarian cancer, VS-6063 can synergistically work with paclitaxel for the treatment of advanced ovarian cancer [93]. Besides, FAK inhibition with TAE-226 re-sensitized resistant ovarian cancer cells to doxorubicin and promoted tumor regression by inhibiting angiogenesis, invasion, and inducing apoptosis levels [94]. The combination of PF562271 and ABT-737 was effective in inducing cell apoptosis in ovarian clear cell carcinoma [42]. In addition, IN10018 in combination with standard chemotherapy for high-grade serous ovarian cancer is currently in phase I clinical trial, this study was designed to evaluate the safety, tolerability and efficacy of IN10018 in combination with standard chemotherapy treatment in high-grade serous ovarian cancer, and commenced in June, 2020 [95].
On the other hand, GSK2256098 in combination with chemotherapies (paclitaxel and topotecan) showed higher sensitivity against uterine Cancer [65]. PF573228 in combination with tamoxifen was able to synergistically inhibit the proliferation of ER-positive breast cancer cells [96].
3.3.5 Other tumors
With regards to the acute leukemia, the combination of TAE226 with Nilotinib showed more significant effects than each single treatment in Ph + ALL [36]. VS-4718 exerts synergistic effects with dasatinib to Ph + B-ALL cell survival, adhesion and improved therapeutic efficacy of Ph + B-ALL in vivo [97]. In addition, VS-4718 significantly improved the efficacy of ABT-199 on inducing cell apoptosis in AML cells (including primary AML CD34 +) and AML cells overexpressing MCL-1 or BCL-XL [98].
In glioblastoma tumors, combination treatment of PF562271 and ganciclovir eliminated the implanted microglioma tumors in mice (GL261 glioma orthotopic model) [99]. Noteworthily, FAK expression and activity are elevated in brain tumor models, suggesting that FAK plays important role in brain tumor. Moreover, the combination of Y15 and temozolomide showed better outcomes than each individual treatment in vivo [78].
Furthermore, VS-6063 reversed the drug resistance of Docetaxel in castration-resistant prostate cancer [100]. Y15 was synergistic with cabozantinib, sorafenib, pazopanib, and sunitinib in treatment of thyroid cancer [76]. C4 cooperating with adriamycin shows synergistic performance in killing neuroblastoma in xenograft models [84]. These results strongly demonstrate the combination potential of FAK inhibitor in cancer therapy.
4 Conclusions and future perspectives
In this review, we highlighted the impact of FAK signaling on cancer progression and elaborated the recent progress in drug development of FAK inhibitors and perspectives on FAK inhibitor therapy. As an intersection target of multiple oncogenic signaling pathways, FAK contributes to tumorigenesis and cancer progression. Accumulating research projects have demonstrated the rationality and effectiveness when employing FAK as a tumor therapeutic target. FAK inhibitors has become a hot spot in cancer drug development.
In recent years, many preclinical studies have confirmed that standard treatment supplemented with FAK-targeted drugs can significantly improve cancer prognosis and reduce chemotherapy resistance [1, 4, 101]. Thus, FAK inhibitors are going to serve as adjuvant drugs that act as chemosensitizers in cancer treatment. Although the importance of FAK inhibition is clear to the cancer therapeutic, the specific mechanism of FAK inhibitor for cancer treatment is still elusive. Further discovery is eagerly needed to elucidate how to improve the efficacy and drug resistance when apply FAK inhibitors with other anti-cancer agents. Besides, Tyr397 is the most common target site in developing FAK inhibitors, other Tyr sites are also deserved to test in the future.
On the other hand, FAK signaling pathway integrates the signal from the extracellular matrix and participates in TME remodeling in turn. Elucidation mechanism of FAK in the regulation of TME component including stroma cells (immune cell, fibroblast cells, endothelial cells) and ECM plasticity would enhance the FAK efficacy and reduce the possibility of acquired drug resistance in cancer therapy. In-depth study the function of FAK mediated signaling network between all the components will bring new ideas and chemotherapeutic strategies for the clinical treatment of tumors. Undoubtedly, highly selective FAK inhibitor combination with the standard therapies will hit cancer cells a second punch, which could benefit patients in the coming future.
Importantly, phenotypes associated with FAK inhibition show that there are multiple regulation for FAK function not only in tumor cells but also in the TME [8]. Since changes in composition and remodeling of TME are one of the most important causes in mediating tumors immune desertification, we speculate that FAK inhibitors in combination with immune checkpoint blockers such as PD-1 antibodies or CTLA-4 antibodies may exhibit impactable prospects in clinical practice. As more studies are going to be conducted in the coming years, the mechanism of FAK-related signaling pathways in the regulation of TME will also be elucidated. This will provide a more fleshed-out and rigorous scientific basis for the improvement of oncology plight.
Data availability
Not applicable.
Abbreviations
- AML:
-
Acute myeloid leukemia
- CAFs:
-
Cancer associated fibroblast
- CR:
-
Complete response
- CRPC:
-
Castration-resistant prostate cancer
- ECM:
-
Extracellular matrix
- ECs:
-
Endothelial cells
- ER+BC:
-
ER+Breast cancer
- EMT:
-
Epithelial to mesenchymal transition
- EOC:
-
Epithelial ovarian cancer
- EWS:
-
Ewing’s sarcoma
- FAK:
-
Focal adhesion kinase
- FERM:
-
Four-point-one, ezrin, radixin, moesin
- FAT:
-
Focal adhesion targeting
- GBM:
-
Glioblastoma multiforme
- MCL-1:
-
Myeloid cell leukemia-1
- MM:
-
Multiple myeloma
- MSC:
-
Mesenchymal stem cell
- NSCLC:
-
Non-small cell lung cancer
- OC:
-
Ovarian cancer
- OCCC:
-
Ovarian clear cell carcinoma
- OSCC:
-
Oral squamous cell carcinoma
- Ph+ALL:
-
Ph+Acute lymphoblastic leukemia
- PC:
-
Pancreatic cancer
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PanNETs:
-
Pancreatic neuroendocrine tumors
- PRR:
-
Proline-rich region
- PTK2:
-
Protein tyrosine kinase 2
- RTKs:
-
Receptor tyrosine kinases
- RMS:
-
Rhabdomyosarcoma
- SCLC:
-
Small cell lung cancer
- TAM:
-
Tumor associated macrophage
- TME:
-
Tumor microenvironment
- VP:
-
Vascular permeability
- 4-MU:
-
4-Methylumbelliferone
- 5-FU:
-
5-Fluorouracil
References
Dawson JC, et al. Targeting FAK in anticancer combination therapies. Nat Rev Cancer. 2021;21(5):313–24.
Lu Y, Sun H. Progress in the development of small molecular inhibitors of focal adhesion kinase (FAK). J Med Chem. 2020;63(23):14382–403.
Jiang H, et al. Development of resistance to FAK inhibition in pancreatic cancer is linked to stromal depletion. Gut. 2020;69(1):122–32.
Lee BY, et al. FAK signaling in human cancer as a target for therapeutics. Pharmacol Ther. 2015;146:132–49.
Diaz OC, et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. Elife. 2019. https://doi.org/10.7554/eLife.47327.
Wang C, et al. Secreted pyruvate kinase M2 promotes lung cancer metastasis through activating the integrin Beta1/FAK signaling pathway. Cell Rep. 2020;30(6):1780-1797.e6.
Chen JS, et al. FAK is involved in invasion and metastasis of hepatocellular carcinoma. Clin Exp Metastasis. 2010;27(2):71–82.
Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14(9):598–610.
Brullo C, Tasso B. New insights on Fak and Fak inhibitors. Curr Med Chem. 2021;28(17):3318–38.
Tapial MP, Lopez NP, Lietha D. FAK structure and regulation by membrane interactions and force in focal adhesions. Biomolecules. 2020. https://doi.org/10.3390/biom10020179.
Cao FY, et al. Chemical structure characteristics and bioactivity of small molecule FAK inhibitors. Anticancer Agents Med Chem. 2016;16(8):934–41.
Dunn KB, Heffler M, Golubovskaya VM. Evolving therapies and FAK inhibitors for the treatment of cancer. Anticancer Agents Med Chem. 2010;10(10):722–34.
Kandil SB, et al. Structure-based virtual screening, synthesis and biological evaluation of potential FAK-FAT domain inhibitors for treatment of metastatic cancer. Molecules. 2020. https://doi.org/10.3390/molecules25153488.
Berger BT, et al. Structure-kinetic relationship reveals the mechanism of selectivity of FAK inhibitors over PYK2. Cell Chem Biol. 2021;28(5):686-698.e7.
Lv P, Chen K, Zhu HL. Recent advances of small molecule focal adhesion kinase (FAK) inhibitors as promising anticancer therapeutics. Curr Med Chem. 2021. https://doi.org/10.2174/0929867328666210331143827.
Brami-Cherrier K, et al. FAK dimerization controls its kinase-dependent functions at focal adhesions. EMBO J. 2014;33(4):356–70.
Goni GM, et al. Phosphatidylinositol 4,5-bisphosphate triggers activation of focal adhesion kinase by inducing clustering and conformational changes. Proc Natl Acad Sci U S A. 2014;111(31):E3177–86.
He M, et al. Focal adhesion kinase is required for KSHV vGPCR signaling. Mol Carcinog. 2012;51(4):339–51.
Choi CH, et al. pH sensing by FAK-His58 regulates focal adhesion remodeling. J Cell Biol. 2013;202(6):849–59.
Murphy JM, et al. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp Mol Med. 2020;52(6):877–86.
Cabrita MA, et al. Focal adhesion kinase inhibitors are potent anti-angiogenic agents. Mol Oncol. 2011;5(6):517–26.
Nguemgo KP, et al. The increased adhesion of tumor cells to endothelial cells after irradiation can be reduced by FAK-inhibition. Radiat Oncol. 2019;14(1):25.
Tavora B, et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature. 2014;514(7520):112–6.
Huang M, et al. Wnt-mediated endothelial transformation into mesenchymal stem cell-like cells induces chemoresistance in glioblastoma. Sci Transl Med. 2020. https://doi.org/10.1126/scitranslmed.aay7522.
Jean C, et al. Inhibition of endothelial FAK activity prevents tumor metastasis by enhancing barrier function. J Cell Biol. 2014;204(2):247–63.
Lei X, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126–33.
Walsh C, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Therapy. 2010;9(10):778–90.
Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11(5):R68.
Stokes JB, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–45.
Roma-Rodrigues C, et al. Targeting tumor microenvironment for cancer therapy. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20040840.
Lagares D, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64(5):1653–64.
Iwatani M, et al. Discovery and characterization of novel allosteric FAK inhibitors. Eur J Med Chem. 2013;61:49–60.
Lv PC, et al. FAK inhibitors in cancer, a patent review. Expert Opin Ther Patents. 2018;28(2):139–45.
Otani H, et al. TAE226, a bis-anilino pyrimidine compound, inhibits the EGFR-mutant kinase including T790M mutant to show anti-tumor effect on EGFR-mutant non-small cell lung cancer cells. PLoS ONE. 2015;10(6): e0129838.
Moritake H, et al. TAE226, a dual inhibitor of focal adhesion kinase and insulin-like growth factor-I receptor, is effective for Ewing sarcoma. Cancer Med. 2019;8(18):7809–21.
Hu Z, Slayton WB. Integrin VLA-5 and FAK are good targets to improve treatment response in the Philadelphia chromosome positive acute lymphoblastic leukemia. Front Oncol. 2014;4:112.
Kurio N, et al. Anti-tumor effect of a novel FAK inhibitor TAE226 against human oral squamous cell carcinoma. Oral Oncol. 2012;48(11):1159–70.
Hao HF, et al. Oral administration of FAK inhibitor TAE226 inhibits the progression of peritoneal dissemination of colorectal cancer. Biochem Biophys Res Commun. 2012;423(4):744–9.
Le Large T, et al. Focal adhesion kinase inhibition synergizes with nab-paclitaxel to target pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res. 2021;40(1):91.
魏玉华, et al. 局部黏着斑激酶抑制剂治疗肿瘤的研究进展. 中华实验外科杂志, 2020. 37(06): 1167–1170.
Al-Ghabkari A, et al. Focal adhesion kinase (FAK) phosphorylation is a key regulator of embryonal rhabdomyosarcoma (ERMS) cell viability and migration. J Cancer Res Clin Oncol. 2019;145(6):1461–9.
Yoon H, et al. Targeted inhibition of FAK, PYK2 and BCL-XL synergistically enhances apoptosis in ovarian clear cell carcinoma cell lines. PLoS ONE. 2014;9(2): e88587.
Stone RL, et al. Focal adhesion kinase: an alternative focus for anti-angiogenesis therapy in ovarian cancer. Cancer Biol Therapy. 2014;15(7):919–29.
Verastem I. Study of PF-00562271, including patients with pancreatic, head and neck, prostatic neoplasms. 2013. https://ClinicalTrials.gov/show/NCT00666926.
Infante JR, et al. Safety, pharmacokinetic, and pharmacodynamic phase I dose-escalation trial of PF-00562271, an inhibitor of focal adhesion kinase, in advanced solid tumors. J Clin Oncol. 2012;30(13):1527–33.
Jones SF, et al. A phase I study of VS-6063, a second-generation focal adhesion kinase inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015;33(5):1100–7.
Shimizu T, et al. A first-in-Asian phase 1 study to evaluate safety, pharmacokinetics and clinical activity of VS-6063, a focal adhesion kinase (FAK) inhibitor in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2016;77(5):997–1003.
Gerber DE, et al. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer. 2020;139:60–7.
Verastem I. Study to investigate the safety, pharmacokinetics, pharmacodynamics and preliminary clinical activity of defactinib in combination with avelumab in epithelial ovarian cancer. 2016. https://ClinicalTrials.gov/show/NCT02943317.
Fennell DA, et al. Maintenance defactinib versus placebo after first-line chemotherapy in patients with merlin-stratified pleural mesothelioma: COMMAND—a double-blind, randomized phase II study. J Clin Oncol. 2019;37(10):790–8.
Mak G, et al. A phase Ib dose-finding, pharmacokinetic study of the focal adhesion kinase inhibitor GSK2256098 and trametinib in patients with advanced solid tumours. Br J Cancer. 2019;120(10):975–81.
Soria JC, et al. A phase I, pharmacokinetic and pharmacodynamic study of GSK2256098, a focal adhesion kinase inhibitor, in patients with advanced solid tumors. Ann Oncol. 2016;27(12):2268–74.
Verastem, I. Dose escalation study in acute myeloid or B-cell acute lymphoblastic leukemia. 2014. https://ClinicalTrials.gov/show/NCT02215629.
Doi T, et al. Phase I study of the focal adhesion kinase inhibitor BI 853520 in Japanese and Taiwanese patients with advanced or metastatic solid tumors. Target Oncol. 2019;14(1):57–65.
Kong DB, Chen F, Sima N. Focal adhesion kinases crucially regulate TGFbeta-induced migration and invasion of bladder cancer cells via Src kinase and E-cadherin. OncoTargets Therapy. 2017;10:1783–92.
Mabeta P. PF573,228 inhibits vascular tumor cell growth, migration as well as angiogenesis, induces apoptosis and abrogates PRAS40 and S6RP phosphorylation. Acta Pharm. 2016;66(3):399–410.
Aboubakar NF, et al. Therapeutic potential of focal adhesion kinase inhibition in small cell lung cancer. Mol Cancer Ther. 2019;18(1):17–27.
Stafman LL, et al. Focal adhesion kinase inhibition contributes to tumor cell survival and motility in neuroblastoma patient-derived xenografts. Sci Rep. 2019;9(1):13259.
Verastem I. A study of VS-6766 v. VS-6766 + defactinib in recurrent G12V or other KRAS-mutant non-small cell lung cancer. 2020. https://ClinicalTrials.gov/show/NCT04620330.
Verastem, I. Phase I dose escalation study of VS-6063 in Japanese subjects with non-hematologic malignancies. 2017. https://ClinicalTrials.gov/show/NCT01943292.
Verastem I. Phase II study of VS-6063 in patients with KRAS mutant non-small cell lung cancer. 2017.
Verastem I. Placebo controlled study of VS-6063 in subjects with malignant pleural mesothelioma. 2017. https://ClinicalTrials.gov/show/NCT01870609.
Thomas Jefferson University Verastem I. Defactinib and VS-6766 for the treatment of patients with metastatic uveal melanoma. 2021. https://ClinicalTrials.gov/show/NCT04720417.
Ghosh AP, et al. Kinomic profiling identifies focal adhesion kinase 1 as a therapeutic target in advanced clear cell renal cell carcinoma. Oncotarget. 2017;8(17):29220–32.
Thanapprapasr D, et al. PTEN expression as a predictor of response to focal adhesion kinase inhibition in uterine cancer. Mol Cancer Ther. 2015;14(6):1466–75.
Zhang J, et al. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle. 2014;13(19):3143–9.
Network UH. A Study of GSK2256098 and trametinib in advanced pancreatic cancer. 2020. https://ClinicalTrials.gov/show/NCT024282708.
Muz B, et al. PYK2/FAK inhibitors reverse hypoxia-induced drug resistance in multiple myeloma. Haematologica. 2019;104(7):e310–3.
Jiang H, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60.
Tiede S, et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis. 2018;7(9):73.
Laszlo V, et al. The FAK inhibitor BI 853520 inhibits spheroid formation and orthotopic tumor growth in malignant pleural mesothelioma. J Mol Med (Berl). 2019;97(2):231–42.
Osipov A, et al. Inhibition of focal adhesion kinase enhances antitumor response of radiation therapy in pancreatic cancer through CD8+ T cells. Cancer Biol Med. 2021;18(1):206–14.
Tomita N, et al. Structure-based discovery of cellular-active allosteric inhibitors of FAK. Bioorg Med Chem Lett. 2013;23(6):1779–85.
Altintop MD, et al. Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors. Eur J Med Chem. 2018;155:905–24.
Golubovskaya VM, et al. A small molecule focal adhesion kinase (FAK) inhibitor, targeting Y397 site: 1-(2-hydroxyethyl)-3, 5, 7-triaza-1-azoniatricyclo [3.3.1.1(3,7)]decane; bromide effectively inhibits FAK autophosphorylation activity and decreases cancer cell viability, clonogenicity and tumor growth in vivo. Carcinogenesis. 2012;33(5):1004–13.
O’Brien S, et al. FAK inhibition with small molecule inhibitor Y15 decreases viability, clonogenicity, and cell attachment in thyroid cancer cell lines and synergizes with targeted therapeutics. Oncotarget. 2014;5(17):7945–59.
Heffler M, et al. FAK and HAS inhibition synergistically decrease colon cancer cell viability and affect expression of critical genes. Anticancer Agents Med Chem. 2013;13(4):584–94.
Golubovskaya VM, et al. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol Cancer Ther. 2013;12(2):162–72.
Zhang H, et al. Efficacy of focal adhesion kinase inhibition in non-small cell lung cancer with oncogenically activated MAPK pathways. Br J Cancer. 2016;115(2):203–11.
Steinestel K, et al. Focal adhesion kinase confers pro-migratory and antiapoptotic properties and is a potential therapeutic target in Ewing sarcoma. Mol Oncol. 2020;14(2):248–60.
Hochwald SN, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–43.
Kandil S, et al. The discovery of new and more potent chloropyramine (C4) analogues for the potential treatment of invasive breast cancer. Chem Biol Drug Des. 2018;91(1):314–21.
Kurenova E, et al. The FAK scaffold inhibitor C4 disrupts FAK-VEGFR-3 signaling and inhibits pancreatic cancer growth. Oncotarget. 2013;4(10):1632–46.
Stewart JE, et al. Inhibition of FAK and VEGFR-3 binding decreases tumorigenicity in neuroblastoma. Mol Carcinog. 2015;54(1):9–23.
Golubovskaya VM, et al. Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth. BMC Cancer. 2013;13:342.
Institute of Cancer Research, U.K.V.I. Phase I Trial of VS-6063 and RO5126766. 2019. https://ClinicalTrials.gov/show/NCT03875820.
Li BQ, et al. WT1 associated protein promotes metastasis and chemo-resistance to gemcitabine by stabilizing Fak mRNA in pancreatic cancer. Cancer Lett. 2019;451:48–57.
Zhao X, et al. Focal adhesion kinase inhibitor PF573228 and death receptor 5 agonist lexatumumab synergistically induce apoptosis in pancreatic carcinoma. Tumour Biol. 2017;39(5):1010428317699120.
Francois RA, et al. Targeting focal adhesion kinase and resistance to mTOR inhibition in pancreatic neuroendocrine tumors. J Natl Cancer Inst. 2015. https://doi.org/10.1093/jnci/djv123.
Hopkins SKCCCaJ. Study of pembrolizumab with or without defactinib following chemotherapy as a neoadjuvant and adjuvant treatment for resectable pancreatic ductal adenocarcinoma. 2018. https://ClinicalTrials.gov/show/NCT03727880.
Heffler M, et al. Focal adhesion kinase autophosphorylation inhibition decreases colon cancer cell growth and enhances the efficacy of chemotherapy. Cancer Biol Therapy. 2013;14(8):761–72.
Wang S, et al. High-throughput chemical screening identifies focal adhesion kinase and aurora kinase B inhibition as a synergistic treatment combination in Ewing sarcoma. Clin Cancer Res. 2019;25(14):4552–66.
Kang Y, et al. Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer. J Natl Cancer Inst. 2013;105(19):1485–95.
Halder J, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2018;67(22):10976–83.
应世生物科技. IN10018联合标准化疗方案治疗高级别浆液性卵巢癌. 2020. http://www.chinadrugtrials.org.cn/CTR20200913.
Hiscox S, et al. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res Treat. 2011;125(3):659–69.
Churchman ML, et al. Synergism of FAK and tyrosine kinase inhibition in Ph(+) B-ALL. JCI Insight. 2016. https://doi.org/10.1172/jci.insight.86082.
Wang X, et al. Combinatorial inhibition of focal adhesion kinase and BCL-2 enhances antileukemia activity of venetoclax in acute myeloid leukemia. Mol Cancer Ther. 2020;19(8):1636–48.
Rolon-Reyes K, et al. Microglia activate migration of glioma cells through a Pyk2 intracellular pathway. PLoS ONE. 2015;10(6): e0131059.
Lin HM, et al. Effect of FAK inhibitor VS-6063 (defactinib) on docetaxel efficacy in prostate cancer. Prostate. 2018;78(4):308–17.
Zhou J, Yi Q, Tang L. The roles of nuclear focal adhesion kinase (FAK) on Cancer: a focused review. J Exp Clin Cancer Res. 2019;38(1):250.
Acknowledgements
We appreciate the guidance and advice of Dr. Yibo Zhang in polishing of manuscript.
Funding
This work was supported by the Natural Science Foundation of Guangdong Province (2017A030313559) and Zhanjiang science and Technology Bureau (NO. 2020A100302).
Author information
Authors and Affiliations
Contributions
YLW and NL wrote the manuscript, CFY and XMJ made diagrams, HL, BZ, YZ contributed substantial advice help to polish the language. QYZ conducted the project and revised the whole manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, Y., Li, N., Ye, C. et al. Focal adhesion kinase inhibitors, a heavy punch to cancer. Discov Onc 12, 52 (2021). https://doi.org/10.1007/s12672-021-00449-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-021-00449-y