
Abstract
The centrosome that functions as a microtubule organizing center of a cell plays a key role in formation of bipolar mitotic spindles. Cells normally have either one (unduplicated) or two (duplicated) centrosomes. However, loss of the mechanisms controlling the numeral integrity of centrosomes leads to centrosome amplification (presence of more than two centrosomes), primarily via overduplication or fragmentation of centrosomes, resulting in defective mitosis and consequentially chromosome instability. Centrosome amplification frequently occurs in various cancers, and is considered as a major cause of chromosome instability. It has recently been found that ROCK2 kinase plays a critical role in promotion of centrosome duplication and amplification. Considering that ROCK2 is activated by Rho protein, and Rho is the immediate downstream target of many growth and hormone receptors, it is possible that such receptors may rather directly affect centrosome duplication and amplification. Indeed, constitutive activation of the receptors known to signal to the Rho pathway leads to promotion of centrosome amplification and chromosome instability in the Rho-ROCK2 pathway-dependent manner. These observations reveal an unexplored, yet important, oncogenic activities of those receptors in carcinogenesis; destabilizing chromosomes through promotion of centrosome amplification via continual activation of the Rho-ROCK2 pathway.
Similar content being viewed by others


Introduction
For a normal cell to become a fully malignant cell, a number of specific genes must be altered, and each genetic alteration alone or in combination renders one or more malignant phenotypes [1]. Destabilization of chromosomes, which is equated to defective mitosis, profoundly affects tumor progression, since loss or gain of even a single chromosome can simultaneously introduce multiple genetic alterations (i.e., loss of tumor suppressor genes, gain of proto-oncogenes, etc.). The centrosome functions to organize microtubules, hence playing a critical role in the formation of proper mitotic spindles [2–4]. The presence of more than two centrosomes (centrosome amplification) leads to formation of defective mitotic spindles and consequentially chromosome segregation errors [2–4]. Many studies have shown the frequent occurrence of centrosome amplification in various types of human cancers [5, 6]. Because of a strong association between the centrosome amplification and aneuploidy, centrosome amplification is believed to be a major cause of chromosome instability in cancer cells [2–7].
Upon cytokinesis, each daughter cell receives only one centrosome, and thus the centrosome, like DNA, must duplicate once during the cell cycle. Aberrant activation of the positive regulators and loss/inactivation of the negative regulators of centrosome duplication lead to centrosome overduplication (more than one duplication in a single cell cycle). As expected from the close relationship between the cell cycle and centrosome duplication, many cell cycle regulators, oncogenic proteins and tumor suppressor proteins have been shown to be involved in the regulation of centrosome duplication either positively or negatively [4]. It has recently been found that ROCK2 kinase (also known as ROKα or Rho-kinase) plays an essential role in the initiation of centrosome duplication, and aberrant activation of ROCK2 results in centrosome amplification [8]. The ROCK2 kinase activity in respect to centrosome duplication and centrosome amplification is regulated by two upstream pathways; the Rho pathway and cyclin-dependent kinase 2 (CDK2)-nucleophosmin (NPM/B23) pathway. These two pathways converge to “superactivate” ROCK2, which in turn rapidly triggers centrosome duplication, and the uncontrolled activity of either pathway leads to centrosome amplification. Although it had been recognized that the oncogenic (constitutive) activation of certain growth, cytokine, and hormone receptors leads to chromosome instability, the phenomenon had not received much attention as it was generally assumed to be an indirect consequence of aberrant growth, cytokine, or hormone receptor activation. However, the finding of the involvement of ROCK2 in the regulation of centrosome duplication and induction of centrosome amplification predicts a critically important oncogenic activity of the Rho-activating receptors, namely destabilizing chromosomes via induction of centrosome amplification through activating ROCK2. Here, such a possibility will be discussed in connection with the induction of centrosome amplification by the other cell cycle regulators, especially the CDK2-NPM/B23 pathway.
Centrosome Amplification, Chromosome Instability, and Cancer
The centrosome is a small non-membranous organelle (1–2 μm in diameter) normally found at the periphery of a nucleus. The primary function of the centrosome is to nucleate and anchor microtubules, and thus the centrosome is often referred to as microtubule organizing center. The centrosome consists of a pair of centrioles and many different proteins surrounding the centrioles (pericentriolar materials or PCM) (Fig. 1a). During interphase, centrosomes organize the cytoplasmic microtubule network (Fig. 1b), while during mitosis, centrosomes form spindle poles and direct the formation of mitotic spindles (Fig. 1c).

Structure and function of centrosome. The centrosome consists of a pair of centrioles and surrounding protein aggregates (PCM) (a). During interphase, centrosomes organize cytoplasmic microtubule networks (b), and during mitosis, centrosomes direct the formation of mitotic spindles
Duplication of centrosomes occurs in coordination with DNA replication [9]. Centrosome duplication is initiated in late G1/early S phases by physical separation of the paired centrioles, followed by formation of a procentriole in the proximity of each pre-existing centriole. During S and G2 phases, the procentrioles elongate and continue to mature by progressively recruiting PCM, and by late G2, two mature centrosomes are generated (Fig. 2). The coupling of initiation of DNA and centrosome duplication is at least in part achieved by the late G1-specific activation of CDK2-cyclin E. CDK2-cyclin E, which is known for its activity to trigger DNA synthesis [10], has been found to also trigger centrosome duplication [11–14], providing the primary ground for the coupling of these two events.

The centrosome (centriole) duplication cycle. CDK2/cyclin E triggers initiation of both DNA replication and centrosome duplication in late G1. Centrosome duplication begins with the physical splitting of paired centrioles, followed by formation of procentrioles near each pre-existing centriole. During S–G2, procentrioles elongate, and centrosomes progressively recruit PCM. In late G2, two mature centrosomes are generated
Recently, the mechanism of how amplified centrosomes are generated has become clear, in which p53 plays a critical role [3, 4]. Proliferating cells, even under an optimal growth condition, often suffer physiological stress that temporarily halts cell cycling irrespective of the p53 status (e.g., imbalance/deprivation of critical molecules such as dNTPs). Cells inflicted with DNA damage (e.g., irradiation, exposure to genotoxic drugs, formation of chemical adducts) also become arrested in S and late G2 phases in a p53-dependent and p53-independent mannor [15]. The cell cycle arrest provides time for the duplicated centrosomes to reduplicate if active CDK2 is available. However, in normal cells, p53 is upregulated in response to the physiological stress from the prolonged arrest by the ARF-mediated inhibition of Mdm2, a protein that promotes p53 degradation [16] as well as in response to DNA damage by the ATM/ATR- and Chk1/Chk2-mediated phosphorylation [17], leading to upregulation of p21Waf1/Cip1, which effectively inhibits CDK2 [18]. Because initiation of centrosome duplication requires the CDK2 activity, initiation of centrosome duplication or reduplication of the duplicated centrosomes is blocked in those arrested cells. In contrast, in cells lacking functional p53, CDK2 activity is unchecked, leading to fortuitous reduplication of centrosomes. Once the stress causing problem is resolved, cells resume cell cycling in the presence of amplified centrosomes, and suffer defective mitoses and chromosome segregation errors.
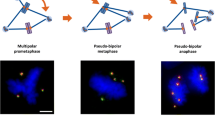
Cells with amplified centrosomes frequently form multiple (>2) spindle poles. Tripolar spindles (Fig. 3a(a)) normally undergo cytokinesis, and some daughter cells are viable yet suffer severe aneuploidy, while some may undergo cell death due to detrimental karyotypic changes. Cells with the spindles with >3 poles (Fig. 3a(b)) usually fail to undergo cytokinesis, and either become binucleated/large mononucleated cells with doubling of genome or die due to mitotic catastrophe. Since the presence of polyploid chromosomes is known to destabilize chromosomes, those cells that have escaped the mitotic catastrophe will suffer chromosome segregation errors due to polyploidy as well as the presence of amplified centrosomes. Cells with amplified centrosomes also frequently form “pseudo-bipolar” spindles (Fig. 3a(c))—all amplified centrosomes position on a bipolar axis, which is known as “centrosome clustering,” forming spindles that structurally resemble the “true” bipolar spindles organized by two centrosomes. Although the cells with pseudo-bipolar spindles usually undergo normal cytokinesis without chromosome segregation errors, some cells with the pseudo-bipolar spindles suffer chromosome segregation errors (Fig. 3a(d), b) [7]: one or a few amplified centrosomes fail to cluster and position on the bipolar axis, yet they are functionally intact, nucleating microtubules which capture chromosomes. Depending on which daughter cell receives these chromosomes, aneuploid cells can be generated.

Abnormal mitosis caused by amplified centrosomes. a p53-null cells were immunostained for γ-tubulin (centrosome) and microtubules; (a) tripolar spindles, (b) multi (>3)-polar spindles, (c) pseudo-bipolar spindles, (d) Pseudo-bipolar spindles with a malpositioned spindle pole (indicated by an arrow). b Risk of chromosome destabilization by pseudo-bipolar spindles. One or a few centrosomes fail to position on the bipolar axis, which nucleate microtubules that capture chromosomes. Depending on whether those chromosomes are segregated into one or the other daughter cell, aneuploid cells may be generated
Numerous studies have shown the frequent occurrence of centrosome amplification in various types of cancers, including breast, lung, head and neck, prostate, colon, brain, liver, pancreas, to name a few [19–25]. Moreover, aberrant mitotic spindles organized by abnormally amplified centrosomes are frequently observed in those tumors, and the occurrence of centrosome amplification is strongly associated with aneuploidy [3, 6]. Through the examination of bladder carcinoma tissue specimens [19], we found a strong association between tumor stages and the degrees of centrosome amplification; the higher the tumor stage, the higher the frequency of centrosome amplification that is observed in the tumors (representative immunostaining images of centrosomes in the bladder cancer tissues are shown as an example in Fig. 4).

Centrosome amplification in human bladder cancer tissues. The touch preparations of bladder cancer specimens and adjacent normal epithelium samples were immunostained with anti-γ-tubulin antibody (centrosome), and counterstained with DAPI (DNA). Original magnification, ×600
CDK2 and Its Downstream Events in the Regulation of Centrosome Duplication and Promotion of Centrosome Amplification
As a key initiator of centrosome duplication, CDK2 also plays a critical role in generation of abnormally amplified centrosomes in cancer cells. For instance, when CDK2 is overactivated by ectopic expression of cyclin E, centrosome amplification occurs at a high frequency, especially in cells with p53 mutation [19, 26]. Several potential targets of CDK2 in the initiation of centrosome duplication have been identified, including nucleophosmin (NPM/B23), Mps1 kinase and CP110 [27–29]. NPM/B23 is a multifunctional protein, often recognized as a molecular chaperoning protein [30] and implicated in diverse cellular functions, including pre-rRNA and pre-mRNA processing, DNA replication, nucleocytoplasmic protein trafficking, and centrosome duplication [31–38]. CDK2-cyclin E phosphorylates NPM/B23 on Thr199, and this phosphorylation is essential for centrosomes to initiate duplication [37]. Through the analysis of the proteins that localize to centrosomes, ROCK2 kinase was found to form a complex with NPM/B23, especially with a high affinity to NPM/B23 phosphorylated on Thr199 [8]. ROCK2 is a member of the ROCK Ser/Thr kinase family and the major effector of small GTPase Rho. ROCK2 is primed for activation by binding of the GTP-bound Rho (Rho-GTP) to the C-terminal region, which disrupts the interaction between the kinase domain and autoinhibitory domain of ROCK2, resulting in 1.5 to twofold increase in the kinase activity [39–41]. However, the binding of NPM/B23 results in further activation of ROCK2 (five to tenfold) [8]. Although ROCK2 forms a complex with unphosphorylated NPM/B23 in vitro, the CDK2-mediated phosphorylation of NPM/B23 (NPM/B23’s acquisition of a high binding affinity to ROCK2) appears to be essential for the ROCK2-NPM/B23 interaction in vivo, likely because of the limited concentration of the proteins, especially of the Rho-bound ROCK2.
We tested the role of ROCK2 in the induction of centrosome amplification by the centrosome reduplication assay, a commonly used assay system for testing the protein of interest for its activity to affect centrosome amplification (Fig. 5). In this assay, cells are arrested at the centrosome duplication permissive stage by exposure to DNA synthesis inhibitors such as aphidicolin (Aph) or hydroxyurea (HU). In these cells, centrosomes continue to duplicate without DNA synthesis, resulting in centrosome amplification. However, this phenomenon occurs preferentially in cells with impaired p53 function as described earlier: in the presence of functional p53, p21 is upregulated in response to the stress associated with exposure to DNA synthesis inhibitors, which inhibits CDK2-cyclin E, and thus centrosome duplication/reduplication cannot be effectively initiated. Thus, the use of the cells compromised for p53 function is important for this assay. In this assay system, either the protein of interest (protein-X) or the siRNA sequence specific for protein-X will be transfected into the cells pre-arrested with Aph or HU. The use of the pre-arresting cells is often critical for circumventing the possibility of the cell cycle arrest at the stage where centrosome duplication is not permissive by overexpression/knockdown of protein-X and for alleviating the potential toxicity associated with overexpression/knockdown of protein-X (arrested cells are known to be more resistant to such toxicity). The transfected cell is further exposed to Aph or HU and the frequency of centrosome amplification is determined. If protein-X promotes centrosome amplification, the protein-X-transfected cells should show a higher frequency of centrosome amplification than the control cells, and the cells silenced for protein-X expression should show a lower frequency of centrosome amplification than the control cells. In contrast, if protein-X suppresses centrosome amplification, the protein-X-transfected cells should show a lower frequency of centrosome amplification than the control cells, and the cells silenced for protein-X expression should show a higher frequency of centrosome amplification than the control cells. It is important to note here that in this assay system, the control cells must be permissive for centrosome amplification, and thus they show the basal level of centrosome amplification frequency (10–40% depending on the cell types). However, the time required for duplicated centrosomes to re-initiate centrosome duplication is consistent in each particular cell line, and thus there is little deviation in the frequency of centrosome amplification from experiment to experiment as long as the same cell line is used.

Centrosome reduplication (amplification) assay. Cells with impaired 53 function are first pre-arrested at centrosome duplication permissive phases of the cell cycle by exposure to either Aph or HU for 24 h. Cells are then transfected with either protein-X (a) or siRNA sequence for protein-X (b). The transfected cells are further cultured in the presence of Aph or HU and examined for the number of centrosomes
Using the centrosome reduplication assay, the constitutively active ROCK2 mutant lacking the autoinhibitory region (CAT mutant), the kinase-dead CAT (CAT-KD), NPM/B23-binding mutant CAT (CATΔNPM), and centrosome localization mutant CAT (CATΔcen) (Fig. 6a) were tested for their activities to promote centrosome amplification (Fig. 6b). When compared with the control cells (~45%), >80% of the CAT-transfected cells underwent centrosome reduplication, demonstrating that ROCK2 possesses the activity to promote centrosome amplification. However, cells transfected with CAT-KD, CATΔNPM, or CATΔcen all showed similar frequencies of centrosome amplification with the control cells, indicating that promotion of centrosome amplification by ROCK2 depends on its kinase activity, NPM/B23 binding, and centrosome localization. We next transfected the pre-arrested cells with either ROCK2 or control siRNA sequence, and the transfected cells were subjected to the centrosome reduplication assay (Fig. 6c). The ROCK2 knockdown cells show a significant reduction in the frequency of centrosome amplification compared with the control cells, indicating that ROCK2 is essential for induction of centrosome amplification.

Role of ROCK2 in centrosome amplification. a Diagram of the CAT and various ROCK2 mutants. CATΔNPM lacks the NPM/B23-interaction domain (a.a. 373–420). CATΔcen lacks the centrosome localization domain (a.a. 457–553). (b) For the centrosome reduplication assay, we used NIH3T3 cells that are partially defective in the p53-dependent checkpoint function. Cells pre-arrested with Aph for 24 h were transfected with CAT, CAT-KD, CATΔNPM or CATΔcen, and were subjected to the centrosome reduplication assay, and the frequencies of centrosome amplification were determined. c Cells pre-arrested with Aph for 24 h were transfected with either ROCK2 siRNA or control siRNA sequence. The inset in the top right shows the immunoblot analysis of ROCK2. The transfected cells were subjected to the centrosome reduplication assay, and the frequencies of centrosome amplification were shown in the graph
The Role of the Upstream Proteins of ROCK2 in the Induction of Centrosome Amplification
The activity of ROCK2 is controlled by Rho GTPases. Rho is active when bound to GTP, while it is inactive when bound to GDP [42]. Rho binds to ROCK2 for activation by disrupting the negative regulatory interaction between the C-terminal autoinhibitory and the N-terminal kinase domains [43]. There are three major Rho isoforms, RhoA, B, and C, which share 85% sequence identity. Because all isoforms are known to activate ROCK2 in vitro [44], we tested which Rho isoform(s) are important for promotion of centrosome amplification in cells [45]. When cells transfected with the siRNA sequence specific for each Rho isoform were tested by the centrosome reduplication assay, we found that silencing of RhoA alone and RhoC alone both resulted in suppression of centrosome reduplication, while depletion of RhoB alone had no effect [45], indicating that both RhoA and RhoC, but not RhoB, are essential for centrosome duplication. This finding raises a question of whether there are any functional differences between RhoA and RhoC to control centrosome duplication. When the constitutively active RhoA and RhoC mutants are overexpressed in cells, both promoted centrosome amplification effectively [45]. However, the constitutively active RhoA and RhoC mutants harboring a point mutation that disrupts the ability of ROCK2 recognition [46, 47] failed to promote centrosome amplification [45], indicating that ROCK2 is a primary target of both RhoA and RhoC to promote centrosome amplification. It is possible that both RhoA and C contribute to the total pool of cellular Rho activity that induces and promotes centrosome amplification, and that depletion of either one reduces the total Rho levels to such an extent that the overall level becomes limiting to drive centrosome amplification. However, it remains possible that, although ROCK2 is the primary target of RhoA and RhoC, they may also possess their own unique functions and targets for induction and/or promotion of centrosome amplification. Considering that overactivation of RhoA as well as RhoC is commonly found in human cancers [48], centrosome amplification may be one of the important factors contributing to carcinogenesis associated with overexpression or overactivation of RhoA and RhoC.
The activity of Rho is controlled by three classes of proteins: guanine nucleotide exchange factors (GEFs), which facilitate the exchange of GDP to GTP, GTPase-activating proteins, which increase the rate of GTP hydrolysis to GDP, and GDP dissociating inhibitors, which inhibit spontaneous GDP–GTP exchange [42]. Many growth, cytokine, and hormone receptors signal to GEFs, which leads to Rho activation [49]. The finding of the involvement of the Rho–ROCK2 pathway in centrosome amplification leads to an unexplored, yet important aspect of the oncogenic activities of those receptors. It has been known that oncogenic activation of many receptors leads to destabilization of chromosomes. However, such a phenomenon has been belittled as an indirect consequence of the continuous firing of a particular biological pathway. Because Rho is one of the immediate effectors of many receptors, activating mutation of those receptors may be more directly involved in destabilization of chromosomes through generation of amplified centrosomes through continual activation of the Rho-ROCK2 pathway. Indeed, it has been shown that activation of epidermal growth factor, which is known to activate the Rho–ROCK2 pathway, promotes centrosome duplication as well as centrosome amplification [50, 51]. We here tested the Met hepatocyte growth factor (HGF) receptor, which is also known to activate the Rho–ROCK2 pathway [49], for its ability to promote centrosome amplification. NIH3T3 do not express Met receptor, but express HGF. Thus, when Met is introduced into NIH3T3 cells, Met is activated in an autocrine fashion [52]. When we stably overexpressed Met in NIH3T3 cells and continuously boosted the Met activity by providing additional HGFs into the media (the autocrine system is not strong enough to ensure the continuous firing of the receptor), an increased frequency of centrosome amplification was observed (Fig. 7). Moreover, when either ROCK2 or RhoA expression was partially silenced in the Met-transfected cells, centrosome amplification was markedly suppressed (Fig. 7). Thus, the oncogenic/constitutive activation of certain receptors promotes centrosome amplification in the Rho–ROCK2 pathway-dependent manner.

Constitutive activation of Met receptor leads to centrosome amplification in the Rho and ROCK2-dependent manner. NIH3T3 cells were stably transfected with Met receptor. In addition, NIH3T3 cells stably expressing Met receptor were transfected with either ROCK2 or RhoA siRNA sequences. The transfected cells were then cultured for 10 days in the media supplemented with HGF, and the frequencies of centrosome amplification were determined. As references, the control cells and Met-expressing cells without added HGF are included in the graph
Conclusion
The findings described here depict that the two pathways—the receptor-Rho and CDK2-NPM/B23 pathways—converge to ROCK2 to control centrosome duplication and to induce centrosome amplification if these pathways are aberrantly activated (Fig. 8). All the proteins involved in these pathways (CDK2, cyclin E, receptors, NPM/B23, and Rho) are known to be frequently mutated, overexpressed or upregulated in human cancers [5]. Considering that centrosome amplification is one of the major causes of chromosome instability in cancers, and chromosome instability drives tumor progression and acquisition of malignant phenotype, induction of centrosome amplification is an important aspect of their overall oncogenic activities. Further understanding of this unique oncogenic activity of these oncogenic proteins will be informative for designing effective cancer intervention protocols targeting centrosome duplication and centrosome amplification. Such an approach may prove effective, since centrosome duplication, like DNA duplication, occurs only in actively proliferating cells. Many chemotherapeutic agents target DNA synthesis and thus selectively kill proliferating cells, yet increase the rate of secondary mutations by interfering with DNA metabolisms, leading to frequent generation of drug-resistant cells and secondary tumors. In contrast, a protocol designed to target centrosome duplication not only is specific to proliferating cells, but also inhibits cell division (and potentially induces cell death) and suppresses chromosome instability.

Model of the regulation of centrosome duplication by the receptor-Rho–ROCK2 pathway and the CDK2-NPM/B23-ROCK2 pathway. Ligand binding to many growth, cytokine, and hormone receptors leads to activation of Rho-GEFs, which in turn activate Rho proteins. Rho-GTP then binds to and activates ROCK2. CDK2-cyclin E, which is temporarily activated by cyclin E expression in late G1 phase of the cell cycle phosphorylates NPM/B23 on Thr199. NPM/B23 acquires a high binding affinity to ROCK2 when Thr199 is phosphorylated and binds to ROCK2. NPM/B23-binding superactivates ROCK2, which in turn rapidly triggers centrosome duplication
References
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61:759–767
Doxsey S (2002) Re-evaluating centrosome function. Nat Rev Mol Cell Biol 2:688–698
Fukasawa K (2005) Centrosome amplification, chromosome instability and cancer development. Cancer Lett 230:6–19
Fukasawa K (2007) Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer 7:911–924
Brinkley BR (2001) Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol 11:18–21
D'Assoro AB, Lingle WL, Salisbury JL (2002) Centrosome amplification and the development of cancer. Oncogene 21:6146–6153
Ganem NJ, Godinho SA, Pellman D (2009) A mechanism linking extra centrosomes to chromosomal instability. Nature 460:278–282
Ma Z, Kanai M, Kawamura K et al (2006) Interaction between ROCK II and nucleophosmin/B23 in the regulation of centrosome duplication. Mol Cell Biol 26:9016–9034
Hinchcliffe EH, Sluder G (2001) It takes two to tango: understanding how centrosome duplication is regulated throughout the cell cycle. Genes Dev 15:1167–1181
Nevins JR (1992) E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 258:424–429
Hinchcliffe EH, Li C, Thompson EA et al (1999) Requirement of Cdk2-cyclin E activity for repeated centrosome reproduction in Xenopus egg extracts. Science 283:851–854
Lacey KR, Jackson PK, Stearns T (1999) Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci USA 96:2817–2822
Matsumoto Y, Hayashi K, Nishida E (1999) Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr Biol 9:429–432
Tarapore P, Okuda M, Fukasawa K (2002) A mammalian in vitro centriole duplication system: evidence for involvement of CDK2/cyclin E and Nucleophosmin/B23 in centrosome duplication. Cell Cycle 1:75–81
Taylor WR, Stark GR (2001) Regulation of the G2/M transition by p53. Oncogene 20:1803–1815
Sherr CJ (2006) Divorcing ARF and p53: an unsettled case. Nat Rev Cancer 6:663–673
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K et al (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 73:39–85
Harper JW (1997) Cyclin dependent kinase inhibitors. Cancer Surv 29:91–107
Kawamura K, Izumi H, Ma Z et al (2004) Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res 64:4800–4809
D'Assoro AB, Lingle WL, Salisbury JL (2004) Centrosome amplification and the origin of chromosomal instability in breast cancer. Mammary Gland Biol Neoplasia 9:275–283
Koutsami MK, Tsantoulis PK, Kouloukoussa M et al (2006) Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol 209:512–521
Weber RG, Bridger JM, Benner A et al (1998) Centrosome amplification as a possible mechanism for numerical chromosome aberrations in cerebral primitive neuroectodermal tumors with TP53 mutations. Cytogenet Cell Genet 83:266–269
Carroll PE, Okuda M, Horn HF et al (1999) Centrosome hyperamplification in human cancer: chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene 18:1935–1944
Pihan GA, Wallace J, Zhou Y et al (2003) Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res 63:1398–1404
Nakajima T, Moriguchi M, Mitsumoto Y et al (2004) Centrosome aberration accompanied with p53 mutation can induce genetic instability in hepatocellular carcinoma. Mod Pathol 17:722–727
Mussman JG, Horn HF, Carroll PE et al (2000) Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 19:1635–1646
Okuda M, Horn HF, Tarapore P et al (2000) Nucleophosmin/B23 is a target of CDK2-cyclin E in centrosome duplication. Cell 103:127–140
Fisk HA, Winey A (2001) The mouse Mps1p-like kinase regulates centrosome duplication. Cell 106:95–104
Chen Z, Indjeian VB, McManus M et al (2002) CP110, a cell cycle-dependent CDK substrate, regulates centrosome duplication in human cells. Dev Cell 3:339–350
Szebeni A, Olson MO (1999) Nucleolar protein B23 has molecular chaperone activities. Protein Sci 8:905–912
Takemura M, Sato K, Nishio M et al (1999) Nucleolar protein B23.1 binds to retinoblastoma protein and synergistically stimulates DNA polymerase alpha activity. J Biochem 125:904–909
Valdez BC, Perlaky L, Henning D et al (1994) Identification of the nuclear and nucleolar localization signals of the protein p120. Interaction with translocation protein B23. J Biol Chem 269:23776–23783
Tarapore P, Shinmura K, Suzuki H et al (2006) Thr199 phosphorylation targets nucleophosmin to nuclear speckles and represses pre-mRNA processing. FEBS Lett 580:399–409
Colombo E, Marine JC, Danovi D et al (2002) Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol 4:529–533
Bertwistle D, Sugimoto M, Sherr CJ (2004) Physical and functional interactions of the Arf tumor suppressor protein with Nucleophosmin/B23. Mol Cell Biol 24:985–996
Korgaonkar C, Hagen J, Tompkins V et al (2005) Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol 25:1258–1271
Tokuyama Y, Horn HF, Kawamura K et al (2001) Specific phosphorylation of nucleophosmin on Thr(199) by cyclin-dependent kinase 2-cyclin E and its role in centrosome duplication. J Biol Chem 276:21529–21537
Grisendi S, Bernardi R, Rossi M et al (2005) Role of Nucleophosmin in embryonic development and tumorigenesis. Nature 437:147–153
Leung T, Chen XQ, Manser E et al (1996) The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol Cell Biol 16:5313–5327
Matsui T, Amano M, Yamamoto T et al (1996) Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. EMBO J 15:2208–2216
Nakagawa O, Fujisawa K, Ishizaki T et al (1996) ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett 392:189–193
Etienne-Manneville S, Hall A (2002) Rho GTPases in cell biology. Nature 420:692–695
Amano M, Ito M, Kimura K et al (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271:20246–20249
Wheeler AP, Ridley AJ (2004) Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res 301:43–49
Kanai M, Crowe MS, Zheng Y et al (2010) RhoA and RhoC are both required for the ROCK II-dependent promotion of centrosome duplication. Oncogene 29(45):6040–6050
Fujisawa K, Madaule P, Ishizaki T et al (1998) Different regions of Rho determine Rho-selective binding of different classes of Rho target molecules. J Biol Chem 273:18943–18949
Sahai E, Alberts AS, Treisman R (1998) RhoA effector mutants reveal distinct effector pathways for cytoskeletal reorganization, SRF activation and transformation. EMBO J 17:1350–1361
Vega FM, Ridley AJ (2008) RhoGTPases in cancer cell biology. FEBS Lett 582:2093–2101
Schiller MR (2006) Coupling receptor tyrosine kinases to Rho GTPases–GEFs what's the link. Cell Signal 18:1834–1843
Sherline P, Mascardo RN (1982) Epidermal growth factor induces rapid centrosomal separation in HeLa and 3T3 cells. J Cell Biol 93:507–511
Balczon R, Bao L, Zimmer WE et al (1995) Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 130:105–115
Shinomiya N, Gao CF, Xie Q, Gustafson M, Waters DJ, Zhang YW, Vande Woude GF (2004) RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res 64:7962–7970
Acknowledgments
This article is supported by the National Institute of Health (NCI and NIGM) (USA) and the State of Florida (USA).
Conflict of Interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fukasawa, K. Aberrant Activation of Cell Cycle Regulators, Centrosome Amplification, and Mitotic Defects. HORM CANC 2, 104–112 (2011). https://doi.org/10.1007/s12672-010-0060-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-010-0060-4