
Abstract
Familial primary hyperparathyroidism (FPHPT) may occur due to an underlying germ-line mutation in the MEN1, CASR, or HRPT2/CDC73 genes. The disease may be undiagnosed in the absence of a history suggestive of FHPT. Young PHPT patients (≤45 years of age) are more likely to harbor occult FPHPT. A total of 1,161 (136 were ≤45 years of age) PHPT patients underwent parathyroidectomy from 2001 to 2009. Thirty-four patients declined participation. Sixteen patients were diagnosed in the clinical routine with FPHPT (11 MEN1, four MEN2A, and one HPT-JT) and were not included in the genetic analysis. Eighty-six young (≤45 years of age) patients with clinically non-syndromic PHPT underwent genetic analysis. Sanger sequencing of all coding regions of the MEN1, CASR, and the HRPT2/CDC73 genes was performed. Eight of 86 (9.3%) young patients with clinically non-familial PHPT displayed deleterious germ-line mutations in the susceptibility genes (4 MEN1, 3 CASR, and 1 HRPT2/CDC73). There was one insertion, one deletion, two nonsense, and four missense mutations, all predicted to be highly damaging to protein function and absent in 3,244 control chromosomes. Germ-line mutations in known susceptibility genes within young patients with PHPT, including those diagnosed in the clinical routine, was 24/102 (23.5%; 15 MEN1, four RET, three CASR, and two HRPT2/CDC73). We demonstrate that germ-line inactivating mutations in susceptibility genes are common in young patients with clinically non-familial PHPT. Thus, enhanced use of genetic analysis may be warranted in clinically non-familial young PHPT patients.
Similar content being viewed by others

Introduction
Primary hyperparathyroidism (PHPT) occurs sporadically in the majority of cases. However, familial PHPT may occur as part of multiple endocrine neoplasia type 1 (MEN1), MEN2A, the HPT-jaw tumor syndrome (HPT-JT), familial isolated hyperparathyroidism (FIHPT), and familial hypocalciuric hypercalcemia (FHH) [1–4].
In patients with a germ-line mutation of the MEN1, CASR, or HRPT2/CDC73 genes, PHPT is the dominant feature and typically occurs at a young age, indicating that the most efficient way to identify index patients with familial PHPT is to screen patients with PHPT [5]. The setting in which HPT occurs in MEN2A is by nature different, and medullary thyroid carcinoma is the dominant feature while HPT is generally diagnosed synchronously or after thyroidectomy [6].
MEN1 is thought to be relatively rare (approximately one in 30,000), and a consensus definition of MEN1 is used widely [7]. A MEN1 case exhibits tumors in two of the three principal organs (parathyroid, enteropancreatic endocrine tissue, and anterior pituitary). A germ-line MEN1 gene mutation is identifiable in 85% to 95% of typical MEN1 families [7]. HPT is the most common and usually the initial endocrine manifestation in patients with MEN1, typically presenting between age 25 to 45 years, although significant variation exist among affected kindreds [8]. The underlying germ-line mutation in the MEN1 gene renders the parathyroid glands more susceptible to expressing a tumor after only one somatic mutation (second hit) [1]. Thus, it is not surprising that the parathyroid glands are usually asymmetrically enlarged. Recognition of the MEN1 syndrome is important if the appropriate operative procedure is to be performed. Surgery for HPT in MEN1 patients occurs in several circumstances, including patients with asymptomatic hypercalcemia with an established diagnosis of MEN1, patients with symptomatic hypercalcemia and confirmed MEN1, or in patients who presents with “sporadic/ non-familial” PHPT harboring occult MEN1 [4]. Some MEN1 patients may have only a single macroscopically abnormal gland and may therefore undergo suboptimal surgical resection leading to recurrence and re-operation. The initial surgical procedure of choice in a MEN1 patient with HPT is either subtotal parathyroidectomy or total parathyroidectomy with heterotopic autotransplantation of a portion of the resected parathyroid tissue [4]. Bilateral transcervical thymectomy is also advocated to prevent or treat thymic carcinoid associated with MEN1 as well as to excise intrathymic supranumerary parathyroid glands [4].
HPT-JT syndrome is due to inactivating mutations in the HRPT2/CDC73 gene encoding parafibromin [2]. Similar to MEN1, HPT is the most common feature of the disease, occurring in approximately 80% of adults, with an early onset (mean age, 32 years) [5]. The syndrome is associated with a high incidence of severe hypercalcemia, risk of parathyroid cancer (about 15%), and may be uniglandular [9]. In general, HPT may be treated with resection of grossly enlarged parathyroid glands unless parathyroid cancer is suspected [4].
HPT in the setting of an underlying CASR mutation may either present as FHH or FIHPT [5, 10]. The diagnosis of FHH as well as its distinction from HPT may be challenging. The combination of hypercalcemia, intact PTH levels in the mid-range, hypermagnesemia, relative hypocalciuria, and a family history of hypercalcemia are suggestive of the disease. A definite diagnosis can be made by the demonstration of germ-line-inactivating mutations in the CASR gene [5]. The majority of patients with FHH are asymptomatic and do not benefit from surgical resection of their mildly enlarged parathyroid glands [11, 12]. However, some individuals with FHH develop symptomatic HPT [13, 14]. Inactivating mutations of the CASR may lead to atypical phenotypic features including hypercalcemia, inappropriately high serum PTH and magnesium levels, and relative hypercalciuria, with nephrolithiasis in a subset of patients which resembles PHPT [10, 15]. Traditionally, a urinary calcium/creatinine (Ca/Cr) clearance ratio is used to distinguish between PHPT and FHH, with the latter having a ratio below 0.010, although modest overlap exists [16].
We demonstrate that germ-line-inactivating mutations in disease-causing genes are common in young patients with clinically non-familial PHPT.
Materials and Methods
The study design is presented in Fig. 1a. A prospective database identified 1,161 primary HPT patients who underwent parathyroidectomy from 2001 to 2009, where follow-up data were available. A total of 136 patients were 45 years old or younger at time of operation and were considered young. This age cut-off was used since most patients with FHPT are diagnosed prior to the age of 45 years [5, 17–19]. Thirty-four patients declined to participate in genetic analysis. Sixteen patients were diagnosed in the clinical routine (genetic testing preformed outside of the study based upon history and physical) with FPHPT preoperatively or during postoperative surveillance (11 MEN1, four MEN2A, and one HPT-JT) and were not included in the genetic analysis. Eighty-six young (≤45 years of age) patients with clinically non-familial PHPT were included and underwent genetic analysis. The diagnosis of clinically non-familial disease was based on a detailed family history obtained by the referring physician (internist/endocrinologist) and operating surgeon prior to surgery. The personal and family history was updated at the time of enrollment in the study (6–120 months after surgery). The family history was obtained to identify FPHPT with questions such as “Are there blood relatives who have had neck surgery, kidney stones, brain tumors (i.e., pituitary), ulcers, high calcium levels, jaw tumors, pancreatic tumors?”. All 86 patients denied a personal or family history suggestive of FPHPT.

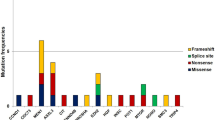
a Study design. b Summary of detected mutations
Patients underwent either minimally invasive parathyroidectomy (MIP) or standard cervical exploration, as described [20]. Intraoperative PTH (IOPTH) was used in all cases, with a greater than 50% reduction from the preoperative baseline with an absolute normalization of the PTH levels being consistent with curative resection. Informed consent was obtained and the study design approved by the local institutional review board.
Amplification of All Coding Regions of MEN1, CASR, and HRPT2
Whole blood was obtained through phlebotomy and genomic DNA from leukocytes was isolated by standard methods [21]. Two hundred nanograms of genomic DNA is polymerase chain reaction (PCR)-amplified using specific primers for exons 1–10 of the MEN1 gene, exons 1–10 of the CASR gene, and exons 1–17 of the HRPT2/CDC73 gene, as described previously [13, 21, 22]. Primer sequences are provided in supplementary Table 1.
DNA Sequencing and Analysis
The PCR products were purified using the Qiagen PCR purification kit (Qiagen, Valencia, CA) according to the instructions from the manufacturer. Each sample (40 ng of DNA) underwent both forward and reverse Sanger sequencing at the Keck DNA Sequencing Facility at Yale University. Data were analyzed with the use of Sequencing Analysis and AutoAssembler software (Applied Biosystems) and publically available Web-based resources (NCBI-Blast, GenBank). All novel DNA variants were verified by re-sequencing using genomic DNA from a separate extraction. All variants were analyzed for predicting damaging effects of missense mutations using the PolyPhen 2 software (http://genetics.bwh.harvard.edu/pph2), as described [23]. The frequency of single-nucleotide polymorphisms (SNP) in the examined cases was compared with the HapMap CEU population (http://www.ncbi.nlm.nih.gov/projects/SNP).
Statistical Analysis
Student’s unpaired t test and Chi2 test were used for statistical evaluation, with p < 0.05 considered to be significant. All results are expressed as mean±SEM (standard error of the mean).
Results
The clinical characteristics of the 86 young patients with clinically non-syndromic PHPT are displayed in Table 1. Compared with PHPT patients presenting at age 45 years or older, young PHPT patients were significantly more often male, had symptoms of nephrolithiasis, and absence of bone disease. The biochemical indices of the disease, frequency of neurocognitive symptoms, type of operative intervention, and histopathological characteristics were similar in young and old patients.
Sanger sequencing of all coding regions of MEN1, CASR, or HRPT2/CDC73 genes identified germ-line mutations (four MEN1, three CASR, and one HRPT2/CDC73) in eight cases of clinically sporadic PHPT (Fig. 1b). There was one insertion, one deletion, two nonsense, and four missense mutations. All four missense mutations were predicted to be highly damaging to protein function with PolyPhen-2 scores ranging between 0.899 and 0.998 (Table 2). None of the missense variants were present in 3,244 control chromosomes. Additionally, two of the MEN1 gene missense mutations (E45G and P193L) have previously been described [24, 25]. The frequency of genotypes in commonly occurring SNPs of the MEN1, CASR, and HRPT2/CDC73 genes in the current cohort (mainly of Western European descent) were similar to the HapMap CEU population (Utah residents of Northern and Western European ancestry; supplementary Table 2).
Of the two patients with novel MEN1 gene mutations, both required multiple gland removal during MIP and an appropriate decrease in IOPTH leading to normocalcemia at present (Table 2). Case #96 presented with hypercalcemic crisis during pregnancy and underwent a subtotal parathyroidectomy (three excised glands) in the second trimester causing significant improvement of hypercalcemia and successful delivery at term. Eighteen months later, she underwent reoperation due to progressive PHPT with removal of two enlarged glands (i.e., glands 4 and 5) with heterotopic autotransplantation and is currently normocalcemic (calcium 8.8 mg/dl; reference range, 8.8–10.2 mg/dl; PTH 12 pg/ml, reference range, 10–65 pg/ml). Case #349 underwent a subtotal parathyroidectomy (3.5 glands) in 2001, followed by reoperation with removal remnant fourth gland and fifth mediastinal gland with heterotopic autotransplantation.
Interestingly, all three patients with germ-line CASR gene mutations displayed biochemical indices consistent with PHPT and 2/3 had osteoporosis. The 24-h urine collections were within the normal range, and the Ca/Cr clearance was 0.010–0.011. Standard four gland exploration was required in all cases and subtotal parathyroidectomy (two glands in two cases and 3.5 glands in one case) caused an appropriate IOPTH decline, normocalcemia, and normal PTH levels during follow up (12–18 months). The 16-year-old male with HRPT2/CDC73 mutation presented with hypercalcemic crisis requiring intravenous saline administration and excision of a single 4,500 mg parathyroid adenoma lead to IOPTH normalization and normocalcemia at present (30 months follow-up).
The overall prevalence of germ-line mutations in known disease-causing genes in young patients with PHPT, including those diagnosed in the clinical routine was 24/102 (23.5%; 15 MEN1, four RET, three CASR, and two HRPT2/CDC73).
Discussion
We demonstrate that germ-line-inactivating mutations in CASR, MEN1, and HRPT2/CDC73 are common in young patients with clinically non-familial PHPT. Parathyroid surgery in patients with MEN1 and HRPT2/CDC73 gene mutations should be conceptualized as a debulking or palliative procedure since recurrence is inevitable if survival is unlimited and is indicated to treat and prevent the complications of HPT. Thus, it is likely that the patients presented here will recur in the future and thus require life-long follow-up. The role of parathyroid surgery in CASR gene carriers is controversial and evolving, and it is unclear whether achieving normocalcemia has any benefit in reducing the complications of HPT. It is noteworthy that all three CASR gene carriers remain normocalcemic. When multiglandular disease is discovered at the time of surgery, the surgeon must suspect familial hyperparathyroidism, although the majority of patients with PHPT and multiglandular disease do not have familial disease.
Current guidelines suggest that DNA sequence testing for mutations of CASR, MEN1, and HRPT2/CDC73 genes is not recommended on a routine basis unless FPHPT is suspected [26]. The prevalence of germ-line mutations in disease-causing genes in young PHPT with apparent non-familial disease was previously unknown. The diagnosis of FPHPT is of crucial importance for a number of reasons. It affects the operative strategy in CASR, MEN1, and HRPT2/CDC73 carriers and standard cervical exploration rather than MIP would be preferred [4]. The risk of parathyroid carcinoma in HRPT2/CDC73 gene carriers and the high incidence of supranumerary parathyroid glands and concomitant thymic carcinoid in MEN1 gene carriers have to be considered [4]. The majority of CASR gene carriers does not benefit from parathyroid surgery and could be avoided. DNA sequencing CASR, MEN1, and HRPT2/CDC73 young PHPT patients with clinically non-familial disease may confirm a syndrome in a proband, and exon-specific gene testing could be offered to family members. Such knowledge would provide crucial information to affected and non-affected family members with regard to further surveillance and treatment of the tumor susceptibility syndromes [26].
The current study represents the most comprehensive evaluation of germline mutations in susceptibility genes for PHPT in young patients with clinically non-familial PHPT. Nevertheless, the study has some weaknesses. Only cases of operatively verified PHPT were included in the study. The prevalence of CASR, MEN1, and HRPT2/CDC73 gene mutations in young PHPT not referred for parathyroidectomy was not assessed in this study. However, we anticipate that most young patients (≤45 years of age) are referred for surgery since the NIH guidelines recommends parathyroidectomy for all PHPT under the age of 50 years, even when asymptomatic [27]. Similarly, the prevalence of CASR, MEN1, and HRPT2/CDC73 gene mutations in non-familial PHPT >45 years of age was not evaluated in the current study. The rationale to use 45 years of age as a cut-off for “young” patients relates to the fact that the mean age of presentation for PHPT of MEN1 is 25 to 45 years of age and 32 years of age for PHPT of HPT-JT syndrome, although patients can present as young as 10–15 years of age [28]. Individuals with inherited inactivating CASR gene mutations display hypercalcemia at birth. However, the mean age of parathyroidectomy in a large family with an atypical CASR gene mutation causing a phenotype which resembles PHPT was 44 years of age [29]. Studies evaluating the frequency of MEN1 gene germline mutations in PHPT patients have previously used an arbitrary cut-off of 30 years of age [25, 30]. These studies, however, did not analyze the prevalence of CASR and HRPT2/CDC73 gene mutations; the number of analyzed subjects was relatively low. In addition, it is well known that the incidence of primary HPT increases with age especially in women at the time of menopause (approximately age 45 years) and when postmenopausal [31]. Thus, although arbitrary, we found age 45 years to be a reasonable cut-off for the definition of “young”. The high prevalence of FPHPT in the current cohort may relate to referral bias of cases to a specialized parathyroid surgery center. This may be true for the 16 cases of FPHPT diagnosed in the clinical routine but is unlikely to have played a role in the 8/86 (9.3%) cases displaying occult CASR, MEN1, and HRPT2/CDC73 gene mutations. In the current study, we did not evaluate presence of germline mutations in the kindreds of carriers and thus, do not know whether the detected mutations represents inherited versus de novo mutations. Further analyses are warranted to determine the prevalence of de novo germline mutations of the CASR, MEN1, and HRPT2/CDC73 genes in young patients with clinically non-familial PHPT. Additionally, the current study did not evaluate genes in the family of cyclin-dependent kinase inhibitors that have been shown to be rarely mutated in MEN1 [32]. The setting in which HPT occurs in MEN2A (due to germline RET mutation) is by nature different, and medullary thyroid carcinoma is the dominant feature. Thus, we found no reason for analyzing the RET gene in the cohort of 86 patients with clinically non-familial young PHPT patients.
Given the high prevalence of germ-line mutations in clinically non-familial young PHPT patients, a more liberal use of DNA testing may be advocated for these patients even in the absence of a suspicious family history. This may aid the operating physician in their planning of the overall operative procedure, potentially preventing the need for recurrent disease and improved evaluation of the kindred.
References
Heppner C, Kester MB, Agarwal SK, Debelenko LV, Emmert-Buck MR, Guru SC, Manickam P, Olufemi S-E, Skarulis MC, Doppman JL, Alexander RH, Kim YS, Saggar SK, Lubensky IA, Zhuang Z, Liotta LA, Chandrasekharappa SC, Collins FS, Speigel AM, Burns AL, Mark SJ (1997) Somatic mutations of the MEN1 gene in parathyroid tumours. Nature Genet 16:375–378
Carpten JDRC, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, Agarwal SK, Sood R, Jones MP, Moses TY, Haven C, Petillo D, Leotlela PD, Harding B, Cameron D, Pannett AA, Hoog A, Heath H, James-Newton LA, Robinson B, Zarbo RJ, Cavaco BM, Wassif W, Perrier ND, Rosen IB, Kristoffersson U, Turnpenny PD, Farnebo LO, Besser GM, Jackson CE, Morreau H, Trent JM, Thakker RV, Marx SJ, Teh BT, Larsson C, Hobbs MR (2002) HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet 32:676–680
Pollak MR, Brown EM, Chou YH, Hebert SC, Marx SJ, Steinmann B, Levi T, Seidman CE, Seidman JG (1993) Mutations in the human Ca(2+)-sensing receptor gene cause familial hypocalciuric hypercalcemia and neonatal severe hyperparathyroidism. Cell 75:1297–1303
Stalberg P, Carling T (2009) Familial parathyroid tumors: diagnosis and management. World J Surg 33:2234-2243
Marx SJSW, Agarwal SK, Burns AL, Weinstein LS, Cochran C, Skarulis MC, Spiegel AM, Libutti SK, Alexander HR Jr, Chen CC, Chang R, Chandrasekharappa SC, Collins FS (2002) Hyperparathyroidism in hereditary syndromes: special expressions and special managements. J Bone Miner Res Suppl 2:N37–N43
Skinner MA, Moley JA, Dilley WG, Owzar K, Debenedetti MK, Wells SA Jr (2005) Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 353:1105–1113
Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ (2001) Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 86:5658–5671
Skogseid B, Rastad J, Oberg K (1994) Multiple endocrine neoplasia type 1. Clinical features and screening. Endocrinol Metab Clin North Am 23:1–18
Wassif WS, Moniz CF, Friedman E, Wong S, Weber G, Nordenskjöld M, Peters TJ, Larsson C (1993) Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab 77:1485–1489
Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, Marx SJ (2002) Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore) 81:1–26
Thorgeirsson U, Costa J, Marx SJ (1981) The parathyroid glands in familial hypocalciuric hypercalcemia. Hum Pathol 12:229–237
Marx SJ, Stock JL, Attie MF, Downs R Jr, Gardner DG, Brown EM, Spiegel AM, Doppman JL, Brennan MF (1980) Familial hypocalciuric hypercalcemia: recognition among patients referred after unsuccessful parathyroid exploration. Ann Intern Med 92:351–356
Carling T, Szabo E, Bai M, Ridefelt P, Westin G, Gustavsson P, Trivedi S, Hellman P, Brown EM, Dahl N, Rastad J (2000) Familial hypercalcemia and hypercalciuria caused by a novel mutation in the cytoplasmic tail of the calcium receptor. J Clin Endocrinol Metab 85:2042–2047
Guarnieri V, Canaff L, Yun FH, Scillitani A, Battista C, Muscarella LA, Wong BY, Notarangelo A, D’Agruma L, Sacco M, Cole DE, Hendy GN (2010) Calcium-sensing receptor (CASR) mutations in hypercalcemic states: studies from a single endocrine clinic over three years. J Clin Endocrinol Metab 95:1819–1829
Carling T, Rastad J, Szabo E, Westin G, Åkerström G (2000) Reduced vitamin D receptor mRNA levels in parathyroid glands of primary and secondary hyperparathyroidism. J Clin Endocrinol 85:2000–2003
Christensen SE, Nissen PH, Vestergaard P, Heickendorff L, Rejnmark L, Brixen K, Mosekilde L (2008) Plasma 25-hydroxyvitamin D, 1,25-dihydroxyvitamin D, and parathyroid hormone in familial hypocalciuric hypercalcemia and primary hyperparathyroidism. Eur J Endocrinol 159:719–727
Sharretts JM, Simonds WF (2010) Clinical and molecular genetics of parathyroid neoplasms. Best Pract Res Clin Endocrinol Metabol 24:491–502
Oucharek JJ, O’Neill CJ, Suliburk JW, Sywak MS, Delbridge LW, Sidhu SB (2011) Durability of focused minimally invasive parathyroidectomy in young patients with sporadic primary hyperparathyroidism. Ann Surg Oncol 18:1290–1292
Skandarajah A, Barlier A, Morlet-Barlat N, Sebag F, Enjalbert A, Conte-Devolx B, Henry JF (2010) Should routine analysis of the MEN1 gene be performed in all patients with primary hyperparathyroidism under 40 years of age? World J Surg 34:1294–1298
Carling T, Udelsman R (2008) Focused approach to parathyroidectomy. World J Surg 32:1512-1517
Carling T, Correa P, Hessman O, Hedberg J, Skogseid B, Lindberg D, Rastad J, Westin G, Akerstrom G (1998) Parathyroid MEN1 gene mutations in relation to clinical characteristics of nonfamilial primary hyperparathyroidism. J Clin Endocrinol Metab 83:2960–2963
Shattuck TM, Costa J, Bernstein M, Jensen RT, Chung DC, Arnold A (2002) Mutational analysis of Smad3, a candidate tumor suppressor implicated in TGF-beta and menin pathways, in parathyroid adenomas and enteropancreatic endocrine tumors. J Clin Endocrinol Metab 87:3911–3914
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S, Albarello L, Doglioni C, Schott C, Capelli P, Chilosi M, Boninsegna L, Becker KF, Falconi M, Scarpa A (2010) MEN1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Canc 17:771–783
Kihara M, Miyauchi A, Ito Y, Yoshida H, Miya A, Kobayashi K, Takamura Y, Fukushima M, Inoue H, Higashiyama T, Tomoda C (2009) MEN1 gene analysis in patients with primary hyperparathyroidism: 10-year experience of a single institution for thyroid and parathyroid care in Japan. Endocr J 56:649–656
Eastell R, Arnold A, Brandi ML, Brown EM, D’Amour P, Hanley DA, Rao DS, Rubin MR, Goltzman D, Silverberg SJ, Marx SJ, Peacock M, Mosekilde L, Bouillon R, Lewiecki EM (2009) Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab 94:340–350
Bilezikian JP, Khan AA, Potts JT Jr (2009) Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab 94:335–339
Howell VM, Zori RT, Stalker HJ, Williams C, Jesse N, Nelson AE, Robinson BG, Marsh DJ (2004) A molecular diagnosis of hyperparathyroidism-jaw tumor syndrome in an adolescent with recurrent kidney stones. J Pediatr 145:567
Szabo E, Carling T, Hessman O, Rastad J (2002) Loss of heterozygosity in parathyroid glands of familial hypercalcemia with hypercalciuria and point mutation in calcium receptor. J Clin Endocrinol Metab 87:3961–3965
Cardinal JW, Bergman L, Hayward N, Sweet A, Warner J, Marks L, Learoyd D, Dwight T, Robinson B, Epstein M, Smith M, Teh BT, Cameron DP, Prins JB (2005) A report of a national mutation testing service for the MEN1 gene: clinical presentations and implications for mutation testing. J Med Genet 42:69–74
Lundgren E, Rastad J, Thrufjell E, Akerström G, Ljunghall S (1997) Population-based screening for primary hyperparathyroidism with serum calcium and parathyroid hormone values in menopausal women. Surgery 121:287–294
Agarwal SK, Mateo CM, Marx SJ (2009) Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab 94:1826–1834
Acknowledgments
We thank participating patients and referring physicians who aided in data collection. R.P.L. is an Investigator of the Howard Hughes Medical Institute. T.C. is a Doris Duke-Damon Runyon Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation and the Doris Duke Charitable Foundation.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 172 kb)
Rights and permissions
About this article
Cite this article
Starker, L.F., Åkerström, T., Long, W.D. et al. Frequent Germ-Line Mutations of the MEN1, CASR, and HRPT2/CDC73 Genes in Young Patients with Clinically Non-familial Primary Hyperparathyroidism. HORM CANC 3, 44–51 (2012). https://doi.org/10.1007/s12672-011-0100-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-011-0100-8