
Abstract
Multiple genetic alterations play a role in the pathogenesis of ovarian cancer. Although many key proteins and pathways involved in ovarian carcinogenesis and metastasis have been discovered, knowledge of the early steps leading to malignancy remains poorly understood. This poor understanding stems from lack of data from early-stage cancers and absence of a well-established premalignant state universal to all ovarian cancer subtypes. Existing evidence suggests that ovarian cancers develop either through a stepwise mutation process (low-grade pathway), through genetic instability resulting in hastened metastasis (high-grade pathway), or more recently through what has been described as the “‘fimbrial-ovarian’ serous neoplasia theory.” In this latter model, ovarian serous cancers evolve from premalignant lesions in the distal fallopian tube called tubal intraepithelial carcinoma. In this manuscript, we review key genetic and molecular changes that occur in cancer cell progression and suggest a model of ovarian cancer pathogenesis involving both tumor cell mutations and microenvironmental factors.
Similar content being viewed by others


Introduction
Ovarian cancer is the most common cause of death from gynecologic malignancies and the second most common gynecologic cancer [1]. Epithelial neoplasms are the most common malignant ovarian neoplasm [2]. Most cases are diagnosed late, resulting in poor clinical outcome. Survival of epithelial ovarian cancer (EOC) patients remains <50% at 5 years in spite of recent advances in cytoreductive surgery and chemotherapeutic agents [1]. Despite the promise for better outcomes in recurrent EOC and advanced-stage ovarian cancer through biologic therapies, a better strategy could be achieved by detecting high-risk patients and offering risk-reducing surgery that has been shown to be successful in patients at high risk due to genetic mutations [3]. Conversely, EOCs are considerably heterogeneous. For instance, subtypes such as endometrioid, serous, mucinous, and clear cell all manifest differently and involve unique pathogenic molecular blueprints [4]. Among serous tumors, low- versus high-grade tumors also have different molecular pathogenesis. Considerable progress has been made in comprehending the genetic makeup and physiological processes involved in ovarian cancer, but its underlying etiology is not well understood. In this review, we will focus on the existing science in characterizing the initial events in EOC.
Etiology of Sporadic Epithelial Ovarian Cancer
During embryogenesis, the coelomic layer develops into peritoneal mesothelium that surrounds the ovary and later differentiates through metaplasia to an epithelial layer [5]. Interestingly, contrary to other malignancies, as the ovarian epithelium acquires a malignant phenotype, it becomes more, rather than less, differentiated. This feature may explain its ability to transform into any cell type found in the Müllerian tract, such as those in the fallopian tube, uterus, cervix, and ovarian stroma [6]. Germline mutations are responsible for about 10% of ovarian malignancies and commonly involve BRCA1, BRCA2, or mismatch repair genes. Here, we will first focus on features of sporadic epithelial ovarian cancer.
For carcinogenesis to occur, the progenitor cell must surmount the physiologic checks and balances in order to become a clinically evident tumor [6]. Examples of such mechanisms include impairment of apoptosis, uncontrolled cell proliferation, angiogenesis, and metastasis. Several hypotheses have been proposed with regard to the possible origins of ovarian cancer (Table 1). The fact that there is a positive correlation between the number of ovulatory cycles with risk of ovarian cancer paved the way to the incessant ovulation hypothesis by Fathalla in 1971 [7]. According to this theory, with every ovulatory cycle, ovarian surface epithelial cells are injured, and subsequently, through repair mechanisms, the cells are predisposed to development of mutations and later malignancies. Coherent with this hypothesis, multiparity [8–10], length of lactation [11], and oral contraceptive usage [8, 12] all decrease the risk of developing ovarian cancer. Moreover, confirmatory data from animal studies support the incessant ovulation hypothesis [13, 14]. But, this hypothesis is somewhat undermined by the fact that progesterone-only oral contraceptives are as effective as ovulation-inhibiting contraceptives despite not inhibiting ovulation [15]. In addition, although patients with polycystic ovarian syndrome (PCOS) are known to have decreased ovulatory or rather anovulatory cycles, these women are still at increased risk of EOC [16].
Pitfalls in the incessant ovulation theory and evidence of increased risk among women using fertility drugs in order to conceive led to the gonadotropin hypothesis. It states that ovarian epithelial cells are more predisposed for tumor transformation after exposure to follicle-stimulating hormone (FSH) and luteinizing hormone (LH). Gonadotropin hormones (FSH, LH, and human chorionic gonadotropin) are known to activate EOCs to proliferate, and this process involves the mitogen-activated kinase pathway [17]. A recent study using human ovarian cancer cell lines (CaOV-3 and SKOV-3) showed that FSH/LH promote PGE2 production through cox-1 and cox-2 upregulation and increase cell invasion. In addition, treatment of these cells with cox inhibitors abrogated the stimulatory effects of gonadotropins on cell motility and cell invasion [18]. In addition, when the FSH receptor was overexpressed in non-tumorigenic SV40 Tag-immortalized ovarian surface epithelium (OSE)-derived cell lines, other proteins such as EGFR, HER-2, and c-myc were also upregulated [19]. The same was also shown when measuring other potential oncogenes in vitro such as cyclin G2, Meis-1, β-catenin, β-1 integrin, and IGF-1[20, 21]. In a case–control study, Whittemore et al. [8] found that infertile women have a higher chance of developing EOC (OR = 2.8) and borderline tumors (OR = 4.0) after taking infertility medications. However, other studies [22] have reported conflicting results, which prompted the idea that it is the infertility state, rather than gonadotropin medical therapy, that increases the risk of EOC [23]. Moreover, it has already been shown that FSH and LH receptors are frequently found on normal ovarian surface epithelial cells and 60% of malignant ovarian cancer cells [24]. Thus far, no evidence has been published showing that gonadotropins promote alterations of OSE to a malignant phenotype. However, animal studies have shown that gonadotropins play a key role in tumor growth [25], angiogenesis [25], vascular endothelial growth factor (VEGF) expression [26], and cell adhesion [27]. Therefore, current studies suggest a role for gonadotropins in ovarian cancer progression, but not necessarily in causality.
Progesterone has been shown to reduce the risk for ovarian cancer [15]. Similarly, progestin-only contraceptives are as valuable as combined OCPs in reducing the risk for ovarian cancer [15, 28]. The mechanism for this effect may be related to reduced ovarian testosterone levels [29]. Moreover, cell proliferation in OSE cells can occur through androgen receptors [30]. Developing follicles contain the highest androgen levels [31], providing an androgen-rich microenvironment around the epithelial cells. However, in vitro studies have not validated the effect of androgen derivatives on cancer cell growth [32]. Known hyperandrogenic diseases such as PCOS, hirsutism, and acne have been shown to be linked with increased EOC risk [16].
Various inflammatory processes are involved with each ovulation and are believed to be the causative process responsible for priming OECs to genetic damage and carcinogenesis. With every ovulatory cycle, inflammation occurs through the action of cytokines and chemotaxis of inflammatory cells and plays an important role in tissue repair [23]. The inflammation theory is supported with the observation that women taking anti-inflammatory medications (NSAIDS, ASA) have lower incidence of ovarian cancer [33]. The molecular pathway in this process involves intracellular effectors implicated in malignant transformation such as VEGF, NF-κB, nitric oxide synthase, and cyclooxygenase-2 [33]. Other pro-inflammatory chemicals such as talc and asbestos have been shown to predispose patients to ovarian cancer [34]. However, no confirmation of the relationship between talc or asbestos exposure and ovarian carcinogenesis in an animal model has ever been established.
All of the aforementioned pathways may play a part in the overall process of ovarian carcinogenesis in certain groups of patients, but their direct relationship may be in doubt and suggests that other mechanisms may be involved. To enable detection of high-risk patients early, more research is required to identify key genetic and epigenetic factors.
Earliest Identifiable Processes in Tumor Progression
It is believed that EOCs originate from a single cell that has become abnormal. This belief is based on studies showing common characteristics between primary and metastatic lesions when analyzing loss of heterozygosity (LOH), X-chromosome inactivation, and DNA mutations [35]. Identification of early processes involved in tumor progression is complex since the tumors usually present late, making it difficult to clearly identify the earliest events.
Comparison of Genetic Makeup of Early- Versus Late-Stage High-Grade Ovarian Cancers
Analyses of high-grade tumors have revealed overexpression of genes potentially responsible for cancer development and predicting clinical outcome (described later). While the earliest genetic events in ovarian cancer remain elusive, with the emergence of new technologies such as microarray expression profiling and comparative genome hybridization (CGH), many potential early genetic incidents have been discovered. At the expression level, early-stage tumors have profound changes in gene expression, which can be surprisingly similar to those found in late-stage tumors [36]. This goes against the hypothesis that advanced-stage tumors originate or progress from early-stage ones. But, when CGH analysis was used in the same study, differences were found at the genome levels that were more congruent with the evolution of cancer theory. Another study analyzing cancer specimens obtained from the ovary and omentum revealed 27 genetic changes that can be used to distinguish primary from metastatic cells [37]. The main pathway found to be altered was the P53 cascade; hence, this study revealed that this pathway may be critical for peritoneal seeding of ovarian cancer cells. Further studies are needed with larger power and microdissected specimens to elucidate differences between stromal and tumoral changes.
Genetic Disorders
In general, the pattern of transmission for familial ovarian cancer is autosomal dominant with variable penetrance. Women with genetic predisposition for ovarian cancer are at high risk to develop the disease on average a decade earlier, with risk of clinical cancer increasing in the fifth decade [38]. BRCA1 and BRCA2 mutations constitute almost 90% of hereditary genetic disorders responsible for ovarian cancer. The lifetime risk of ovarian cancer in this population is estimated to be as high as 56% and 27%, respectively [39]. BRCA genes are the housekeepers of the cell’s genome by mediating signals related to DNA injury, alerting and activating DNA repair mechanisms, monitoring the cell cycle though checks and balances, and promoting apoptosis [40–42]. BRCA has a close relationship with an array of important transcription factors, including p53, STAT1, c-Myc, JunB, ATF-1, and others [43]. If the BRCA gene malfunctions, the cell will have an increased number of centrosomes, inadequate repair of damaged DNA, and will succumb to aneuploidy and will be predisposed to mutations [43]. Although cancers positive for these mutations will proliferate faster, better prognosis is observed [44]. Genetic evaluation revealed that BRCA1 and BRCA2 mutant tumors each have different gene expression profiles, while sporadic tumors had a profile similar to both BRCA1 and BRCA2 mutant tumors [45]. In the sporadic ovarian cancer population, mutations in the BRCA gene are uncommon [46]. Meanwhile, in the Jewish population, BRCA1 and BRCA2 mutations were only found in 4% of borderline tumors compared to early-stage ovarian tumors (24.2%), implicating a role for BRCA mutations in cancer initiation [47]. Factors affecting BRCA function include alternate splicing, epigenetic factors, and other genetic causes [48–50].
The remaining 10% of hereditary ovarian cancers consist of Lynch syndrome or hereditary nonpolyposis colorectal cancer (HNPCC). This syndrome is associated with alterations in DNA mismatch repair genes. It affects 5% of the population, and patients with this syndrome carry a risk of developing ovarian cancer of about 12% [51]. When the cell DNA repair apparatus malfunctions, DNA instability occurs and the cell becomes predisposed to unchecked mutations, leading to cell proliferation. Although ovarian carcinogenesis occurs in women with HNPCC, it has not been well studied. Other familial syndromes associated with stromal ovarian tumors include Peutz–Jeghers syndrome, which consists of hamartomatous polyposis (mutation in the STK11 gene, 21% lifetime risk), and Gorlin syndrome (mutation in PTCH, 20% lifetime risk), respectively.
Animal Models
Several animal models have been designed in order to explore the pathogenesis of early ovarian carcinoma. To study the cell impact of various oncogenes, Orsulic et al. [52] used transgenic ovarian surface epithelial cells (p53-deficient) expressing the avian receptor TVA. After exposure to a variety of oncogenes, cells became tumorigenic when two out of three genes (c-myc, k-ras, Akt) were overexpressed. The transformed cells were then implanted into the bursal sac around the ovary of mice and later developed into peritoneal carcinomatosis similar to the metastatic pattern observed in human ovarian tumors. Connolly et al. [53] used genetically modified mice that express the transforming region of SV40 T-antigen controlled by the Müllerian inhibitory substance type II receptor gene promoter, resulting in de novo ovary-specific oncogenesis. In this model, the tumors were of high grade and were found in 50% of the mice along with carcinomatosis and ascites. Dinulesco et al. [54] established a model of endometrioid ovarian carcinogenesis. In this study, ovarian bursal cavity was injected with adenoviral vectors expressing Cre recombinase, which resulted in the overexpression of k-ras and conditional PTEN deletion. Cells that overexpressed k-ras alone resulted in lesions histologically consistent with endometriosis, whereas cells that overexpressed k-ras along with PTEN knockout underwent rapid transformation to endometrioid carcinomas. Wu et al. [55] have introduced mouse models of ovarian endometrioid cancer containing deletion of APC and PTEN tumor suppressor genes resulting in a profile very similar to human cancer. A recent study used a syngeneic mouse model of human epithelial ovarian cancer model by orthotopically injecting murine ovarian carcinoma cells into transgenic mice [56]. This study allows research of the spread of disease to the peritoneum. Due to their ability to develop spontaneous ovarian tumors, the laying hen has also been a model animal used for ovarian cancer research. The CA125 ovarian cancer marker is also expressed in chicken ovarian cancer cells while absent in normal chicken ovarian cells [57]. Molecular studies of ovarian cancer have taken advantage of Drosophila melanogaster models. This model has given insight into genes involved in border cell migration and motility [58]. Recent studies in Drosophila have elucidated the role of transcriptional coactivator Yap as an ovarian cancer oncogene [59]. Wistar and Sprague–Dawley rats have also been found to develop spontaneous ovarian tumors and thus have served as models for cancer research [60]. Collectively, these models are aiding scientists to uncover the oncogenes responsible for early cancer development. Foreseen limitations to the application of these models include the difficulty in the delivery of genetically modified cells and the unclear role of SV40 T-antigen in human cancer.
Models of Ovarian Cancer
Clinical, translational, as well as genetic studies have elucidated two major categories of ovarian carcinogenesis based on the idea that tumors are heterogeneous: high-grade malignancies tend to be fast growing and chemosensitive, and the low-grade neoplasms typically grow slowly, but are less sensitive to chemotherapy [61]. Gershenson et al. presented a study comprising 112 low-grade ovarian serous carcinoma patients which identified the average age at diagnosis to be 43, much lower than the age for women with high-grade cancer. Women with low-grade ovarian cancer had prolonged median survival of 81 months compared to 57–65 months in those with high-grade epithelial ovarian cancer [62–64]. Pathologic findings show that 60% of low-grade ovarian serous carcinomas are associated with a low malignant potential (LMP) neoplasm, whereas it was found in only 2% of high-grade ovarian carcinomas. Moreover, recurrent serous ovarian tumors of LMP are mostly low-grade lesions and have slow progression [65, 66]. Genetic and protein alterations in tumor cells also support the idea that the different types of ovarian cancers have unique pathogenesis (Table 2). Gene analyses have shown that b-raf, k-ras, and PTEN mutations occur more often in LMP tumors compared to high-grade tumors (30–50% versus 20%, respectively) [61, 67–69]. Conversely, HLA-G, HER2, and AKT levels are increased in high-grade tumors (61%, 20–66%, and 12–30%, respectively) as compared to LMP tumors [70, 71, 72]. While P53 is found to be mutated in >80% of high-grade tumors, it is rarely mutated in LMP tumors [73–75].
Assessment of whole genome profiles of ovarian cancers of various grades has shed light on the developmental association amongst the tumors. In such an approach, LMP tumors show closer similarity to normal ovarian epithelial cells rather than invasive cancers [4, 76]. Low-grade invasive cancers were also similar to borderline tumors. For example, wild-type P53 was found in LMP and low-grade tumors [4], suggesting that p53 dysfunction may not be required for these tumors. Moreover, developmental origin of these tumors was elucidated in loss of heterozygosity [77] and CGH [78] studies where LMP and benign adenomas share common genetic aberrations compared to high-grade tumors. Benign cystadenomas, LMP tumors, and mucinous adenocarcinomas have similar gene expression, which may be indicative of the sequential progression from adenoma to LMP and then to adenocarcinoma [79, 80].
In a recent study, Pothuri et al. [81] elucidated potential early aberrations in ovarian tumorigenesis. In this study, molecular, genetic, and morphologic analyses of early ovarian tumors and normal ovarian tissues revealed that EOCs originate from dysplastic precursor lesions within ovarian inclusion cysts. In addition, laser microdissection and gene expression profiling of ovarian cystic epithelium revealed a quasi-neoplastic signature involving signal transduction, cell cycle control, and mitotic spindle formation. Moreover, these cells had a high proliferation index and aneuploidy. These findings suggest that at least some EOCs may arise in ovarian cystic inclusion sites, and these tumors may be preceded by identifiable dysplastic precursor lesions.
Compelling data offer evidence for a model where the distal fallopian tube is believed to be an early precursor for pelvic serous tumors. This theory emerged when pathology of specimens obtained from prophylactic bilateral salpingo-oophorectomy in BRCA-positive patients revealed the presence of premalignant lesions/early serous carcinomas in the distal fallopian tubes. Moreover, the same findings were found in more than 50% of patients with unknown BRCA status with pelvic serous carcinoma, establishing a potential link between the fallopian tube and pelvic serous cancer. Further analysis of tubal intraepithelial carcinoma (TIC) and their adjacent serous carcinoma showed shared P53 mutations [82], thus supporting an origin in the distal fallopian tube [83, 84]. Przybycin et al. [85] showed that TIC was found in 59% of high-grade serous carcinomas and 92% were found in the fimbriated end of the tube. When concomitant invasive serous carcinoma was present, TIC was found in 71% of cases. Additional work is needed to provide the mechanistic links between these potential precursor lesions and ovarian cancer.
Genetic and Protein Alterations in Ovarian Cancer
Most of the information about genetic and protein changes in ovarian cancer is based on advanced-stage tumor studies. However, it is important to examine low-stage cancers to understand some of the earliest genetic lesions. Therefore, based on the attributes that cancer cells need to obtain in order to become immortal [86], we will discuss these processes in the context of ovarian cancer (Table 3).
Independence from Growth Signals
Several oncogenes have been studied in ovarian pathology that enable cells to proliferate. One example is src, a tyrosine kinase that plays a functional role in cell proliferation, adhesion, angiogenesis, cell survival [87–89], and in chemotherapeutic drug resistance [90]. Src overexpression is found in 93% of late-stage ovarian cancers and in more than 80% of cell lines [91]. Inhibiting this proto-oncogene with antisense oligonucleotides or small molecule inhibitors results in the diminished growth of ovarian cancer in mouse models through inhibition of angiogenesis [87]. Another group of proteins that are altered in tumorigenesis include the type I tyrosine kinase receptor family HER (Erb). It comprises four monomers: epidermal growth factor receptor (aka EGFR/Erb1/HER-1), HER-2 (proto-oncogene neu), HER-3, and HER-4. EGFR is the most common of the four and is overexpressed in 35–70% of epithelial ovarian cancers [92]. HER-2 lacks an extracellular ligand-binding domain, yet it dimerizes with other type I receptors. HER-2 is overexpressed in 20–30% of ovarian cancer cases [93].
Another well-known oncoprotein is RAS. It is a G-protein involved in cell proliferation. Activated RAS turns on a series of serine/threonine and tyrosine non-receptor kinases, leading to phosphorylation of Erk1 and Erk2 transcription factors, which function in cell growth and proliferation. K-ras mutations have been detected in 61% of borderline tumors, 68% of low-grade tumors, 50% of mucinous adenocarcinomas, and only in 5% of high-grade serous carcinomas [69, 94]. However, Wong et al. [95] analyzed 91 human ovarian tumor samples and found that among the low-grade serous carcinomas, only 19% expressed a KRAS mutation.
Non-responsiveness to Anti-growth Signals
For early malignant transformation to occur, anti-growth pathways must be surpassed. Although not much is known about the genetic mechanisms or the sequence of events, anomalies in cell cycle proteins have been noted. Examples of cell cycle mediators include cyclins, cyclin-dependent kinases (CDK), CDK inhibitors (block the binding of cyclins to CDK), and transcription factors such as pRb, TP53, and E2F. The G1 phase checkpoint (the restriction point), after which the cells are committed to enter S phase, is controlled by cyclins D and E’s phosphorylation of Rb and release of E2F. Cyclin E is associated with poor patient outcome and is expressed in borderline and malignant tumors (40% and 70%, respectively), whereas it is expressed in only 9% of benign tumors [96]. Cdk2, another protein involved in G1-S phase transition with cyclin E, is also found to be elevated in high-grade malignant ovarian tumors as compared to low-grade or even benign tumors [96]. Cyclin D1 is abundantly found in ovarian cancer cell cytoplasm (89%) and nucleus (30%) and scarcely found in normal epithelial ovarian cells [97]. Entry to the M phase is regulated by CDK1/cyclin B complex, which is highly expressed in 80% of malignant cells, but largely undetectable in normal ovarian epithelial cells [98].
Another pathway important in cell cycle control is the RAS-Raf pathway, which involves the myc oncogene/transcription factor. Myc is overexpressed in 30% of ovarian cancers. Moreover, AHRI (aka NOEY2, a GTPase tumor suppressor gene) [99] is a protein found in normal epithelial ovarian cells, but is completely absent in cancer cells. Yu et al. [100] suppressed the clonogenic growth of cancer cells after re-expression of NOEY2 by transfection. Therefore, we can conclude that various genetic anomalies are involved in the disruption of the cell cycle and may give ovarian cancer cells the advantage needed to expand and grow without host supervision.
Surviving Apoptosis
Many proponents of this mechanism strongly believe that avoiding cell death is the most critical step in order to achieve carcinogenesis rather than cell proliferation alone. Various genes and proteins play a part in the mechanism of cell death evasion. One example is the p53 gene and its protein TP53. In normal physiology, it functions by detecting damaged DNA, arresting the cell cycle, initiating mechanisms of repair, and directing the cell cycle into the path of apoptosis [101, 102]. When endogenous p53 is knocked down with small interfering RNA, serous ovarian borderline cancer cells show increased cell invasion and cell survival [102]. Most p53 aberrations are missense mutations [103]. P53 null mutations tend to be associated with a more aggressive disease pattern in ovarian cancer patients and may play a role in the metastatic process [104]. When p53 mutations are absent, its function can also be altered through degradation by ubiquitination, or through translation of TP53 binding protein. Moreover, pathology of ovaries obtained from prophylactic surgery in BRCA1 patients revealed that p53 mutations are detected in inclusion cysts adjacent to cystadenocarcinomas and in microscopic ovarian malignant cells [105]. Collective data suggest that p53 mutations and dysfunction may be one of the critical early steps toward tumor transformation and progression.
Another pathway involved in apoptosis evasion is the PI3-kinase/Akt pathway. It is present in 30% of ovarian pathology [70]. It is also involved in neoangiogenesis, cell invasion, and resistance to chemotherapy [106]. PTEN is the major regulator in this pathway, which acts as a tumor suppressor gene by dephosphorylating PIP3 back to PIP2 and leading the cell to apoptosis. Animal models have shown that PTEN mutations may be an important early event in the pathogenesis of the endometrioid subtype of ovarian cancer [54].
BAF250A, the protein encoded by ARID1A [the AR-rich interactive domain 1A (SWI-Like) gene], is one of the accessory subunits of the SWI–SNF complex that is believed to be responsible for providing specificity in gene expression regulation. The SWI–SNF complex is ubiquitous in eukaryotic cells and is involved in cellular development, differentiation, proliferation, DNA repair, and tumor suppression [107]. It consists of ATP-dependent motion of nucleosomes, therefore controlling the accessibility of transcription genes to promoters. RNA sequencing revealed that ARID1A mutations were found in 46% of ovarian clear cell carcinomas and in 30% of endometrial cancers [108]. These findings correlated with BAF250A loss by immunohistochemistry and were absent in high-grade serous ovarian serous ovarian carcinomas. Hence, ARID1A is a new potential tumor suppressor gene that may play a part in early events leading to ovarian carcinogenesis.NF-KappaB (NFκB), a pleiotropic transcription factor, inhibits cell apoptosis and promotes cancer cell survival. Deregowski et al. [109] used RT-PCR and DNA hybridization microarrays to show that when NFκB is upregulated in transfected cells, BCL-2 family members are also upregulated along with apoptotic inhibitors and other genes important for cell survival. Another study also showed that when NFκB was blocked in mice, the levels of interleukin-8 (IL-8) and VEGF were also decreased, resulting in diminished malignant potential in these ovarian cancer cell lines [110].
Cell Immortality
Cell senescence followed by apoptosis is achieved after the normal cell divides a number of times. The ends of the chromosomes contain DNA/protein complexes that protect the DNA from degradation. If the telomeres are lost, the chromosomes are exposed to defects, which allow p53 and other proteins to initiate apoptosis. Using telomere-specific fluorescence in situ hybridization, 82% of serous tubal intraepithelial carcinomas (STIC), a putative precursor of ovarian high-grade serous carcinoma (HGSC), were found to have short telomeres compared to normal tubal epithelium. In contrast, HGSC had longer telomeres than STIC. These findings suggest that telomerase activity is an important occurrence in the initiation of ovarian tumorigenesis [111]. Most ovarian cancer cells (81–86%) overcome the apoptosis pathway by producing telomerase, which is a reverse transcriptase made of RNA (hTR) and protein (hTERT) [112]. The major subunit linked to tumorigenesis is the overexpression of hTERT [113]. Expression of hTERT and p53 knockdown can transform ovarian surface epithelial cells [114]. Inhibition of telomerase activity is achieved via a functional BRCA [115]. These findings suggest that telomerase activation is an important occurrence in the initiation of ovarian tumorigenesis.
Early Events in the Tumor Microenvironment: Angiogenesis, Invasion, and Metastasis
The tumor microenvironment consists of tumor cells, matrix components, inflammatory cells, and stromal cells. The communication between these various components allows for angiogenesis, invasion into stroma, and metastatic growth in distant organs. The concept of metastasis usually is synonymous with advanced stage of disease, but it has been shown in breast cancer that early tumors may also have subclinical metastasis [116]. In ovarian cancer, changes in the stroma and peritoneum may allow the tumor to spread [117]. Oxygen and nutrients are essential for all cells, including malignant, benign, or normal cells. For cells to receive this basic need, they must reside within 100 μm of a capillary [118]. For a malignant cell to increase in size beyond 1 mm3, genesis of new vessels around its premises must occur. Angiogenesis consists of a complex balance between pro- and anti-angiogenic factors in the cellular microenvironment. VEGF-A is the primary regulator of angiogenesis [119, 120]. It augments vascular permeability, acts as a positive effector for endothelial cell proliferation and migration, modifies endothelial cell gene expression, and allows the cell to evade apoptosis [121, 122]. Studies have shown that VEGF expression induces ovarian cancer cell lines to metastasize and produce ascites and carcinomatosis [123]. The clinical outcomes of ovarian cancer patients are correlated with VEGF levels [124, 125].
Mediators of angiogenesis can either originate from the host or from the tumor cells. Among these, IL-8 is elevated in ovarian cancer patients [126] and is believed to be a key factor for cancer growth and new vessel formation [127]. Integrin subunits also play a key role in invasion and angiogenesis. Davidson et al. [128] studied the expression of αv and β3 integrin subunits in cancer cells and endothelial cells. The αvβ3 integrin was primarily located on newly developing vascular endothelial cells and on ovarian cancer cells. Another group concluded that EphA2, a tyrosine kinase receptor involved in oncogenesis, is overexpressed in 75% of ovarian cancers [129]. When EphA2 is inhibited, cancer growth was also slowed down by anti-angiogenic mechanisms [130, 131]. The patient-specific tumor microenvironment is an important feature that will regulate tumor angiogenesis and modulate the outcome of anti-angiogenic therapy [132].
For metastasis to occur, the basement membrane must be invaded. This involves interactions between the invading cell and the surrounding stroma. To allow endothelial cell migration during angiogenesis, remodeling of the extracellular matrix is necessary and key proteins in this process include the matrix metalloproteinases (MMPs). MMPs are zinc-dependent endopeptidases that degrade collagen and other ECM constituents. They also have the capability to promote angiogenesis through VEGF [133]. Ovarian cancers have been shown to express high levels of MMP2 and MMP9 [18, 134] and at the same time are associated with clinical stage [135] and patient outcome [131]. Huang et al. [136] showed that host-derived MMP-9 expression appears to play a major role in angiogenesis and progression of human ovarian tumors compared to MMPs from tumor cells. Another theory that may potentiate invasion is the psychoneuroimmunomodulation hypothesis where catecholamines are released due to chronic stress. Abundant preclinical evidence supports the role of chronic stress in activating the cholinergic/sympathetic pathways, which can lead to increased invasion and metastasis [137, 138]. Moreover, epidemiological studies show that patients with poor social support status and chronic stress may be at greater risk for worse cancer outcome [138].
The tumor microenvironment consists of inflammatory cells that have the ability to recognize foreign components of the tumor cell and promote tumor cell apoptosis. In an effort to evade recognition by the immune system, which would eventually lead to their destruction, tumor cells have acquired the ability to produce Fas ligand that induces apoptosis in lymphocytes [139] as well as secretion of HLA-G that can inhibit NK cell activity [72, 140]. Cytokine production by mesenchymal cells as well as by the tumor cells helps the tumors to grow and prevent apoptosis [141, 142]. Immunohistochemistry in 186 specimens showed that increased numbers of T cells correlate with an improvement in survival [143]. The role of specific immune cell populations in controlling versus promoting tumor growth remains to be fully defined [144].
An advanced stage of tumor usually is associated with metastasis, which may occur earlier than once thought [116]. Although cells escape from the primary tumor into the vasculature, this does not necessarily mean that they will anchor and grow in different distant organs [145]. Only 30% of stage I cancers showed positive peritoneal cytology [146]. Due to the shedding capability of ovarian cancer, an early role may be played by cell survival promoters such as focal adhesion kinase (FAK) and E-cadherin [147–150]. Moreover, E-cadherin is expressed only in tumor cells (LMP, benign, or low and high grade) and notably in ovarian inclusion cysts, but absent in normal ovarian epithelial cells [151].
Proposed Model of Ovarian Carcinogenesis and Concluding Remarks
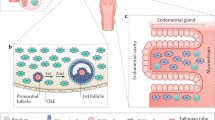
The understanding and identification of key players in the development of ovarian cancer is important in order to provide better targeted therapies. In the broad dual-pathway model of ovarian carcinogenesis, we propose that not only are the early genetic events important but also that the stroma plays a significant role in tumorigenesis (Fig. 1). The sequence of events is quite variable, but important genetic alterations may lead to specific tumor types. For example, KRAS mutations can lead to a LMP tumor, whereas a p53 or BRCA mutation can lead to cancer development through the high-grade pathway. Both pathways include certain characteristics such as evasion from the immune system and invasion into the stroma and peritoneal cavity while continuing to grow and to vascularize. Despite similarities in both pathways, clinically and histologically, each cancer is distinct and a large spectrum of unknown genetic transformations or pathways may play a role in the early steps in carcinogenesis. Therefore, strife exists to identify these early factors responsible for the initiation of oncogenesis in ovarian pathology, enabling clinicians to detect cancer early and to target specific factors to improve clinical therapy outcomes.

Proposed model of ovarian carcinogenesis (adapted from JCO 2008:995–1005 (Feb 20) with permission). Normal ovarian epithelium is exposed to physiologic processes that may predispose to malignant transformation, such as prolonged androgen exposure. A number of characteristics must be obtained, primarily through mutations or other genetic changes, to be transformed to a malignant state. These include unregulated growth, resistance to anti-growth signals, inhibition of apoptosis, evasion of recognition by the immune system, achieving limitless replicative potential, induction of angiogenesis, and invasion of the basement membrane. Examples of specific proteins known to play a role in each of these processes in ovarian cancer are listed in italics. The order in which these mutations may occur is not well understood, but the timing and specific protein affected may be significant in producing different histological subtypes and grades of ovarian cancer. For example, if mutations favoring growth and resistance to apoptosis occurred early, prior to achieving the potential for invasion and metastasis, an intermediate pathologic subtype would be noted more often, such as k-ras mutations in LMP tumors. A mutation leading to genetic instability, such as p53, that occurred early would predispose cells to other mutations and rapid progression to a metastatic phenotype, as seen in high-grade malignancies. Permissive or contributing factors of the microenvironment, such as production of MMPs by fibroblasts (pictured in red), infiltration of inflammatory cells (pictured in blue), and proliferation of endothelial cells for angiogenesis, may be just as important as mutations in the tumor cells
References
Jemal A, Siegel R, Xu J, Ward E (2010) Cancer statistics, 2010. CA Cancer J Clin 60:277–300.
Koonings PP, Campbell K, Mishell DR Jr, Grimes DA (1989) Relative frequency of primary ovarian neoplasms: a 10-year review. Obstet Gynecol 74(6):921–926.
Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’t Veer L, Garber JE, Evans G et al (2002) Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 346(21):1616–1622. doi:10.1056/NEJMoa012158
Bonome T, Lee JY, Park DC, Radonovich M, Pise-Masison C, Brady J, Gardner GJ et al (2005) Expression profiling of serous low malignant potential, low-grade, and high-grade tumors of the ovary. Cancer Res 65(22):10602–10612
Auersperg N, Wong AS, Choi KC, Kang SK, Leung PC (2001) Ovarian surface epithelium: biology, endocrinology, and pathology. Endocr Rev 22(2):255–288
Naora H (2005) Developmental patterning in the wrong context: the paradox of epithelial ovarian cancers. Cell Cycle 4(8):1033–1035
Fathalla MF (1971) Incessant ovulation—a factor in ovarian neoplasia? Lancet 2(7716):163
Whittemore AS, Harris R, Itnyre J (1992) Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case–control studies. II. Invasive epithelial ovarian cancers in white women. Collaborative Ovarian Cancer Group. Am J Epidemiol 136(10):1184–1203
Risch HA, Marrett LD, Howe GR (1994) Parity, contraception, infertility, and the risk of epithelial ovarian cancer. Am J Epidemiol 140(7):585–597
Riman T, Dickman PW, Nilsson S, Correia N, Nordlinder H, Magnusson CM, Persson IR (2002) Risk factors for invasive epithelial ovarian cancer: results from a Swedish case–control study. Am J Epidemiol 156(4):363–373
Gwinn ML, Lee NC, Rhodes PH, Layde PM, Rubin GL (1990) Pregnancy, breast feeding, and oral contraceptives and the risk of epithelial ovarian cancer. J Clin Epidemiol 43(6):559–568
Nasca PC, Greenwald P, Chorost S, Richart R, Caputo T (1984) An epidemiologic case–control study of ovarian cancer and reproductive factors. Am J Epidemiol 119(5):705–713
Fredrickson TN (1987) Ovarian tumors of the hen. Environ Health Perspect 73:35–51
Land JA (1993) Ovulation, ovulation induction and ovarian carcinoma. Baillières Clin Obstet Gynaecol 7(2):455–472
Risch HA (1998) Hormonal etiology of epithelial ovarian cancer, with a hypothesis concerning the role of androgens and progesterone. J Natl Cancer Inst 90(23):1774–1786
Schildkraut JM, Schwingl PJ, Bastos E, Evanoff A, Hughes C (1996) Epithelial ovarian cancer risk among women with polycystic ovary syndrome. Obstet Gynecol 88(4 Pt 1):554–559
Choi KC, Kang SK, Tai CJ, Auersperg N, Leung PC (2002) Follicle-stimulating hormone activates mitogen-activated protein kinase in preneoplastic and neoplastic ovarian surface epithelial cells. J Clin Endocrinol Metab 87(5):2245–2253
Lau MT, Wong AS, Leung PC (2010) Gonadotropins induce tumor cell migration and invasion by increasing cyclooxygenases expression and prostaglandin E(2) production in human ovarian cancer cells. Endocrinology 151(7):2985–2993. doi:10.1210/en.2009-1318
Choi JH, Choi KC, Auersperg N, Leung PC (2004) Overexpression of follicle-stimulating hormone receptor activates oncogenic pathways in preneoplastic ovarian surface epithelial cells. J Clin Endocrinol Metab 89(11):5508–5516
Tashiro H, Katabuchi H, Begum M, Li X, Nitta M, Ohtake H, Okamura H (2003) Roles of luteinizing hormone/chorionic gonadotropin receptor in anchorage-dependent and -independent growth in human ovarian surface epithelial cell lines. Cancer Sci 94(11):953–959
Ji Q, Liu PI, Chen PK, Aoyama C (2004) Follicle stimulating hormone-induced growth promotion and gene expression profiles on ovarian surface epithelial cells. Int J Cancer 112(5):803–814
Brinton LA, Lamb EJ, Moghissi KS, Scoccia B, Althuis MD, Mabie JE, Westhoff CL (2004) Ovarian cancer risk after the use of ovulation-stimulating drugs. Obstet Gynecol 103(6):1194–1203
Ness RB, Cottreau C (1999) Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst 91(17):1459–1467
Zheng W, Lu JJ, Luo F, Zheng Y, Feng Y, Felix JC, Lauchlan SC, Pike MC (2000) Ovarian epithelial tumor growth promotion by follicle-stimulating hormone and inhibition of the effect by luteinizing hormone. Gynecol Oncol 76(1):80–88
Schiffenbauer YS, Abramovitch R, Meir G, Nevo N, Holzinger M, Itin A, Keshet E, Neeman M (1997) Loss of ovarian function promotes angiogenesis in human ovarian carcinoma. Proc Natl Acad Sci USA 94(24):13203–13208
Wang J, Luo F, Lu JJ, Chen PK, Liu P, Zheng W (2002) VEGF expression and enhanced production by gonadotropins in ovarian epithelial tumors. Int J Cancer 97(2):163–167
Schiffenbauer YS, Meir G, Maoz M, Even-Ram SC, Bar-Shavit R, Neeman M (2002) Gonadotropin stimulation of MLS human epithelial ovarian carcinoma cells augments cell adhesion mediated by CD44 and by alpha(v)-integrin. Gynecol Oncol 84(2):296–302
Rosenberg L, Palmer JR, Zauber AG, Warshauer ME, Lewis JL Jr, Strom BL, Harlap S, Shapiro S (1994) A case–control study of oral contraceptive use and invasive epithelial ovarian cancer. Am J Epidemiol 139(7):654–661
Gaspard UJ, Romus MA, Gillain D, Duvivier J, Demey-Ponsart E, Franchimont P (1983) Plasma hormone levels in women receiving new oral contraceptives containing ethinyl estradiol plus levonorgestrel or desogestrel. Contraception 27(6):577–590
Edmondson RJ, Monaghan JM, Davies BR (2002) The human ovarian surface epithelium is an androgen responsive tissue. Br J Cancer 86(6):879–885
McNatty KP, Smith DM, Makris A, Osathanondh R, Ryan KJ (1979) The microenvironment of the human antral follicle: interrelationships among the steroid levels in antral fluid, the population of granulosa cells, and the status of the oocyte in vivo and in vitro. J Clin Endocrinol Metab 49(6):851–860
Seeger H, Wallwiener D, Mueck AO (2006) Is there a protective role of progestogens on the proliferation of human ovarian cancer cells in the presence of growth factors? Eur J Gynaecol Oncol 27(2):139–141
Altinoz MA, Korkmaz R (2004) NF-kappaB, macrophage migration inhibitory factor and cyclooxygenase-inhibitions as likely mechanisms behind the acetaminophen- and NSAID-prevention of the ovarian cancer. Neoplasma 51(4):239–247
Heller DS, Westhoff C, Gordon RE, Katz N (1996) The relationship between perineal cosmetic talc usage and ovarian talc particle burden. Am J Obstet Gynecol 174(5):1507–1510
Duggan BD, Dubeau L (1998) Genetics and biology of gynecologic cancer. Curr Opin Oncol 10(5):439–446
Shridhar V, Lee J, Pandita A, Iturria S, Avula R, Staub J, Morrissey M et al (2001) Genetic analysis of early- versus late-stage ovarian tumors. Cancer Res 61(15):5895–5904
Lancaster JM, Dressman HK, Clarke JP, Sayer RA, Martino MA, Cragun JM, Henriott AH et al (2006) Identification of genes associated with ovarian cancer metastasis using microarray expression analysis. Int J Gynecol Cancer 16(5):1733–1745
Palma M, Ristori E, Ricevuto E, Giannini G, Gulino A (2006) BRCA1 and BRCA2: the genetic testing and the current management options for mutation carriers. Crit Rev Oncol Hematol 57(1):1–23. doi:10.1016/j.critrevonc.2005.05.003
Sogaard M, Kjaer SK, Gayther S (2006) Ovarian cancer and genetic susceptibility in relation to the BRCA1 and BRCA2 genes. Occurrence, clinical importance and intervention. Acta Obstet Gynecol Scand 85(1):93–105
Venkitaraman AR (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108(2):171–182
Yoshida K, Miki Y (2004) Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci 95(11):866–871
Boulton SJ (2006) Cellular functions of the BRCA tumour-suppressor proteins. Biochem Soc Trans 34(Pt 5):633–645
Rosen EM, Fan S, Pestell RG, Goldberg ID (2003) BRCA1 gene in breast cancer. J Cell Physiol 196(1):19–41
Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY (2003) Improved survival in women with BRCA-associated ovarian carcinoma. Cancer 97(9):2187–2195
Jazaeri AA, Yee CJ, Sotiriou C, Brantley KR, Boyd J, Liu ET (2002) Gene expression profiles of BRCA1-linked, BRCA2-linked, and sporadic ovarian cancers. J Natl Cancer Inst 94(13):990–1000
Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, Weber BL, Collins FS, Johnston C, Frank TS (1995) Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet 9(4):439–443. doi:10.1038/ng0495-439
Gotlieb WH, Chetrit A, Menczer J, Hirsh-Yechezkel G, Lubin F, Friedman E, Modan B, Ben-Baruch G (2005) Demographic and genetic characteristics of patients with borderline ovarian tumors as compared to early stage invasive ovarian cancer. Gynecol Oncol 97(3):780–783
Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY (2000) BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res 60(19):5329–5333
Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E et al (2000) Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92(7):564–569
Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE (2002) Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst 94(18):1396–1406
Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, Peltomaki P, Mecklin JP, Jarvinen HJ (1999) Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 81(2):214–218
Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE (2002) Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1(1):53–62
Connolly DC, Bao R, Nikitin AY, Stephens KC, Poole TW, Hua X, Harris SS, Vanderhyden BC, Hamilton TC (2003) Female mice chimeric for expression of the simian virus 40 TAg under control of the MISIIR promoter develop epithelial ovarian cancer. Cancer Res 63(6):1389–1397
Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T (2005) Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med 11(1):63–70
Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, Hanash S et al (2007) Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell 11(4):321–333. doi:10.1016/j.ccr.2007.02.016
Quinn BA, Xiao F, Bickel LE, Martin LP, Hua X, Klein-Szanto AJ, Connolly DC (2010) Development of a syngeneic mouse model of epithelial ovarian cancer. J Ovarian Res 3(1):24. doi:10.1186/1757-2215-3-24
Jackson E, Anderson K, Ashwell C, Petitte J, Mozdziak PE (2007) CA125 expression in spontaneous ovarian adenocarcinomas from laying hens. Gynecol Oncol 104(1):192–198. doi:10.1016/j.ygyno.2006.07.024
Montell DJ (2003) Border-cell migration: the race is on. Nat Rev Mol Cell Biol 4(1):13–24. doi:10.1038/nrm1006
Hall CA, Wang R, Miao J, Oliva E, Shen X, Wheeler T, Hilsenbeck SG, Orsulic S, Goode S (2010) Hippo pathway effector yap is an ovarian cancer oncogene. Cancer Res 70(21):8517–8525. doi:10.1158/0008-5472.CAN-10-1242
Gregson RL, Lewis DJ, Abbott DP (1984) Spontaneous ovarian neoplasms of the laboratory rat. Vet Pathol 21(3):292–299
Shih Ie M, Kurman RJ (2004) Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol 164(5):1511–1518
Gershenson DM, Sun CC, Lu KH, Coleman RL, Sood AK, Malpica A, Deavers MT, Silva EG, Bodurka DC (2006) Clinical behavior of stage II–IV low-grade serous carcinoma of the ovary. Obstet Gynecol 108(2):361–368
Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R (2003) Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol 21(17):3194–3200
Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, Copeland LJ, Walker JL, Burger RA (2006) Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med 354(1):34–43
Malpica A, Deavers MT, Lu K, Bodurka DC, Atkinson EN, Gershenson DM, Silva EG (2004) Grading ovarian serous carcinoma using a two-tier system. Am J Surg Pathol 28(4):496–504
Crispens MA, Bodurka D, Deavers M, Lu K, Silva EG, Gershenson DM (2002) Response and survival in patients with progressive or recurrent serous ovarian tumors of low malignant potential. Obstet Gynecol 99(1):3–10
Mok SC, Bell DA, Knapp RC, Fishbaugh PM, Welch WR, Muto MG, Berkowitz RS, Tsao SW (1993) Mutation of K-ras protooncogene in human ovarian epithelial tumors of borderline malignancy. Cancer Res 53(7):1489–1492
Teneriello MG, Ebina M, Linnoila RI, Henry M, Nash JD, Park RC, Birrer MJ (1993) p53 and Ki-ras gene mutations in epithelial ovarian neoplasms. Cancer Res 53(13):3103–3108
Singer G, Oldt R 3rd, Cohen Y, Wang BG, Sidransky D, Kurman RJ, Shih Ie M (2003) Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 95(6):484–486
Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, Hamilton TC, Tsichlis PN, Testa JR (1992) AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc Natl Acad Sci USA 89(19):9267–9271
Ross JS, Yang F, Kallakury BV, Sheehan CE, Ambros RA, Muraca PJ (1999) HER-2/neu oncogene amplification by fluorescence in situ hybridization in epithelial tumors of the ovary. Am J Clin Pathol 111(3):311–316
Singer G, Rebmann V, Chen YC, Liu HT, Ali SZ, Reinsberg J, McMaster MT et al (2003) HLA-G is a potential tumor marker in malignant ascites. Clin Cancer Res 9(12):4460–4464
Kohler MF, Marks JR, Wiseman RW, Jacobs IJ, Davidoff AM, Clarke-Pearson DL, Soper JT, Bast RC Jr, Berchuck A (1993) Spectrum of mutation and frequency of allelic deletion of the p53 gene in ovarian cancer. J Natl Cancer Inst 85(18):1513–1519
Kupryjanczyk J, Thor AD, Beauchamp R, Merritt V, Edgerton SM, Bell DA, Yandell DW (1993) p53 gene mutations and protein accumulation in human ovarian cancer. Proc Natl Acad Sci USA 90(11):4961–4965
Skilling JS, Sood A, Niemann T, Lager DJ, Buller RE (1996) An abundance of p53 null mutations in ovarian carcinoma. Oncogene 13(1):117–123
Ouellet V, Guyot MC, Le Page C, Filali-Mouhim A, Lussier C, Tonin PN, Provencher DM, Mes-Masson AM (2006) Tissue array analysis of expression microarray candidates identifies markers associated with tumor grade and outcome in serous epithelial ovarian cancer. Int J Cancer 119(3):599–607
Dodson MK, Hartmann LC, Cliby WA, DeLacey KA, Keeney GL, Ritland SR, Su JQ, Podratz KC, Jenkins RB (1993) Comparison of loss of heterozygosity patterns in invasive low-grade and high-grade epithelial ovarian carcinomas. Cancer Res 53(19):4456–4460
Meinhold-Heerlein I, Bauerschlag D, Hilpert F, Dimitrov P, Sapinoso LM, Orlowska-Volk M, Bauknecht T et al (2005) Molecular and prognostic distinction between serous ovarian carcinomas of varying grade and malignant potential. Oncogene 24(6):1053–1065
Heinzelmann-Schwarz VA, Gardiner-Garden M, Henshall SM, Scurry JP, Scolyer RA, Smith AN, Bali A et al (2006) A distinct molecular profile associated with mucinous epithelial ovarian cancer. Br J Cancer 94(6):904–913
Wamunyokoli FW, Bonome T, Lee JY, Feltmate CM, Welch WR, Radonovich M, Pise-Masison C et al (2006) Expression profiling of mucinous tumors of the ovary identifies genes of clinicopathologic importance. Clin Cancer Res 12(3 Pt 1):690–700
Pothuri B, Leitao MM, Levine DA, Viale A, Olshen AB, Arroyo C, Bogomolniy F et al (2010) Genetic analysis of the early natural history of epithelial ovarian carcinoma. PLoS ONE 5(4):e10358. doi:10.1371/journal.pone.0010358
Crum CP (2009) Intercepting pelvic cancer in the distal fallopian tube: theories and realities. Mol Oncol 3(2):165–170. doi:10.1016/j.molonc.2009.01.004
Kindelberger DW, Lee Y, Miron A, Hirsch MS, Feltmate C, Medeiros F, Callahan MJ et al (2007) Intraepithelial carcinoma of the fimbria and pelvic serous carcinoma: evidence for a causal relationship. Am J Surg Pathol 31(2):161–169. doi:10.1097/01.pas.0000213335.40358.47
Levanon K, Crum C, Drapkin R (2008) New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol 26(32):5284–5293. doi:10.1200/JCO.2008.18.1107
Przybycin CG, Kurman RJ, Ronnett BM, Shih Ie M, Vang R (2010) Are all pelvic (nonuterine) serous carcinomas of tubal origin? Am J Surg Pathol 34(10):1407–1416. doi:10.1097/PAS.0b013e3181ef7b16
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100(1):57–70
Han LY, Landen CN, JGHalder Trevino J, Lin YG, Kamat AA, Kim TJ, Merritt WM et al (2006) Anti-angiogenic and anti-tumor effects of Src inhibition in ovarian carcinoma. Cancer Res 66(17):8633–8639
Ishizawar R, Parsons SJ (2004) c-Src and cooperating partners in human cancer. Cancer Cell 6(3):209–214
Silva CM (2004) Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene 23(48):8017–8023
Pengetnze Y, Steed M, Roby KF, Terranova PF, Taylor CC (2003) Src tyrosine kinase promotes survival and resistance to chemotherapeutics in a mouse ovarian cancer cell line. Biochem Biophys Res Commun 309(2):377–383
Wiener JR, Windham TC, Estrella VC, Parikh NU, Thall PF, Deavers MT, Bast RC, Mills GB, Gallick GE (2003) Activated SRC protein tyrosine kinase is overexpressed in late-stage human ovarian cancers. Gynecol Oncol 88(1):73–79
Bartlett JM, Langdon SP, Simpson BJ, Stewart M, Katsaros D, Sismondi P, Love S et al (1996) The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br J Cancer 73(3):301–306
Leary JA, Edwards BG, Houghton CR, Kefford RF, Friedlander ML (1992) Amplification of HER-2/neu oncogene in human ovarian cancer. Int J Gynecol Cancer 2(6):291–294
Suzuki M, Saito S, Saga Y, Ohwada M, Sato I (2000) Mutation of K-RAS protooncogene and loss of heterozygosity on 6q27 in serous and mucinous ovarian carcinomas. Cancer Genet Cytogenet 118(2):132–135
Wong KK, Tsang YT, Deavers MT, Mok SC, Zu Z, Sun C, Malpica A, Wolf JK, Lu KH, Gershenson DM (2010) BRAF mutation is rare in advanced-stage low-grade ovarian serous carcinomas. Am J Pathol 177:1611–1617. doi:10.2353/ajpath.2010.100212
Sui L, Dong Y, Ohno M, Sugimoto K, Tai Y, Hando T, Tokuda M (2001) Implication of malignancy and prognosis of p27(kip1), cyclin E, and Cdk2 expression in epithelial ovarian tumors. Gynecol Oncol 83(1):56–63
Dhar KK, Branigan K, Parkes J, Howells RE, Hand P, Musgrove C, Strange RC, Fryer AA, Redman CW, Hoban PR (1999) Expression and subcellular localization of cyclin D1 protein in epithelial ovarian tumour cells. Br J Cancer 81(7):1174–1181
Barrette BA, Srivatsa PJ, Cliby WA, Keeney GL, Suman VJ, Podratz KC, Roche PC (1997) Overexpression of p34cdc2 protein kinase in epithelial ovarian carcinoma. Mayo Clin Proc 72(10):925–929
Bao R, Connolly DC, Murphy M, Green J, Weinstein JK, Pisarcik DA, Hamilton TC (2002) Activation of cancer-specific gene expression by the survivin promoter. J Natl Cancer Inst 94(7):522–528
Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y, Cuevas B et al (1999) NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc Natl Acad Sci USA 96(1):214–219
Michalovitz D, Halevy O, Oren M (1991) p53 mutations: gains or losses? J Cell Biochem 45(1):22–29
Cheng JC, Auersperg N, Leung PC (2010) Inhibition of p53 induces invasion of serous borderline ovarian tumor cells by accentuating PI3K/Akt-mediated suppression of E-cadherin. Oncogene. doi:10.1038/onc.2010.486
Skilling JS, Squatrito RC, Connor JP, Niemann T, Buller RE (1996) p53 gene mutation analysis and antisense-mediated growth inhibition of human ovarian carcinoma cell lines. Gynecol Oncol 60(1):72–80
Marks JR, Davidoff AM, Kerns BJ, Humphrey PA, Pence JC, Dodge RK, Clarke-Pearson DL, Iglehart JD, Bast RC Jr, Berchuck A (1991) Overexpression and mutation of p53 in epithelial ovarian cancer. Cancer Res 51(11):2979–2984
Kerner R, Sabo E, Gershoni-Baruch R, Beck D, Ben-Izhak O (2005) Expression of cell cycle regulatory proteins in ovaries prophylactically removed from Jewish Ashkenazi BRCA1 and BRCA2 mutation carriers: correlation with histopathology. Gynecol Oncol 99(2):367–375
Hu L, Hofmann J, Lu Y, Mills GB, Jaffe RB (2002) Inhibition of phosphatidylinositol 3′-kinase increases efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Cancer Res 62(4):1087–1092
Reisman D, Glaros S, Thompson EA (2009) The SWI/SNF complex and cancer. Oncogene 28(14):1653–1668. doi:10.1038/onc.2009.4
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J et al (2010) ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 363(16):1532–1543. doi:10.1056/NEJMoa1008433
Deregowski V, Delhalle S, Benoit V, Bours V, Merville MP (2002) Identification of cytokine-induced nuclear factor-kappaB target genes in ovarian and breast cancer cells. Biochem Pharmacol 64(5–6):873–881
Huang S, Robinson JB, Deguzman A, Bucana CD, Fidler IJ (2000) Blockade of nuclear factor-kappaB signaling inhibits angiogenesis and tumorigenicity of human ovarian cancer cells by suppressing expression of vascular endothelial growth factor and interleukin 8. Cancer Res 60(19):5334–5339
Kuhn E, Meeker A, Wang TL, Sehdev AS, Kurman RJ, Shih Ie M (2010) Shortened telomeres in serous tubal intraepithelial carcinoma: an early event in ovarian high-grade serous carcinogenesis. Am J Surg Pathol 34(6):829–836. doi:10.1097/PAS.0b013e3181dcede7
Landen CN, Klingelhutz A, Coffin JE, Sorosky JI, Sood AK (2004) Genomic instability is associated with lack of telomerase activation in ovarian cancer. Cancer Biol Ther 3(12):1250–1253
Poremba C, Heine B, Diallo R, Heinecke A, Wai D, Schaefer KL, Braun Y et al (2002) Telomerase as a prognostic marker in breast cancer: high-throughput tissue microarray analysis of hTERT and hTR. J Pathol 198(2):181–189
Yang G, Rosen DG, Mercado-Uribe I, Colacino JA, Mills GB, Bast RC Jr, Zhou C, Liu J (2007) Knockdown of p53 combined with expression of the catalytic subunit of telomerase is sufficient to immortalize primary human ovarian surface epithelial cells. Carcinogenesis 28(1):174–182
Zhou C, Liu J (2003) Inhibition of human telomerase reverse transcriptase gene expression by BRCA1 in human ovarian cancer cells. Biochem Biophys Res Commun 303(1):130–136
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98(19):10869–10874
Wang E, Ngalame Y, Panelli MC, Nguyen-Jackson H, Deavers M, Mueller P, Hu W et al (2005) Peritoneal and subperitoneal stroma may facilitate regional spread of ovarian cancer. Clin Cancer Res 11(1):113–122
Folkman J (1995) Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1(1):27–31
Folkman J (1992) The role of angiogenesis in tumor growth. Semin Cancer Biol 3(2):65–71
Frumovitz M, Sood AK (2007) Vascular endothelial growth factor (VEGF) pathway as a therapeutic target in gynecologic malignancies. Gynecol Oncol 104:768–778
Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J (1989) Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest 84(5):1470–1478
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985
Yoneda J, Kuniyasu H, Crispens MA, Price JE, Bucana CD, Fidler IJ (1998) Expression of angiogenesis-related genes and progression of human ovarian carcinomas in nude mice. J Natl Cancer Inst 90(6):447–454
Paley PJ, Staskus KA, Gebhard K, Mohanraj D, Twiggs LB, Carson LF, Ramakrishnan S (1997) Vascular endothelial growth factor expression in early stage ovarian carcinoma. Cancer 80(1):98–106
Cooper BC, Ritchie JM, Broghammer CL, Coffin J, Sorosky JI, Buller RE, Hendrix MJ, Sood AK (2002) Preoperative serum vascular endothelial growth factor levels: significance in ovarian cancer. Clin Cancer Res 8(10):3193–3197
Lokshin AE, Winans M, Landsittel D, Marrangoni AM, Velikokhatnaya L, Modugno F, Nolen BM, Gorelik E (2006) Circulating IL-8 and anti-IL-8 autoantibody in patients with ovarian cancer. Gynecol Oncol 102(2):244–251
Xu L, Fidler IJ (2000) Interleukin 8: an autocrine growth factor for human ovarian cancer. Oncol Res 12(2):97–106
Davidson B, Goldberg I, Reich R, Tell L, Dong HP, Trope CG, Risberg B, Kopolovic J (2003) AlphaV- and beta1-integrin subunits are commonly expressed in malignant effusions from ovarian carcinoma patients. Gynecol Oncol 90(2):248–257
Thaker PH, Deavers M, Celestino J, Thornton A, Fletcher MS, Landen CN, Kinch MS, Kiener PA, Sood AK (2004) EphA2 expression is associated with aggressive features in ovarian carcinoma. Clin Cancer Res 10(15):5145–5150
Landen CN Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, Sood AK (2005) Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res 65(15):6910–6918
Landen CN Jr, Lu C, Han LY, Coffman KT, Bruckheimer E, Halder J, Mangala LS et al (2006) Efficacy and antivascular effects of EphA2 reduction with an agonistic antibody in ovarian cancer. J Natl Cancer Inst 98(21):1558–1570
Jung YD, Ahmad SA, Akagi Y, Takahashi Y, Liu W, Reinmuth N, Shaheen RM, Fan F, Ellis LM (2000) Role of the tumor microenvironment in mediating response to anti-angiogenic therapy. Cancer Metastasis Rev 19(1–2):147–157
Belotti D, Paganoni P, Manenti L, Garofalo A, Marchini S, Taraboletti G, Giavazzi R (2003) Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: implications for ascites formation. Cancer Res 63(17):5224–5229
Naylor MS, Stamp GW, Davies BD, Balkwill FR (1994) Expression and activity of MMPS and their regulators in ovarian cancer. Int J Cancer 58(1):50–56
Herrera CA, Xu L, Bucana CD, el Silva VG, Hess KR, Gershenson DM, Fidler IJ (2002) Expression of metastasis-related genes in human epithelial ovarian tumors. Int J Oncol 20(1):5–13
Huang S, Van Arsdall M, Tedjarati S, McCarty M, Wu W, Langley R, Fidler IJ (2002) Contributions of stromal metalloproteinase-9 to angiogenesis and growth of human ovarian carcinoma in mice. J Natl Cancer Inst 94(15):1134–1142
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB et al (2006) Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 12(8):939–944
Antoni MH, Lutgendorf SK, Cole SW, Dhabhar FS, Sephton SE, McDonald PG, Stefanek M, Sood AK (2006) The influence of bio-behavioural factors on tumour biology: pathways and mechanisms. Nat Rev Cancer 6(3):240–248
Ben-Hur H, Gurevich P, Huszar M, Ben-Arie A, Berman V, Tendler Y, Zinder O et al (2000) Apoptosis and apoptosis-related proteins (Fas, Fas ligand, Blc-2, p53) in lymphoid elements of human ovarian tumors. Eur J Gynaecol Oncol 21(1):53–57
Paul P, Rouas-Freiss N, Khalil-Daher I, Moreau P, Riteau B, Le Gal FA, Avril MF, Dausset J, Guillet JG, Carosella ED (1998) HLA-G expression in melanoma: a way for tumor cells to escape from immunosurveillance. Proc Natl Acad Sci USA 95(8):4510–4515
Bukovsky A (2006) Immune system involvement in the regulation of ovarian function and augmentation of cancer. Microsc Res Tech 69(6):482–500
Nash MA, Ferrandina G, Gordinier M, Loercher A, Freedman RS (1999) The role of cytokines in both the normal and malignant ovary. Endocr-Relat Cancer 6(1):93–107
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A et al (2003) Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348(3):203–213
Coukos G, Conejo-Garcia JR, Roden RB, Wu TC (2005) Immunotherapy for gynaecological malignancies. Expert Opin Biol Ther 5(9):1193–1210
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458
Emerich J, Konefka T, Dudziak M, Sobol A (1997) The value of peritoneal cytology in the staging of ovarian cancer. Ginekol Pol 68(2):74–77
Hood JD, Cheresh DA (2002) Role of integrins in cell invasion and migration. Nat Rev Cancer 2(2):91–100
Carreiras F, Rigot V, Cruet S, Andre F, Gauduchon P, Marvaldi J (1999) Migration properties of the human ovarian adenocarcinoma cell line IGROV1: importance of alpha(v)beta3 integrins and vitronectin. Int J Cancer 80(2):285–294
Sundfeldt K (2003) Cell–cell adhesion in the normal ovary and ovarian tumors of epithelial origin; an exception to the rule. Mol Cell Endocrinol 202(1–2):89–96
Halder J, Kamat AA, Landen CN Jr, Han LY, Lutgendorf SK, Lin YG, Merritt WM et al (2006) Focal adhesion kinase targeting using in vivo short interfering RNA delivery in neutral liposomes for ovarian carcinoma therapy. Clin Cancer Res 12(16):4916–4924
Sundfeldt K, Piontkewitz Y, Ivarsson K, Nilsson O, Hellberg P, Brannstrom M, Janson PO, Enerback S, Hedin L (1997) E-cadherin expression in human epithelial ovarian cancer and normal ovary. Int J Cancer 74(3):275–280
Acknowledgments
The authors would like to acknowledge support from the NIH (CA 110793, CA 109298, P50 CA083639, P50 CA098258, CA128797, RC2GM092599, U54 CA151668); the Ovarian Cancer Research Fund, Inc. (Program Project Development Grant); the DOD (OC073399, W81XWH-10-1-0158, BC085265); the Zarrow Foundation; the Marcus Foundation; the Kim Medlin Fund; the Betty Anne Asche Murray Distinguished Professorship; and the Meyer and Ida Gordon Foundation 2.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Saad, A.F., Hu, W. & Sood, A.K. Microenvironment and Pathogenesis of Epithelial Ovarian Cancer. HORM CANC 1, 277–290 (2010). https://doi.org/10.1007/s12672-010-0054-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-010-0054-2