
Abstract
Cancer-associated fibroblasts have been shown to inhibit or stimulate tumor growth depending on stage, grade, and tumor type. It remains unclear, however, the effect of endometrial-cancer-associated fibroblasts on hormone-driven responses in endometrial cancer. In this study, we investigated the effect of normal and cancer-associated stromal cells from patients with and without endometrial cancer on endometrial tumor growth in response to estradiol (E2) and progesterone (P4). Compared to benign endometrial stromal cells, the low-grade and high-grade cancer-associated stromal cells exhibited a blunted hormone response for proliferation as well as IGFBP1 secretion. Additional analysis of the influence of stromal cells on hormone-driven tumor growth was done by mixing stromal cells from benign, low-grade, or high-grade tumors, with Ishikawa cells for subcutaneous tumor formation. The presence of both benign and high-grade cancer-associated stromal cells increased estradiol-driven xenografted tumor growth compared to Ishikawa cells alone. Low-grade cancer-associated stromal cells did not significantly influence hormone-regulated tumor growth. Addition of P4 attenuated tumor growth in Ishikawa + benign or high-grade stromal cells, but not in Ishikawa cells alone or with low-grade stromal cells. Using an angiogenesis focused real-time array TGFA, TGFB2 and TGFBR1 and VEGFC were identified as potential candidates for hormone-influenced growth regulation of tumors in the presence of benign and high-grade stromal cells. In summary, endometrial-cancer-associated cells responded differently to in vitro hormone treatment compared to benign endometrial stromal cells. Additionally, presence of stromal cells differentially influenced hormone-driven xenograft growth in vivo depending on the disease status of the stromal cells.
Similar content being viewed by others


Introduction
Endometrial cancer is the most common gynecologic malignancy in the USA. The American Cancer Society estimates that in 2014, there will be 52,630 new uterine corpus cancer diagnoses and 8590 estimated deaths [3]. This represents a consistent increase in both the estimated incidence and mortality of patients with uterine corpus cancers. The most common cause of endometrial cancer is unopposed estrogen-induced epithelial proliferation leading to endometrial hyperplasia followed by cancer. Progestins are used in patients who desire to preserve their future fertility, in patients who are not operative candidates due to medical morbidity, and as therapy in women with advanced or recurrent disease. In early stage disease, a variety of progestin formulations have been utilized with an overall response rate of 73 %, but many patients recur off therapy [8]. Most of what is known about the mechanisms of action of progestin therapy in endometrial cancer has come from preclinical studies focusing on the epithelial carcinoma cells. These types of studies often ignore the key role of the tumor microenvironment in the pathogenesis of endometrial cancer.
While the details of the reciprocal, multistep, heterotypic signaling between carcinoma cells and the tumor microenvironment (including stromal fibroblasts) that results in the histopathological transformation of normal tissue into malignancy and the progression to metastatic disease remain to be fully elucidated, the importance of these interactions is increasingly being recognized [18, 14, 13, 34]. In the case of endometrial cancer, the development of the endometrium provides insights into these interactions. The endometrial mesenchymal cells play a necessary role for appropriate differentiation and function of endometrial epithelial cells. Reciprocal paracrine signaling driven by fluctuating sex steroid hormones, estrogen and progesterone, determines the epithelial cell identity, morphology, functional expression patterns, proliferation state, and rate of apoptosis [26, 22, 23]. Furthermore, tissue recombination experiments using hormone receptor knockouts have demonstrated the necessity of stromal estrogen and progesterone receptors in modulating the proliferation of endometrial epithelial cells through paracrine signals [10, 26].
In this study, we report that primary stromal cells isolated from patients with endometrial cancer respond differently to estrogen and progestin exposure compared to cells isolated from cancer-free controls. Benign stromal cell isolates exposed to estrogen and progesterone demonstrated decreased proliferation and produced high levels of IGFBP-1. Both of these responses were blunted in cells derived from endometrial cancer patients. Additionally, in a subcutaneous xenograft model, we showed that the addition of endometrial stroma from patients with or without endometrial cancer had the capacity to influence hormone-regulated growth of a low-grade endometrial cancer cell line.
Materials and Methods
Ethics Statement
All animal experiments were approved by the Northwestern University Animal Care Committee. Endometrial tumors were obtained from women undergoing hysterectomies at Northwestern Memorial Hospital. Patients provided written informed consent before surgery, and these studies were approved by the Northwestern Institutional Review Board in accordance with US Department of Health regulations.
Endometrial Cancer Tissues and Cell Cultures
Ishikawa cells were obtained from S. Bulun (Northwestern University). These cells originated from a well-differentiated endometrial adenocarcinoma with a known PTEN mutation [31, 32]. Ishikawa cells were authenticated by the DNA Sequencing and Analyses Core at the University of Colorado (Christopher Korch) using DNA profiling. Ishikawa cells were maintained in DMEM/F12 (1:1) supplemented with 100 U/ml penicillin, 100 μ Fungizone, and 10 % fetal bovine serum (FBS).
Stromal cells were obtained from normal endometrial tissues and endometrial cancer tissues from surgical specimens (Supplementary Table S3). Normal endometrial tissues were obtained from premenopausal women undergoing hysterectomy for uterine leiomyoma or pelvic organ prolapse, from both the proliferative and secretory phases of the menstrual cycle. Endometrial cancer tissues were obtained from premenopausal or postmenopausal women undergoing hysterectomy for biopsy proven endometrial cancer. The tumors collected from patients who consented to participate in this study were greater than 1.5 cm in largest dimension. Hospital pathologist reviewed each case prior to providing a 0.5–1-cm segment of the tumor. The stromal cells that were isolated were taken from these tumors excised from the uterus by pathology personnel. None of the subjects received any preoperative hormonal therapy within 6 months of surgery.
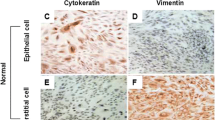
The tissue was minced thoroughly in calcium- and magnesium-free Hanks’ Balanced Salt Solution (HBSS). The minced tissue was placed in an enzyme solution containing 0.5 % (w/v) collagenase and 0.02 % DNase (w/v) and incubated for 20 min at 37 °C in a shaking water bath. The supernatant was recovered and placed at 4 °C. The remaining tissue was further digested in an enzyme solution consisting of 0.5 % (w/v) collagenase, 0.02 % (w/v) DNase, 0.1 % (w/v) hyaluronidase, and 0.1 % (w/v) pronase and processed as described above. The cell suspensions from the first and second digestions were centrifuged at 2000×g for 5 min, and the pellet was resuspended in HBSS. The suspensions were then passed through a 70-μm filter. Cells were cultured in DMEM/F12 (1:1) (Invitrogen, Carlsbad, CA) containing 10 % FBS and penicillin (100 U/ml) and streptomycin (100 U/ml) at 37 °C in humidified atmosphere with 5 % CO2. Culture medium was changed every 3 days. Cell purity was assessed by immunocytochemistry using antibodies against cytokeratin (Cell Signaling Technology) and vimentin (Calbiochem). The purity of the stromal cell preparations used in these studies was >95 % (Fig. 1). All the experiments described were performed within six passages from isolation.

Patient tissues and stromal cell cultures from benign endometrium and low-grade and high-grade cancer. Formalin-fixed samples from hysterectomy specimens were immunohistochemically stained for cytokeratin to highlight epithelial cells from the stromal cells. Benign endometrium is demonstrated in (a). Endometrial cancer may be diagnosed by areas of back-to-back glands in the absence of intervening stroma (as is seen in a low-grade specimen, b [arrows]) or irregular infiltration of glands in altered fibroblastic stroma (as is seen in a high-grade specimen, c). d–f. Representative primary stromal cell isolate cultures were stained with antibodies for vimentin (red) and cytokeratin (green). d Benign stromal cells, e low-grade cancer-associated stromal cells, and f high-grade cancer-associated stromal cells (Color figure online)
Cell Proliferation
To assess endometrial stromal cell proliferation, BrdU assay (Roche) was used. Endometrial stromal cells were seeded in a 96-well tissue culture treated plate at 1500 cells per well in a 37 °C incubator. The cells were allowed to attach 6 h to overnight, washed with phosphate-buffered saline (PBS), and then serum-starved 6 h to overnight in serum-free DMEM/F12 (1:1). The cells were then treated with DMEM/F12 (1:1) supplemented with 1 % FBS depleted of steroids by treatment with dextran-coated charcoal (S-FBS), 10 nM estradiol (E2), and 1 uM medroxyprogesterone acetate (MPA) or ethanol control (final concentration 0.5 % v/v) for 48 h. BrdU was added to each well for a final concentration of 10 μM BrdU 12 h prior to completion of 48-h incubation. Cells were then processed per manufacturer instructions. Each treatment was conducted in quadruplicate.
ELISA Assays
To assess endometrial stromal cell IGFBP-1 production, the Human IGFBP-1 ELISA Kit (RayBio) was used. Endometrial stromal cells were seeded in a 24-well tissue culture treated plate at 1250 cells per well in a 37 °C incubator. The cells were allowed to attach overnight and treated with 10 nM E2 and 1 uM MPA or vehicle for 14 days, with media changes every 2 days. Supernatants were harvested and IGFBP-1 concentration was determined in appropriate serial dilutions per manufacturer’s instructions. Each treatment was conducted in quadruplicate.
Immunofluorescent Staining
Cells were fixed with 4 % paraformaldehyde (Sigma), and coverslips were then washed with PBS and permeablized with 0.1 % Triton–0.1 % deoxycholate (Sigma). Cells were blocked with 5 % bovine serum albumin (BSA, Sigma) made in PBS. Subsequently, antibodies to progesterone receptor (PR; Dako), cytokeratin (Cell Signaling Technology), and vimentin (Calbiochem) were added to each sample and incubated overnight at 4 °C. A fluorescence-conjugated antimouse or antirabbit secondary antibodies (Vector Labs) were incubated for 1 h. After multiple washings, coverslips were mounted with mounting media (Invitrogen) for fluorescence on glass slides, and cells were visualized using a fluorescent inverted microscope, Axiovert 200 (Zeiss).
Western Blot
Whole cell lysates were obtained on ice using the M-PER Mammalian lysis solution (Thermo Scientific) supplemented with protease and phosphatase inhibitors (Sigma). The concentration of protein in the lysates was measured using the Micro BCA kit (Thermo Scientific). Isolated protein samples were run on 8 % acrylamide gels and then transferred onto polyvinylidene difluoride membranes (Whatman). Membranes were blocked in 5 % bovine serum albumin (BSA, Sigma) in TBS-T at room temperature for 1 h. Membranes were then incubated overnight at 4 °C with primary antibodies against PR (Dako). The blots were washed four times in TBS-T at room temperature and then incubated with secondary peroxidase-conjugated goat antimouse antibody for 1 h at room temperature. The membranes were developed with the ECL Super Signal West Femto detection kit (Thermo Scientific). The membranes were stripped using Restore Western Blot Stripping Buffer (Pierce) and probed with an antibody to beta-actin (Sigma) for a loading control.
Tumor Xenografts
Four- to 6-week-old CD-1 nude female mice were purchased from Charles River Laboratories. All mice were ovariectomized 2 days prior to xenografting. One million Ishikawa cells with or without two million stromal cells were suspended in ice-cold 1:2 PBS/Matrigel (BD Biosciences) in a total volume of 100 μl and subcutaneously injected in the right and left dorsum of each mouse. Each stromal cell group was composed of a mixture of cells isolated from two different patients. A total of 32 mice were injected in order to have eight mice per group. Four days prior to xenografting, all the mice were implanted subcutaneously with an estrogen pellet (0.1 mg, 60-day release for 1.6 ug/day; Innovative Research of America). Half of the mice additionally had progesterone pellets (150 mg, 60-day release for 2.5 mg/day; Innovative Research of America) implanted subcutaneously. Mice were weighed regularly and tumor sizes were measured. Tumor sizes were measured with calipers through the course of the experiment. Tumor volumes were calculated using the formula: tumor volume = ½(length × width)2 [19]. After 32 days, tumors were excised and fixed for immunohistochemistry or flash frozen for RNA extraction.
Real-Time PCR Array
RNA was isolated from tumors using Qiagen RNeasy plus mini kit (Qiagen) according to the manufacturer’s protocol. Concentration and purity of extracted RNA were determined using the ND-1000 Spectrophotometer (NanoDrop, Wilmington, DE). Total RNA samples were DNase-treated to remove any contamination using DNA-free RNA kit (ZymoResearch). All RNA samples were evaluated for quality. One microgram of total RNA was reverse transcribed in a total volume of 20 μl for the synthesis of cDNA (SABiosciences) according to the manufacturer’s protocol. A focused real-time PCR array was used to compare the expression of genes associated with Angiogenesis (SABiosciences). Specifically, the Human Angiogenesis PCR Array that contains 84 key genes involved in modulating the biological processes of angiogenesis was used and performed according to manufacturer’s instructions using RT2 SYBR® Green ROX qPCR Mastermix. Each well in the 96-well plate contained specific primers for each gene. Each group (Ishikawa, Ishikawa+benign, Ishikawa+LG-CA, Ishikawa+HG-CA) treated with E2 or E2+P4 was done in triplicate (n = 3 tumors). All reactions were carried out on an ABI QuantStudio 12K Flex Sequence Detection System (Applied Biosystems, Foster City, CA) for 40 cycles (95 °C for 15 s, 60 °C for 1 min) after 10-min incubation at 95 °C. Analyses of the raw data were done through the Superarray Data Analysis Web Portal (SuperArray Bioscience Corp.).
Immunohistochemistry
Tissues were fixed in formalin and paraffin-embedded. Five to 6-μm tissue sections were placed on glass slides. Sections were deparaffinized and stained using the Envision DAB HRP kit (Dako) or hematoxylin and eosin (H&E). Heat-induced epitope retrieval was performed in a 10 nM sodium citrate buffer with 0.05 % Tween (Sigma) at pH 6.0 pre-heated to 99 °C. Slides were placed in sodium citrate buffer for 45 min. Slides were cooled to 30 min at room temperature and washed in 1× TBS-T for 5 min. The Dako EnVision HRP IHC kit was used according to manufacturer specifications. Tissue sections were incubated with primary antibodies 4 °C overnight in a humidified chamber. Antibodies used were cytokeratin (Cell Signaling), CD10 (Pierce), Ki67 (Abcam), cleaved caspase-3 (Cell Signalling Technology), PR (Dako), and CD31 (Santa Cruz). Slides were washed in 1× TBST with gentle agitation. The appropriate antimouse or antirabbit secondary antibodies were applied to the tissue sections. The DAB solution was applied. The length of the reaction varied with each antibody but was consistent across all xenografted tumors. Mayer’s hematoxylin was used as counterstain. The sections were then placed in a solution of 28 % ammonium hydroxide and then rinsed. The sections were dehydrated via two changes of 95 % ethanol, two changes of 100 % ethanol, and two changes of Xylene. The sections were then mounted onto coverslips using Cytoseal XYL mounting media (Richard-Allan Scientific). The slides were then visualized and images captured using the Leica DM5000B Microscope. CD31 quantitation was done by counting the number of CD31 stained vessels in three random fields per tumor. Four tumors from each treatment group were quantified.
Statistical Analysis
Statistical analysis was performed using Prism software (v5; GraphPad, San Diego, CA). p values ≤0.05 were considered significant. Data from the BrdU incorporation and IGFBP1 assays were analyzed using the unpaired t test. For tumor growth curves, linear regression analysis was done and the slope and intercept were compared between growth curves. Statistical significance of tumor sizes at specific time points was determined with multiple t tests using the Holm-Sidak method, with alpha = 5.000 %. Statistical significance for CD31 staining was determined using the Holm-Sidak multiple t test comparisons, with alpha = 5.000 %.
Results
Differential Response of Stromal Cells to Hormones
Human endometrial stromal cells were isolated from patients with histopathology-confirmed benign endometrium (benign), low-grade endometrial cancer (LG-CA), and high-grade endometrial cancer (HG-CA) (Fig. 1). The stromal cell isolates were >95 % free of endometrial epithelial cells as demonstrated by immunofluorescence microscopy utilizing vimentin to identify stromal cells and cytokeratin to identify the epithelial cells (Fig. 1). No significant differences in morphology were observed among the benign, LG-CA, and HG-CA stromal cells.
The stromal cell response to hormone exposure was examined by culturing stromal cells in E2 and MPA. Treatment of benign stromal cells with E2 and MPA significantly inhibited stromal cell proliferation as measured by BrdU incorporation compared to vehicle alone (Fig. 2a). In contrast, HG-CA stromal cell proliferation was minimally affected by the addition of E2+MPA while LG-CA stromal cells exposed to E2 and MPA exhibited a more varied proliferative response. The proliferative response of HG-CA stromal cell isolates was significantly different when compared to responses of either benign or LG-CA cells (p < 0.01). Chronic exposure to progestins in vivo causes endometrial stromal cells to decidualize. In vitro, decidualization has been characterized, in part, by the secretion of IGFBP-1 by endometrial stromal cells [15]. Compared to vehicle control, exposure to E2 and MPA resulted in significantly increased production of IGFBP-1 by benign stromal cells (p < 0.01) but not stromal cells from endometrial cancer patients (Fig. 2b). In order to determine if decreased progesterone receptor expression was responsible for this impaired response to hormonal exposure, we performed immunofluorescence microscopy for PR. PR was present in all three isolates at similar levels (Fig. 2c).

Effect of E2 and MPA on cancer-associated stromal cells. a Primary stromal cells were treated with a combination of 10 nM of E2 and 1 uM MPA or vehicle control 48 h, and proliferation was measured by BrdU incorporation. Data are expressed as the % decrease of no hormone control. b Stromal cells were treated with 10 nM E2 and 1 uM MPA for 14 days. IGFBP-1 protein in the supernatant was quantified using ELISA assay. The mean ± SEM are shown. Asterisk denotes p < 0.01. c PR was detected by immunofluorescence in benign (ESC), LG-CA, and HG-CA stromal cells
Influence of Stromal Cells on Endometrial Tumor Growth
It has been demonstrated that endometrial stromal cells release important paracrine factors that mediate the hormone response of epithelial cells [22, 5]. The impact of stromal cells from LG-CA and HG-CA as well as from benign endometrium on endometrial tumor growth was further explored using a subcutaneous xenograft model.
Nude mice were ovariectomized, and a mixed population of Ishikawa cells with stromal cells from benign, LG-CA, or HG-CA were grafted subcutaneously. Mice were treated with exogenous E2 (1.6 μg/day) or E2 and P4 (2.5 mg/day) and growth of tumors was monitored. For this experiment, stromal cells derived from two patients were combined and mixed with Ishikawa cells prior to injection. In response to E2, all tumors grew over a period of 32 days. In response to E2 alone, the presence of benign and HG-CA stromal cells and, to a lesser extent, LG-CA stromal cells increased tumor size compared to Ishikawa cells alone (Fig. 3a). Statistical analysis revealed a significant difference in the slopes and intercepts of the growth curves of tumors from Ishikawa alone compared to Ishikawa + benign stromal cells (slope, 2.67 ± 0.95 vs 8.31 ± 1.7, p < 0.01), indicating an increased growth rate in the presence of stromal cells. This difference was significant by day 18. Similarly, the presence of HG-CA stromal cells increased tumor growth significantly in the presence of E2 compared to Ishikawa cells alone demonstrated by increased slope of growth curves (2.67 ± 0.95 vs 9.48 ± 2.2, p < 0.01; Fig. 3a). The presence of LG-CA cells appeared to increase tumor growth (slope, 2.67 ± 0.95 vs 6.71 ± 2.1, p = 0.06); however, the difference in slope as well as tumor sizes at each time point compared to Ishikawa (no stromal cells) tumors did not reach statistical significance, due to variability between tumors.

Effect of stromal cells on hormone driven growth of Ishikawa cell tumors. Both flanks of ovariectomized, nude mice were injected with Ishikawa cells alone (Ishi alone), or a combination of Ishikawa and benign stromal cell, Ishikawa and LG-CA cells, or Ishikawa and HG-CA cells. On the day of xenografting, mice were implanted subcutaneously with E2 or E2+P4 releasing pellets. Tumors were measured two times weekly until mice were killed and tumors were collected. a Tumor volumes in xenografts treated with estradiol alone. b–e Tumor volumes with or without stromal cells in response to E2 or E2+P4
The addition of P4 influenced tumor growth as well, and this was dependent on the type of stromal cells present. First, the addition of P4 did not affect growth of xenografted tumors from Ishikawa cells alone, compared to E2 treatment during the 32-day period of growth (Fig. 3b). Similarly, the presence of LG-CA stromal cells mixed with Ishikawa cells did not significantly affect tumor volume in response to E2+P4 (Fig. 3d). In contrast, treatment with E2+P4 attenuated the growth of tumors when Ishikawa cells were mixed with HG-CA or benign stromal cells, compared to E2 alone (Fig. 3c, e). Comparison of growth curves in response to E2 versus E2+P4 of tumors with benign stromal cells demonstrated a difference in the slopes of the growth curves (8.31 ± 1.7 vs 5.81 ± 1.4; p = 0.27) that did not reach statistical significance; however, there was a significant difference in elevation or intercept (y-intercept when x = 0.0: −5.436 ± 40.45 vs −17.54 ± 34.89; p < 0.0001; Fig. 3c). This indicates that the lines are indeed different and that P4 alters growth of the tumors when benign stromal cells are present. P4 attenuated tumor growth as tumor sizes were significantly smaller at each time point with the exception at the last time point, day 32. The presence of HG-CA stromal cells showed similar effects to that of the benign stromal cells in that the differences in slopes were not significant (9.48 + 2.2 vs 5.35 + 1.4; p = 0.14) but the elevations or intercepts were significantly different (y-intercept when x = 0.0: −10.07 ± 51.52 vs −14.12 ± 32.38; p < 0.0001; Fig. 3e) demonstrating a significant effect of P4 on tumors. Comparisons of tumor sizes at each time point showed significantly smaller tumors with P4.
H&E staining and immunohistochemical analysis were done on representative xenografted tumor sections for cytokeratin, CD10, Ki67, progesterone receptor, and CD31 (Fig. 4; Supplementary Fig. S2). Ishikawa cells stained intensely for cytokeratin (Fig. 4a), as well as Ki67 (Fig. 4c), indicative of active proliferation. Stromal cells stained with CD10 (Fig. 4b) and revealed low density of stromal cells in all the mixed xenografted tumors examined. There was no obvious staining for Ki67 in the stromal cells, indicating no active proliferation in these cells. In all tumors, progesterone receptor expression was seen in the Ishikawa cells and also evident in most CD10+ stromal cell elements (Fig. 4d). The staining patterns of CD10, Ki67, and PR did not differ significantly between the three tumor isolates (benign, LG-CA, HG-CA). CD31 which is a marker for endothelial cells was evident within the tumors and surrounding the tumor grafts. Quantitation of CD31 vessels within tumors revealed lower numbers of stained vessels in tumors treated with E2 with LG-CA and HG-CA stromal cells compared to tumors with benign stromal cells. CD31 levels in tumors treated with E2+P4 did not differ from E2 treatment for any of the tumors. Areas of necrosis were observed in the tumors (Supplementary Fig. S2) that may be indicative of insufficient access to nutrients.

Immunohistochemical staining of tumors. Tumors were fixed, paraffin-embedded and sectioned. Representative micrographs of immunohistochemical staining of tumors are shown for a cytokeratin, b CD10, c Ki67, d PR, and e CD31. Tumor (T), stromal cells (S), endothelial cells (E)
Role of Stromal Cells on Angiogenesis-Associated Genes
Since it has been shown that stromal cells can secrete paracrine factors to regulate angiogenesis [7], we hypothesized that factors that govern angiogenesis may be responsible, in part, for the clear differences in hormonal responsiveness observed when Ishikawa cells were co-cultured with benign and HG-CA stromal cells. We therefore looked at the expression pattern of genes known to influence angiogenesis using an RT-PCR-based focused array. Tumors from four groups were assessed in triplicate: Ishikawa alone, Ishikawa+benign, Ishikawa+LG-CA, and Ishikawa+HG-CA. Two comparisons were done. The first was a comparison in fold changes of gene expression between Ishikawa only and the presence of stromal cells. Significant fold differences (p < 0.05) of 1.5 or larger are shown in Supplementary Table S1. Of the 84 genes represented in this array, a total of 26 genes were significantly altered when stromal cells were present (benign 16, LG-CA 11, HG-CA 21). All three stromal cell isolates shared six genes in common and three of these genes were members of the TGF family (TGFA, TGFB2, and TGFBR1), which were significantly decreased when stromal cells were present (Fig. 5a).

Expression of angiogenesis-associated genes in endometrial tumors. Tumors were flash-frozen and RNA-extracted from tumors. Expression of genes using an angiogenesis focused real-time PCR array was measured. a Genes significantly regulated by the presence of stromal cell type are shown. b Genes significantly regulated by E2+P4 compared to E2 alone in each tumor type are shown
A second comparison was made between E2 and E2+P4 treatments within each tumor group (Fig. 5b). Of the 84 genes represented in this array, a total of 32 genes were significantly altered with E2+P4 compared to E2 treatment (Supplementary Table S2). Tumors from Ishikawa cells alone exhibited 11 genes significantly decreased with P4 treatment, despite no significant alteration in tumor growth from E2, suggesting that these genes may not be directly associated with growth of tumors. Tumors with benign stromal cells exhibited alteration of 24 genes by P4, the highest number, among the tumor groups. Tumors with benign and HG-CA stromal cells exhibited a growth-restricted response to P4, and the one common gene significantly altered in these two groups that was not in the Ishikawa only or in the LG-CA groups was VEGFC. Moreover, tumors with HG-CA stromal cells were smaller with E2+P4 compared to E2 alone, and 10 genes were significantly decreased. However, when P4 responsive genes were compared between HG-CA and the non-responding groups, Ishikawa only and LG-CA, only VEGFC was differentially regulated in HG-CA. These data suggest that VEGFC may be a hormonally responsive gene that affects tumor growth in the presence of benign or HG-CA stromal cells.
Discussion
The endometrium is one of the most responsive tissues to sex steroid hormones, estrogen and progesterone. Type 1 endometrial cancer is a hormone-dependent disease and thus, in order to understand the biology of endometrial cancer growth and progression, it is important not only to determine how hormones drive the cancer cells but also to study the important paracrine actions of the microenvironment. This is especially relevant for the endometrium since stromal cells are highly responsive to hormones and they have been shown to secrete key paracrine factors that affect the epithelial cells [26]. Our study demonstrated that stromal cells from the benign and cancerous endometrium respond differently to estradiol and progesterone. Furthermore, stromal cells significantly influenced hormone-driven growth of endometrial tumors in vivo.
Our in vitro studies investigating hormone response of benign and cancer-associated stromal cells from the endometrium demonstrated that cancer-associated stromal cells respond differently to E2 and MPA. The decrease in proliferation did not occur and IGFBP1 secretion was blunted in the cancer-associated stromal cells. Factors which contribute to blunted progesterone response in cancer-associated endometrial stromal cells remain to be elucidated. We and others have demonstrated that activated signaling pathways promote progesterone resistance that is observed in endometrial stromal cells, specifically from the disease of endometriosis [1, 12, 21, 25, 46]. This includes blunted decidualization in response to progestin treatment. The tumor microenvironment has been deemed an inflammatory site with dysregulated secretion of cytokines and inflammatory mediators [24], leading to hyperactivation of signaling pathways. In addition, chronic inflammation could lead to epigenetic changes within the chromatin and transcriptional machinery affecting the progesterone receptor transcriptional function in these cells. This could explain the blunted proliferation and decidualization in the low grade and high grade–associated stromal cells in response to estradiol and progestin treatment.
The tumor growth promoting properties of benign stromal cells were demonstrated in this study. One of the reasons why stromal cells from benign endometrium are able to promote tumor growth in response to E2 may be because stromal cells are responding to exogenous E2 and P4 and secreting the necessary growth-promoting paracrine signals. In a normal setting, this would allow proliferation of the endometrium as a whole, including epithelial cells. When mixed with tumor cells, however, we would expect the same paracrine signals to promote growth of cancer cells. In the same context, P4 would attenuate E2-driven growth, which is what was observed in this study. LG-CA stromal cells were not as responsive to E2 and P4 for reasons discussed above and thus insufficiently released the paracrine factors that promote growth, or attenuation of growth. The response to hormones in the presence of HG-CA, however, was different. We observed that these tumors behaved similarly to those that contained benign endometrial stromal cells, for reasons that remain unknown. It is possible that other pathways aside from those that directly control proliferation and decidualization are involved in controlling tumor growth through indirect mechanisms. It is obvious, however, from these data that stromal cells from a low-grade tumor are different from those from a high-grade tumor underscoring the biological difference between low-grade and high-grade tumors. Identification of mutational status, hormone response genes, and pathways between the low-grade and high-grade endometrial cancers will help us to better understand the complex paracrine interactions between cancerous epithelial cells and the underlying stroma. It is important to note that tumors were treated with hormones from the time of xenografting for a duration of 32 days. Whether longer treatment with hormones is necessary to observe differences of the tumors with LG stromal cells remains to be tested.
Our findings are novel as examination of endometrial cancer epithelial-stromal cell interactions have to date been limited to in vitro culture systems, co-culture with benign stromal cells only, or paracrine activity of cancer-associated stromal cells in the absence of hormonal influence. Arnold et al. demonstrated that conditioned media collected from benign endometrial stromal cells decreased proliferation of Ishikawa cells [6]. Subramaniam et al. showed that conditioned media from cancer-associated stromal cells promoted proliferation of two endometrial cancer cell lines and primary epithelial cell isolates [41]. Our in vivo experiments show that the presence of stromal cells from benign, HG-CA, and LG-CA increases growth of tumors. It is important to note not only the difference in the model systems (in vitro vs in vivo) but that exogenous estradiol was given to ovariectomized mice. Arnold et al. demonstrated that stromal cell isolates from cancer-free patients exhibited greater proliferation when grown on plastic surfaces compared to growth on a basement membrane extract (Matrigel) or in contact with epithelial cells [5]. Similarly, stromal cells in our xenografts exhibited no visible Ki67 staining indicative of minimal proliferation. The relatively small population of stromal cells remaining at the end of the hormone treatments were supportive of minimal proliferation. Consistent with this observation, Hu et al. demonstrated that fibroblasts, while important for establishing xenograft tumor initiation, do not survive long in xenograft tumors [20]. It does appear that in this system, an ongoing stromal cell presence may be required to maintain hormonal regulation, such that once the stromal compartment has become so small, the Ishikawa-alone growth program may be re-established in all tumors and growth then becomes regulated by other microenvironmental interactions, such as angiogenesis.
It is clear from various model systems that reciprocal interactions between tumor cells and their microenvironment drive the processes of tumorigenesis including proliferation, invasion, and metastasis [37]. For example, in a prostate cancer xenograft model, normal stromal cells do not stimulate SV40-initiated epithelial cells to proliferate when co-cultured in collagen gel within the male mouse renal capsule. In contrast, cancer-associated stroma did stimulate the same epithelial cells to proliferate [33]. Similar findings have been described in other xenograft and tissue culture models suggesting that both stromal and epithelial compartments must be abnormal to promote tumorigenesis [10]. It is interesting that despite altered in vitro responses to hormone exposure, high-grade endometrial cancer-associated stromal cells continued to exhibit hormone responsiveness to the endometrial cancer cell line. This model may be further explored to elucidate the mechanism of action of systemic hormone therapy with the goal of optimizing response rates that have been relatively poor [38, 35, 36].
Quantitation of CD31 staining differed in tumors treated with E2 with LG-CA or HG-CA stromal cells compared to tumors with benign stromal cells. Interestingly, CD31 staining varied considerably in Ishikawa tumors without stromal cells which made it difficult to compare levels with other tumors. CD31 vessel staining was also not uniformly dispersed within the tumors and vessel sizes differed. Whether the presence of stromal cells affect vessel branching and localization which ultimately affects growth of the tumor remains to be determined. The real-time PCR array of genes associated with angiogenesis revealed that numerous genes were differentially regulated depending on the type of stromal cell isolates present, or in response to P4. If genes were grouped according to differential expression associated with tumor growth, for example, the influence of stromal cells on estradiol-driven tumor growth, six genes—EPHB4, PECAM1, SERPINE1, TGFA, TGFB2, and TGFBR1—emerged. TGFB function varies widely in a context-dependent manner [27]. The exact contextual elements dictate whether TGFBs function to suppress or promote tumorigenesis [28]. In the human uterus, TGFB mRNA and protein expression can be detected in both glandular and stromal cell types but are expressed at higher concentrations in the stromal compartment [17, 9]. Studies that characterize the effect of TGFBs in Ishikawa cells have primarily used TGFB1; exposure of Ishikawa cells to TGFB1 in most of these studies resulted in an inhibition of proliferation [30, 39, 4]. Tanwar et al. [43] demonstrated that deletion of APC in the stromal compartment of the endometrium in mice resulted in the development of glandular hyperplasia that progresses to endometrial carcinoma. They also demonstrated that the mutant APC resulted in the suppression of TGFB and BMP signaling supporting the involvement of TGFB in the stromal/epithelial paracrine communication.
VEGFC was the one gene that correlated with significantly altered expression and decreased tumor growth in response to P4. VEGFC has been shown to be associated with promoting aggressive behaviors of tumors including lymphangiogenesis and lymph node metastasis [40, 47, 2, 44]. In addition, hormones can regulate VEGFC expression [40, 47]. VEGFC is an angiogenic growth factor that is expressed in the endometrium [29, 16]. VEGFC has been associated with promoting endothelial cell functions, vascular permeability and angiogenesis in endometriosis [45, 42], and endometrial cancer [11]. It is possible that VEGFC, through stromal cells, may promote angiogenesis to promote tumor growth and that treatment with P4 decreases the production of this growth factor, decreases angiogenesis, and thereby attenuates tumor growth.
In summary, we have demonstrated that cancer-associated stromal cells of the endometrium respond differently to progesterone and influence tumor growth depending on the grade of the tumor (low vs high) they were associated with. Furthermore, stromal cells from benign, LG-CA, and HG-CA potentiated tumor growth in response to estradiol. Angiogenic genes are significantly altered in tumors depending on the presence of stromal cells and the hormone treatment. The identification and study of important paracrine factors will shed light on the mediators of tumor progression and allow us to identify novel targeted therapies specific for this disease.
References
Aghajanova L, Hamilton A, Kwintkiewicz J, Vo KC, Giudice LC (2009) Steroidogenic enzyme and key decidualization marker dysregulation in endometrial stromal cells from women with versus without endometriosis. Biol Reprod 80(1):105–114. doi:10.1095/biolreprod.108.070300
Akagi K, Ikeda Y, Miyazaki M, Abe T, Kinoshita J, Maehara Y, Sugimachi K (2000) Vascular endothelial growth factor-C (VEGF-C) expression in human colorectal cancer tissues. Br J Cancer 83(7):887–891. doi:10.1054/bjoc.2000.1396
American Cancer Society (2014) Cancer Facts Fig
Anzai Y, Gong Y, Holinka CF, Murphy LJ, Murphy LC, Kuramoto H, Gurpide E (1992) Effects of transforming growth factors and regulation of their mRNA levels in two human endometrial adenocarcinoma cell lines. J Steroid Biochem Mol Biol 42(5):449–455
Arnold JT, Kaufman DG, Seppala M, Lessey BA (2001) Endometrial stromal cells regulate epithelial cell growth in vitro: a new co-culture model. Hum Reprod 16(5):836–845
Arnold JT, Lessey BA, Seppala M, Kaufman DG (2002) Effect of normal endometrial stroma on growth and differentiation in Ishikawa endometrial adenocarcinoma cells. Cancer Res 62(1):79–88
Bausero P, Cavaille F, Meduri G, Freitas S, Perrot-Applanat M (1998) Paracrine action of vascular endothelial growth factor in the human endometrium: production and target sites, and hormonal regulation. Angiogenesis 2(2):167–182
Bovicelli A, D’Andrilli G, Giordano A, De Iaco P (2013) Conservative treatment of early endometrial cancer. J Cell Physiol 228(6):1154–1158. doi:10.1002/jcp.24292
Chegini N, Zhao Y, Williams RS, Flanders KC (1994) Human uterine tissue throughout the menstrual cycle expresses transforming growth factor-beta 1 (TGF beta 1), TGF beta 2, TGF beta 3, and TGF beta type II receptor messenger ribonucleic acid and protein and contains [125I]TGF beta 1-binding sites. Endocrinology 135(1):439–449. doi:10.1210/endo.135.1.8013382
Cunha GR, Cooke PS, Kurita T (2004) Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol 67(5):417–434
Donoghue JF, Lederman FL, Susil BJ, Rogers PA (2007) Lymphangiogenesis of normal endometrium and endometrial adenocarcinoma. Hum Reprod 22(6):1705–1713. doi:10.1093/humrep/dem037
Eaton JL, Unno K, Caraveo M, Lu Z, Kim JJ (2013) Increased AKT or MEK1/2 activity influences progesterone receptor levels and localization in endometriosis. J Clin Endocrinol Metab 98(12):E1871–E1879. doi:10.1210/jc.2013-1661
Egeblad M, Nakasone ES, Werb Z (2010) Tumors as organs: complex tissues that interface with the entire organism. Dev Cell 18(6):884–901. doi:10.1016/j.devcel.2010.05.012
Felix AS, Weissfeld J, Edwards R, Linkov F (2010) Future directions in the field of endometrial cancer research: the need to investigate the tumor microenvironment. Eur J Gynaecol Oncol 31(2):139–144
Ganeff C, Chatel G, Munaut C, Frankenne F, Foidart JM, Winkler R (2009) The IGF system in in-vitro human decidualization. Mol Hum Reprod 15(1):27–38. doi:10.1093/molehr/gan073
Girling JE, Rogers PA (2009) Regulation of endometrial vascular remodelling: role of the vascular endothelial growth factor family and the angiopoietin-TIE signalling system. Reproduction 138(6):883–893. doi:10.1530/REP-09-0147
Gold LI, Saxena B, Mittal KR, Marmor M, Goswami S, Nactigal L, Korc M, Demopoulos RI (1994) Increased expression of transforming growth factor beta isoforms and basic fibroblast growth factor in complex hyperplasia and adenocarcinoma of the endometrium: evidence for paracrine and autocrine action. Cancer Res 54(9):2347–2358
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y et al (2010) MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9(7):1956–1967. doi:10.1158/1535-7163.MCT-09-1012
Hu M, Yao J, Carroll DK, Weremowicz S, Chen H, Carrasco D, Richardson A et al (2008) Regulation of in situ to invasive breast carcinoma transition. Cancer Cell 13(5):394–406. doi:10.1016/j.ccr.2008.03.007
Kim TH, Yu Y, Luo L, Lydon JP, Jeong JW, Kim JJ (2014) Activated AKT pathway promotes establishment of endometriosis. Endocrinology 155(5):1921–1930. doi:10.1210/en.2013-1951
Kurita T, Medina R, Schabel AB, Young P, Gama P, Parekh TV, Brody J et al (2005) The activation function-1 domain of estrogen receptor alpha in uterine stromal cells is required for mouse but not human uterine epithelial response to estrogen. Differentiation 73(6):313–322. doi:10.1111/j.1432-0436.2005.00033.x
Kurita T, Wang YZ, Donjacour AA, Zhao C, Lydon JP, O’Malley BW, Isaacs JT, Dahiya R, Cunha GR (2001) Paracrine regulation of apoptosis by steroid hormones in the male and female reproductive system. Cell Death Differ 8(2):192–200. doi:10.1038/sj.cdd.4400797
Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014:149185. doi:10.1155/2014/149185
Lee II, Kim JJ (2014) Influence of AKT on progesterone action in endometrial diseases. Biol Reprod 91(3):63. doi:10.1095/biolreprod.114.119255
Li Q, Kannan A, DeMayo FJ, Lydon JP, Cooke PS, Yamagishi H, Srivastava D, Bagchi MK, Bagchi IC (2011) The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 331(6019):912–916. doi:10.1126/science.1197454
Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13(10):616–630. doi:10.1038/nrm3434
Massague J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103(2):295–309
Moller B, Lindblom B, Olovsson M (2002) Expression of the vascular endothelial growth factors B and C and their receptors in human endometrium during the menstrual cycle. Acta Obstet Gynecol Scand 81(9):817–824
Murphy LJ, Gong Y, Murphy LC (1992) Regulation of transforming growth factor gene expression in human endometrial adenocarcinoma cells. J Steroid Biochem Mol Biol 41(3–8):309–314
Nishida M (2002) The Ishikawa cells from birth to the present. Hum Cell 15(3):104–117
Nishida M, Kasahara K, Oki A, Satoh T, Arai Y, Kubo T (1996) Establishment of eighteen clones of Ishikawa cells. Hum Cell 9(2):109–116
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR (1999) Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 59(19):5002–5011
Pietras K, Ostman A (2010) Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res 316(8):1324–1331. doi:10.1016/j.yexcr.2010.02.045
Piver MS, Barlow JJ, Lurain JR, Blumenson LE (1980) Medroxyprogesterone acetate (Depo-Provera) vs. hydroxyprogesterone caproate (Delalutin) in women with metastatic endometrial adenocarcinoma. Cancer 45(2):268–272
Podratz KC, O’Brien PC, Malkasian GD Jr, Decker DG, Jefferies JA, Edmonson JH (1985) Effects of progestational agents in treatment of endometrial carcinoma. Obstet Gynecol 66(1):106–110
Polyak K, Haviv I, Campbell IG (2009) Co-evolution of tumor cells and their microenvironment. Trends Genet 25(1):30–38. doi:10.1016/j.tig.2008.10.012
Quinn MA, Cauchi M, Fortune D (1985) Endometrial carcinoma: steroid receptors and response to medroxyprogesterone acetate. Gynecol Oncol 21(3):314–319
Ripley D, Tang XM, Ma C, Chegini N (2001) The expression and action of granulocyte macrophage-colony stimulating factor and its interaction with TGF-beta in endometrial carcinoma. Gynecol Oncol 81(2):301–309. doi:10.1006/gyno.2001.6161
Sapoznik S, Cohen B, Tzuman Y, Meir G, Ben-Dor S, Harmelin A, Neeman M (2009) Gonadotropin-regulated lymphangiogenesis in ovarian cancer is mediated by LEDGF-induced expression of VEGF-C. Cancer Res 69(24):9306–9314. doi:10.1158/0008-5472.CAN-09-1213
Subramaniam KS, Tham ST, Mohamed Z, Woo YL, Mat Adenan NA, Chung I (2013) Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS One 8(7):e68923. doi:10.1371/journal.pone.0068923
Takehara M, Ueda M, Yamashita Y, Terai Y, Hung YC, Ueki M (2004) Vascular endothelial growth factor A and C gene expression in endometriosis. Hum Pathol 35(11):1369–1375. doi:10.1016/j.humpath.2004.07.020
Tanwar PS, Zhang L, Roberts DJ, Teixeira JM (2011) Stromal deletion of the APC tumor suppressor in mice triggers development of endometrial cancer. Cancer Res 71(5):1584–1596. doi:10.1158/0008-5472.CAN-10-3166
Wu QW, She HQ, Liang J, Huang YF, Yang QM, Yang QL, Zhang ZM (2012) Expression and clinical significance of extracellular matrix protein 1 and vascular endothelial growth factor-C in lymphatic metastasis of human breast cancer. BMC Cancer 12:47. doi:10.1186/1471-2407-12-47
Xu H, Zhang T, Man GC, May KE, Becker CM, Davis TN, Kung AL et al (2013) Vascular endothelial growth factor C is increased in endometrium and promotes endothelial functions, vascular permeability and angiogenesis and growth of endometriosis. Angiogenesis 16(3):541–551. doi:10.1007/s10456-013-9333-1
Yin X, Pavone ME, Lu Z, Wei J, Kim JJ (2012) Increased activation of the PI3K/AKT pathway compromises decidualization of stromal cells from endometriosis. J Clin Endocrinol Metab 97(1):E35–E43. doi:10.1210/jc.2011-1527
Zhang H, Muders MH, Li J, Rinaldo F, Tindall DJ, Datta K (2008) Loss of NKX3.1 favors vascular endothelial growth factor-C expression in prostate cancer. Cancer Res 68(21):8770–8778. doi:10.1158/0008-5472.CAN-08-1912
Acknowledgments
We are grateful to Marshall Caraveo and Dr. Susan Olalekan who assisted with experiments for this study. We are grateful to the Gynecologic Oncology Team, Doreine Carson, Cary Passaglia, and Racher Bers, for their help in consenting patients and obtaining tissues, and the Mouse Histology and Phenotyping Core facilities at the Robert Lurie Cancer Center at Northwestern University for Ki67 and CD31 IHC staining of the tumors. This work was funded by a grant from the National Institute of Health, National Cancer Institute, Grant # RO1CA155513.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Novelty & Impact
This is the first study that demonstrates the influence of benign and cancer-associated stromal cells from the endometrium on hormone-regulated endometrial tumor growth. Given the hormonal dependence of endometrial cancer and the endometrium as a whole, understanding the paracrine actions of stromal cells on tumor cells is key to identifying new targets of therapy, especially for high-grade cancers.
Rights and permissions
About this article

Cite this article
Pineda, M.J., Lu, Z., Cao, D. et al. Influence of Cancer-Associated Endometrial Stromal Cells on Hormone-Driven Endometrial Tumor Growth. HORM CANC 6, 131–141 (2015). https://doi.org/10.1007/s12672-015-0223-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0223-4