
Abstract
Purpose
We recently showed that the quaternary lidocaine derivative, QX-314, produces long-lasting local anesthesia with a slow onset in animal models in vivo. As quaternary agents do not rapidly penetrate biological membranes or the blood-brain barrier, QX-314 may represent a local anesthetic with decreased systemic toxicity compared with conventional tertiary aminoamines. To test this hypothesis, we conducted an in vivo animal study in mice to compare QX-314 with lidocaine in terms of its relative central nervous system (CNS) and cardiac toxicity.
Methods
With approval from the institutional Animal Care Committee, we used the “up-and-down” method to determine the relative potencies (ED50) of lidocaine and QX-314 for CNS and cardiac toxicity in adult CD-1 mice (weight, 20 to 35 g). The animals were administered either intravenous lidocaine or QX-314 (dose range, 7.5 to 30 mg·kg−1) and were observed for signs of CNS toxicity (convulsions, ataxia, loss of righting reflex, and/or death). We also observed animals for electrocardiographic evidence of toxic effects on cardiac automaticity, conductivity, and rhythmicity.
Results
The ED50 of lidocaine for CNS toxicity as determined by the “up-and-down” method was 19.5 mg·kg−1 (95% confidence interval [CI], 17.7 to 21.3 mg·kg−1; n = 6) compared with 10.7 mg·kg−1 for QX-314 (95% CI, 9.1 to 12.3 mg·kg−1; n = 6) (potency ratio, 1.8). Similarly, the ED50 of lidocaine for electrocardiographic evidence of cardiac toxicity was significantly higher than that of QX-314 (ED50 of lidocaine, 21.2 mg·kg−1; 95% CI, 19.0 to 23.4 mg·kg−1; n = 6 vs ED50 of QX-314, 10.6 mg·kg−1; 95% CI, 8.4 to 12.8 mg·kg−1; n = 6) (potency ratio, 2.0).
Conclusions
In this in vivo animal study, the relative potencies of QX-314 for systemic CNS and cardiac toxicity were significantly higher than those of lidocaine. These data do not support the hypothesis that QX-314 is a safer local anesthetic compared with lidocaine in terms of systemic toxicity. Whereas our results do not exclude the possibility that QX-314 may represent a clinically useful agent to produce long-lasting local anesthesia and nociceptive blockade after a single shot in humans, its systemic toxicity relative to conventional tertiary aminoamide local anesthetics and the underlying mechanisms warrant further study.
Résumé
Objectif
Nous avons récemment démontré que le dérivé quaternaire de la lidocaïne, le QX-314, produisait une anesthésie locale prolongée, avec début d’action lent, chez les modèles animaux in vivo. Étant donné que les agents quaternaires pénètrent lentement les membranes biologiques et la barrière hémato-encéphalique, le QX-314 pourrait constituer un anesthésique local avec une toxicité systémique réduite par rapport aux amino-amines tertiaires conventionnels. Afin de tester cette hypothèse, nous avons réalisé une étude animale in vivo chez la souris afin de comparer le QX-314 à la lidocaïne en termes de leurs toxicités relatives au niveau du système nerveux central (SNC) et du cœur.
Méthode
Après avoir reçu l’approbation du Comité de recherche sur les animaux de notre institution, nous avons utilisé la méthode des suites croissantes et décroissantes afin de déterminer les puissances relatives (ED50) de la lidocaïne et du QX-314 en matière de toxicité au niveau du SNC et du cœur chez des souris adultes CD-1 (poids : 20-35 g). Nous avons administré de la lidocaïne intraveineuse ou du QX-314 aux souris (doses entre 7,5 et 30 mg·kg−1), et nous avons observé l’apparition de signes de toxicité systémique (convulsions, ataxie, perte du réflexe de redressement et/ou décès). Nous avons également examiné les animaux afin de déterminer la présence de signes électrocardiographiques d’effets toxiques sur l’automaticité, la conductivité et la rythmicité cardiaque.
Résultats
La dose efficace à 50 % (ED50) de la lidocaïne pour la toxicité systémique telle que déterminée par la méthode des suites croissantes et décroissantes était de 19,5 mg·kg−1 [intervalle de confiance (IC) 95 %, 17,7-21,3 mg·kg−1; n = 6], par rapport à 10,7 mg·kg−1 (IC 95 %, 9,1-12,3 mg·kg−1; n = 6) pour le QX-314 (rapport de puissance, 1,8). De même, la ED50 de la lidocaïne pour les signes électrocardiographiques de toxicité cardiaque était significativement plus élevée que celle du QX-314 (lidocaïne, 21,2 mg·kg−1; IC 95 %, 19,0-23,4 mg·kg−1; n = 6 vs QX-314, 10,6 mg·kg−1; IC 95 %, 8,4-12,8 mg·kg−1; n = 6; rapport de puissance, 2,0).
Conclusion
Dans cette étude animale in vivo, les puissances relatives du QX-314 pour la toxicité systémique au niveau du SNC et du cœur étaient significativement plus élevées que pour la lidocaïne. Ces données ne confirment pas l’hypothèse que le QX-314 est un anesthésique local plus sécuritaire que la lidocaïne en termes de toxicité systémique. Bien que nos résultats n’excluent pas la possibilité que le QX-314 constitue un agent utile d’un point de vue clinique à réaliser une anesthésie locale prolongée et un bloc nociceptif après une injection unique chez l’humain, sa toxicité systémique par rapport aux anesthésiques locaux amino-amides tertiaires conventionnels et les mécanismes sous-jacents nécessitent des études plus approfondies.
Similar content being viewed by others


According to conventional pharmacological wisdom, QX-314 (N-[2,6-dimethylphenyl carbamoylmethyl] triethylammonium), a quaternary derivative of lidocaine (Fig. 1), produces local anesthetic effects only when administered intracellularly to neurons.1 – 3 This supposition relates to the intracellular location of the local anesthetic binding site in the pore of voltage-gated Na+ channels4 and QX-314’s relatively poor penetrability through biological membranes due to its permanent positive charge. However, we have recently shown in animal models in vivo that QX-314 produces long-lasting nociceptive, sensory, and motor blockade with a slow onset when administered extracellularly as a peripheral local anesthetic.5 , 6 As quaternary agents do not rapidly penetrate biological membranes or the blood-brain barrier, QX-314 may represent a local anesthetic with decreased systemic toxicity compared with conventional tertiary aminoamines. In order to test this hypothesis, we conducted an in vivo animal study in mice to compare QX-314 with lidocaine in terms of its relative potencies for systemic central nervous system (CNS) and cardiac toxicity.

Molecular structure of lidocaine and QX-314. QX-314 (N-ethyl lidocaine; N-[2,6-dimethylphenyl carbamoylmethyl] triethylammonium) is a lidocaine derivative whose sole structural difference to the mother compound is the presence of an additional N-ethyl group. This renders the amino group quaternary, i.e., permanently positively charged. As a result, the agent traditionally has been considered membrane-impermeant
Methods
The experimental protocol was approved by The University of British Columbia Committee on Animal Care (Vancouver, BC, Canada). All efforts were made to minimize the suffering of animals and the number of animals used. All animals used in this experiment were adult CD-1 mice (weight, 20 to 35 g). The mice were housed 12 to a cage with a 12-hr light-dark cycle and free access to food and water. All animals were naïve to drug applications, and each animal was used only once for an experiment.
Drugs and chemicals
Lidocaine hydrochloride and N-(2,6-dimethylphenyl carbamoylmethyl) triethylammonium chloride (QX-314) were purchased from Sigma-Aldrich Canada Ltd. (Oakville, ON, Canada). Lidocaine and QX-314 were dissolved in normal saline (NaCl solution of 0.9% weight per volume). Both lidocaine and QX-314 solutions were freshly prepared on each experimental day immediately before the start of the experiments.
“Up-and-down method”
Traditional dose-response studies typically require a minimum of six mice at each dose and approximately five doses in order to construct the dose-response curve and determine the effective (ED50) and maximum tolerated doses. Designs utilizing a substantial number of animals for toxicity studies and the determination of lethal doses (such as LD50s, or ED50s for toxicity outcomes other than death) are no longer deemed acceptable by many Animal Care Committees, including that at our institution; however, an alternative approach, the “up-and-down method” (also known as the “staircase bioassay”), has been shown to be equally effective at determining an ED50 while reducing the number of animals to approximately one-third.7 – 9 This method has been used effectively for a variety of experimental settings and endpoints, including toxicity studies/lethal dose determinations and antinociceptive efficacy studies.10 , 11 Whereas the “up-and-down” method does not fully characterize the shape and slope of dose-response curves, it is associated with proven utility and reliability in determining the ED50 with appropriate statistical significance and excellent correlation with results from traditional dose-response studies.11 In brief, it involves administering a dose that is estimated to approximate the ED50 for the experimental endpoint of interest. If no effect is observed at this dose, then the dose for the subsequent drug application will be increased by a defined (log) dosing increment. If there is a positive effect at this increased dose, then the following dose will be decreased by the dosing increment. This procedure is repeated until data from a sufficient number of animals (i.e., a minimum of five additional animals after the first change from positive to negative or negative to positive effect) has been obtained. In this study, we used the “up-and-down” method to test two experimental endpoints, i.e., CNS toxicity and cardiac toxicity.
Central nervous system toxicity
Mice were placed gently into a holder and the tail was cleaned with an alcohol swab. With the use of a 30G 1/2 inch hypodermic needle and a 0.3 mL insulin syringe, the animals were injected intravenously in the tail vein with lidocaine or QX-314 and observed for generalized convulsions as a primary endpoint of CNS toxicity. We also monitored and tested the animals for other behavioural evidence of CNS toxicity (ataxia, loss of righting reflex, and death). If any of these endpoints were observed, then the experiment was considered a positive result for the “up-and-down” method. If a positive result occurred, then the subsequent dose in the series was decreased (see below). If a negative result occurred, then the dose for the next animal in the series was increased. The process was repeated with a new animal (naïve to drug application) injected with the new dose. Starting doses and dosing increments were determined from prior pilot experiments as estimations of ED50s and their standard deviations, respectively.7 For lidocaine, the starting dose was 15 mg·kg−1 with a log dosing increment of 0.222; for QX-314, the starting dose was 10 mg·kg−1 with a log dosing increment of 0.176 (Table 1). After each experiment, all mice were euthanized with an overdose of sevoflurane.
Cardiac toxicity
Animals were briefly anesthetized with sevoflurane in an anesthetic chamber (sevoflurane 0.2 mL in a 1.5-L chamber for two minutes). The animals were then taken out of the anesthetic chamber and placed in a Plexiglas holder equipped with predrilled access holes to permit the insertion of three electrocardiogram (ECG) leads. The distal ends (2 mm) of Teflon-coated stainless steel electrode wire (Medwire 316 SS5T; Leico Industries Inc., New York, NY, USA) were stripped of their coating, shaped in the form of a hook, and placed subcutaneously using a 25G hypodermic needle. One lead each was placed at the base of the neck, over the anterior abdomen subcostally, and in the midclavicular line; a ground electrode was placed in the right flank. Electrocardiographic data were acquired and displayed with a MacLab® recording system (MacLab/8 MK3, Version 3.4, ADInstruments Pty Ltd, Bella Vista, NSW, Australia) equipped with AC and 50 Hz filters and connected to a Macintosh computer (Apple, Cupertino, CA, USA) running the Chart software component of MacLab.
The animals were injected intravenously in the tail vein with lidocaine or QX-314 as described above, and they were observed for electrocardiographic evidence of toxic effects on cardiac automaticity, conductivity, and rhythmicity. We examined ECG traces at 30 sec before and after injection and determined heart rate from the number of QRS complexes within a one second interval. The primary endpoint for a positive result was a decrease in baseline heart rate ≥ 30%. We also considered the following secondary endpoints as a positive result: any evidence of atrioventricular conduction blockade, QRS complex widening, and/or other arrhythmia; for this purpose, ECG traces were assessed by the primary experimenter and a certified specialist anesthesiologist. If any of the above changes were observed, the study drug dose was decreased by the dosing increment (cf. below); for a negative result, the dose was increased. The process was repeated with a new drug-naïve animal at each new dose. As for the CNS toxicity experiments, starting doses and dosing increments were determined from prior pilot studies. For lidocaine, the starting dose was 30 mg·kg−1 with a log dosing increment of 0.301 (Table 2). For QX-314, the starting dose was 15 mg·kg−1 with a log dosing increment of 0.301. All animals were euthanized with sevoflurane after each experiment.
Data analysis
The data were analyzed as described for the “up-and-down method” (cf. above) by Lichtman11 and using the statistical methods and tables for ED50 determination originally described by Dixon in 1965.7 In brief, ED50s were calculated using the equation:
where Xf is the last dose administered; k is the tabular value determined based on the number of trials and the resulting test series, and d is the dosing increment. The 95% confidence intervals (CIs) were then calculated using the following equation:
where 95% CL denotes the 95% confidence limit calculated as follows:
where d is the dosing increment (cf. above), n is the number of trials (or animals), and 1.96 reflects an α level of 0.05.11 The 95% CIs were then determined according to equation (2) by adding or subtracting the 95% CL, equation (3), from the ED50, equation (1). Differences in ED50 between lidocaine and QX-314 were considered significant if the 95% CIs did not overlap.11
In the systemic cardiac toxicity experiments, we used descriptive statistics for baseline heart rates and expressed the data as mean (SD). The data were analyzed with the aid of Microsoft Excel version 2003 (Microsoft Corporation, Redmond, WA, USA) and Prism version 5 software (GraphPad, San Diego, CA, USA).
Results
The ED50 of lidocaine for CNS toxicity as determined by the “up-and-down” method was 19.5 mg·kg−1 (95% CI, 17.7 to 21.3 mg·kg−1; n = 6), compared with 10.7 mg·kg−1 for QX-314 (95% CI, 9.1 to 12.3 mg·kg−1; n = 6) (potency ratio, 1.8) (Fig. 2A). All mice (n = 3) that exhibited a positive result for CNS toxicity after lidocaine administration recovered completely. Of the three mice that tested positive for CNS toxicity after QX-314 administration, only one recovered completely; the other two animals died within three minutes of injection. The time to onset of generalized convulsions ranged from two to five seconds in the lidocaine group and from 25 to 30 sec in the QX-314 group. Table 1 provides details on the “up-and-down” test series results from the CNS toxicity experiments.

Systemic central nervous system and cardiac toxicity in mice of intravenous lidocaine and QX-314. The graphs show the respective potencies as expressed by ED50 values with their 95% confidence intervals as determined by the “up-and-down” method (each group, n = 6; for details, see body text). QX-314 was significantly more potent than lidocaine in terms of systemic central nervous system (CNS) toxicity (panel [A]; potency ratio, 1.8) as well as in terms of systemic toxic effects on cardiac automaticity, conductivity, and rhythmicity (panel [B]; potency ratio, 2.0)
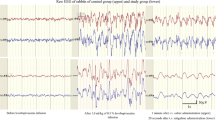
In the systemic cardiac toxicity experiments, the mice had baseline heart rates of 490 (88) beats·min−1 in the lidocaine group and 550 (45) beats·min−1 in the QX-314 group (each group, n = 6). Similar to the CNS toxicity experiments, the ED50 of lidocaine for electrocardiographic evidence of cardiac toxicity was significantly higher than that of QX-314 (ED50 of lidocaine, 21.2 mg·kg−1; 95% CI, 19.0 to 23.4 mg·kg−1; n = 6 vs ED50 of QX-314, 10.6 mg·kg−1; 95% CI, 8.4 to 12.8 mg·kg−1; n = 6) (potency ratio, 2.0) (Fig. 2B). In both groups, all animals with a positive result exhibited significant bradycardia consistent with conduction blockade, with a maximum observed reduction in baseline heart rate of 56% in both groups. In one animal injected with lidocaine, a Q wave amplitude increase to a magnitude approximating that of the R wave was noted. In one animal injected with QX-314, heart rate determination at 30 sec was not possible due a transient ECG baseline shift out of the visible monitor field. The ECG tracing reappeared at 39 sec, at which time the heart rate determination was performed (positive result). All mice died that tested positive for cardiac toxicity after lidocaine or QX-314 administration. In Fig. 3, examples are shown of the electrocardiographic effects of lidocaine (A) and QX-314 (B) on cardiac automaticity, conductivity, and rhythmicity. Table 2 summarizes the results from the “up-and-down” test series from the cardiac toxicity experiments.

Electrocardiographic (ECG) tracings are shown before and after intravenous administration of (A) lidocaine 15 mg·kg−1 and (B) QX-314 15 mg·kg−1 in mice. The ECG tracings were captured at 30 sec before (baseline) and 30 sec after the injection. The animal injected with lidocaine exhibited no significant ECG changes, with the heart rate remaining stable at ~600 beats·min−1. The animal injected with QX-314 developed marked conduction blockade and bradycardia, with a 56% decrease in heart rate from ~540 to ~240 beats·min−1
Discussion
In this in vivo animal study in mice, the relative potencies for systemic CNS and cardiac toxicity of the quaternary lidocaine analog, QX-314, were significantly higher than those of lidocaine, by a factor of approximately 2. These results were contrary to what one might expect intuitively, and they do not support the hypothesis that QX-314 per se is a safer local anesthetic compared with lidocaine in terms of systemic toxicity.
Our findings that lidocaine produces systemic CNS and cardiac toxicity in mice with an ED50 of 19.5 and 21.2 mg·kg−1, respectively, corresponds well with published observations from mice and other species. For example, the intravenous convulsant dose (CD100) of lidocaine is approximately 22 mg·kg−1 in monkeys and dogs12 , 13; in mice, the intravenous CD50 was previously found to be 21.5 mg·kg−1.14 In addition to serving as a strong external validation of our results, these findings re-emphasize the utility, accuracy, and reliability of the “up-and-down” method to determine drug potencies while greatly reducing the number of required experimental animals compared with traditional pharmacological dose-response and toxicity methods.
Consistent with the traditionally held notion that the cardiovascular system is more resistant to systemic local anesthetic toxicity than the CNS,15 the ED50 value of lidocaine in the present study was slightly lower for CNS effects than for cardiac effects; QX-314 was not associated with a difference in potency. However, it is important to note in this regard that the relative resistance of the cardiovascular system to local anesthetic toxicity relates to cardiovascular depression or LD100 as experimental endpoints.16 , 17 In our study, we did not look at cardiovascular collapse or death as “up-and-down method” endpoints for cardiac toxicity; rather, we looked at effects on automaticity, conductivity, and rhythmicity in the ECG, known to occur in a lower concentration range.18 While these details—combined with the fact that lidocaine has long been known to affect cardiac Na+ channels at lower concentrations than in myelinated nerve19—could account for the relative closeness in observed potency for CNS vs cardiac toxicity in the present investigation, we noted that all animals in both groups died that tested positive for cardiac toxicity.
For our systemic cardiac toxicity experiments, we used sevoflurane anesthesia to produce immobility to aid placement of the ECG leads in the animals, as we found in pilot experiments that the active movement of fully conscious mice inside the holder impairs accurate ECG lead placement, dislodges leads, and/or produces an unacceptable level of recording noise. Unfortunately, sevoflurane itself depresses both the CNS and the cardiovascular system. Hence, we cannot completely rule out the possibility that this may have affected the experimental results. However, sevoflurane is associated with rapid induction and elimination times, and all the animals were conscious during the intravenous injection. Hence, any residual sevoflurane concentrations in the animals during the actual ECG observations were likely small. Furthermore, all animals (i.e., both lidocaine and QX-314 groups at all doses) received equal treatment with respect to sevoflurane anesthesia.
Although we investigated the systemic CNS and cardiac effects independently in separate lines of experiments, they are not necessarily physiologically independent one from the other, as signs of cardiac toxicity may occur secondary to CNS toxicity and vice versa. For example, in 1986, Heavner reported that cardiac dysrhythmias occurred when local anesthetics were administered into the lateral cerebral ventricle of cats.20 It has also been demonstrated that administration of local anesthetics, such as lidocaine and bupivacaine, directly into the nucleus tractus solitarius of the rat medulla can lead to hypotension, bradycardia, and arrhythmias.21 – 23 Similarly, CNS effects may occur due to cardiovascular toxicity. For example, cardiac dysrhythmia or depression may lead to cerebral hypoxia resulting in seizure activity or other adverse CNS effects; this is particularly the case for loss of righting reflex and death as endpoints. However, no animal in our study tested positive for CNS toxicity on the basis of these endpoints alone, i.e., without convulsions or ataxia. That said, the above is still possible and, in fact, may be more probable with QX-314 than with lidocaine. Several authors have suggested that QX-314 does not cross the blood-brain barrier, or if it does, it does so extremely slowly.24 , 25 Since the CNS effects in response to QX-314 still occurred relatively quickly after injection, it is theoretically possible that observed convulsions may have been due in part to QX-314 having caused cerebral hypoxia secondary to cardiac toxicity rather than due to direct action of QX-314 in the brain. Supportive of this possibility would be our findings that the majority of animals with evidence of CNS toxicity following QX-314 injection died within three minutes, whereas all animals injected with lidocaine recovered fully.
This is the second study in which we have unexpectedly found the quaternary lidocaine derivative, QX-314, to be significantly more toxic than its tertiary mother compound. While the present experiments focus on systemic toxicity, we recently reported that administering QX-314 in the lumbar intrathecal space as a spinal anesthetic produces marked irritation and death in mice – at concentrations lower than those associated with robust motor blockade and in contrast to lidocaine.26 Although these results do not categorically rule out the possibility that QX-314 may still represent a useful peripheral local anesthetic agent with the potential to produce long-lasting local anesthesia and nociceptive blockade after a single dose, our previous results indicate that QX-314’s potency for peripheral sensory and motor blockade is equal or lower than that of lidocaine.5 , 6 Combined with our findings of a higher toxic potency, these observations would collectively suggest that QX-314 has a lower therapeutic index than lidocaine and is less safe. That said, the routine safe clinical practice of administering a local anesthetic that is longer acting but more systemically toxic than lidocaine is commonplace and well illustrated by the example of bupivacaine. However, many important questions regarding the clinical toxicity of QX-314 relative to the shorter-acting conventional tertiary aminoamide local anesthetics and its underlying mechanisms remain unanswered and require further investigation.
For example, from a pharmacokinetic point of view, we studied systemic toxicity in the present experiments in the context of intravenous injection (which clinically may be inadvertent or deliberate). However, it is not known how QX-314 would compare with lidocaine in terms of systemic toxicity due to systemic absorption from a peripheral/tissue injection site; in this scenario, it is conceivable that QX-314 might represent an advantage due to its inherently slower absorption kinetics. In turn, it is possible that the higher intravenous toxic potency of QX-314 compared with lidocaine has a pharmacokinetic component due to slower redistribution from the central compartment, i.e., plasma. That said, our present and previous results strongly support the notion that QX-314 as a quaternary agent can penetrate biological membranes and the blood-brain barrier (albeit more slowly than lidocaine), and it is “effective” extracellularly27 without co-administration of other pharmacological tools that might facilitate intracellular flux, such as transient receptor potential vanilloid (TRPV1) receptor agonists.5 , 6 , 28 , 29 It should be noted in this context that the concept of administering quaternary drugs in general is very familiar to clinical anesthesiologists, as illustrated by the examples of neuromuscular blocking drugs and glycopyrrolate.
From the point of view of cellular and molecular actions, the precise mechanisms by which QX-314 produced its systemic toxic effects in the present study and the intrathecal adverse effects in our recent report26 remain unknown. However, our observations that QX-314 produces systemic and intrathecal adverse effects at a higher potency than lidocaine despite its inherent property of slower diffusibility indicate that the quaternary amine function likely confers specific structure-activity relationship properties to this compound that are not shared by lidocaine. The fact that relatively small variations of a local anesthetic molecule can significantly effect potency with regard to specific molecular targets is well-known and impressively exemplified by the differential stereoselecive actions of levo- and dextrobupivacaine on various human K+ channels.30 , 31 While a body of future research is required to define the different actions of QX-314, our recent observations that this agent itself at clinically relevant concentrations activates TRPV1 channels32 may provide a mechanism for some of its adverse effects.
Finally, we are well aware that our toxicological data stem from animal studies in small rodents and cannot necessarily be extrapolated directly to the clinical domain in humans due to inter-species differences in dose-response, biotransformation, and target biological substrates,33 again emphasizing the imperative for further study to include different species.
In summary, in an in vivo animal study using the “up-and-down” method in mice, the quaternary lidocaine derivative, QX-314, produced systemic CNS and cardiac toxicity with intravenous potencies approximately twice as high as lidocaine. On the basis of the data available, we conclude that QX-314 appears to have a relatively narrow therapeutic range compared with lidocaine, and, despite its inherently lower diffusibility, does not represent a safer local anesthetic agent in terms of systemic toxicity.
References
Frazier DT, Narahashi T, Yamada M. The site of action, active form of local anesthetics. II. Experiments with quaternary compounds. J Pharmacol Exp Ther 1970; 171: 45-51.
Narahashi T, Frazier DT, Moore JW. Comparison of tertiary and quaternary amine local anesthetics in their ability to depress membrane ionic conductances. J Neurobiol 1972; 3: 267-76.
Strichartz GR. The inhibition of sodium currents in myelinated nerve by quaternary derivatives of lidocaine. J Gen Physiol 1973; 62: 37-57.
Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994; 265: 1724-8.
Lim TKY, MacLeod BA, Ries CR, Schwarz SKW. The quaternary lidocaine derivative, QX-314, produces long lasting local anesthesia in animal models in vivo. Anesthesiology 2007; 107: 305-11.
Ries CR, Pillai R, Chung CCW, Wang JTC, MacLeod BA, Schwarz SKW. QX-314 produces long-lasting local anesthesia modulated by transient receptor potential vanilloid receptors in mice. Anesthesiology 2009; 111: 122-6.
Dixon WJ. The up-and-down method for small samples. J Am Stat Assoc 1965; 60: 967-78.
Bruce RD. An up-and-down procedure for acute toxicity testing. Fundam Appl Toxicol 1985; 5: 151-7.
Dixon WJ. Staircase bioassay: the up-and-down method. Neurosci Biobehav Rev 1991; 15: 47-50.
Lipnick RL, Cotruvo JA, Hill RN, et al. Comparison of the up-and-down, conventional LD50, and fixed-dose acute toxicity procedures. Food Chem Toxicol 1995; 33: 223-31.
Lichtman AH. The up-and-down method substantially reduces the number of animals required to determine antinociceptive ED50 values. J Pharmacol Toxicol Methods 1998; 40: 81-5.
Munson ES, Tucker WK, Ausinsch B, Malagodi MH. Etidocaine, bupivacaine, and lidocaine seizure thresholds in monkeys. Anesthesiology 1975; 42: 471-8.
Liu PL, Feldman HS, Giasi R, Patterson MK, Covino BG. Comparative CNS toxicity of lidocaine, etidocaine, bupivacaine, and tetracaine in awake dogs following rapid intravenous administration. Anesth Analg 1983; 62: 375-9.
de Jong RH, Bonin JD. Deaths from local anesthetic-induced convulsions in mice. Anesth Analg 1980; 59: 401-5.
Scott DB. Toxicity caused by local anaesthetic drugs. Br J Anaesth 1981; 53: 553-4.
Liu P, Feldman HS, Covino BM, Giasi R, Covino BG. Acute cardiovascular toxicity of intravenous amide local anesthetics in anesthetized ventilated dogs. Anesth Analg 1982; 61: 317-22.
Covino BG. Toxicity and systemic effects of local anesthetic agents. In: Strichartz GR (Ed.). Handbook of Experimental Pharmacology, Vol. 81: Local Anesthetics. Berlin: Springer-Verlag; 1987: 187-212.
Covino BG. Ultralong-acting local anesthetic agents. Anesthesiology 1981; 54: 263-4.
Courtney KR, Strichartz GR. Structural elements which determine local anesthetic activity. In: Strichartz GR (Ed.). Handbook of Experimental Pharmacology, Vol. 81: Local Anesthetics. Berlin: Springer-Verlag; 1987: 53-94.
Heavner JE. Cardiac dysrhythmias induced by infusion of local anaesthetics into the lateral cerebral ventricle of cats. Anesth Analg 1986; 65: 133-8.
Thomas RD, Behbehani MM, Coyle DE, Denson DD. Cardiovascular toxicity of local anesthetics: an alternative hypothesis. Anesth Analg 1986; 65: 444-50.
Denson DD, Behbehani MM, Gregg RV. Effects of an intravenously administered arrhythmogenic dose of bupivacaine at the nucleus tractus solitarius in the conscious rat. Reg Anesth 1990; 15: 76-80.
Denson DD, Behbehani MM, Gregg RV. Enantiomer-specific effects of an intravenously administered arrhythmogenic dose of bupivacaine on neurons of the nucleus tractus solitarius and the cardiovascular system in the anesthetized rat. Reg Anesth 1992; 17: 311-6.
Omana-Zapata I, Khabbaz MA, Hunter JC, Bley KR. QX-314 inhibits ectopic nerve activity associated with neuropathic pain. Brain Res 1997; 771: 228-37.
Kawamata M, Sugino S, Narimatsu E, et al. Effects of systemic administration of lidocaine and QX-314 on hyperexcitability of spinal dorsal horn neurons after incision in the rat. Pain 2006; 122: 68-80.
Schwarz SKW, Cheung HMC, Ries CR, et al. Lumbar intrathecal administration of the quaternary lidocaine derivative, QX-314, produces irritation and death in mice. Anesthesiology 2010; 113: 438-44.
Longobardo M, Gonzalez T, Navarro-Polanco R, et al. Effects of a quaternary bupivacaine derivative on delayed rectifier K+ currents. Br J Pharmacol 2000; 130: 391-401.
Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature 2007; 449: 607-10.
Gerner P, Binshtok AM, Wang CF, et al. Capsaicin combined with local anesthetics preferentially prolongs sensory/nociceptive block in rat sciatic nerve. Anesthesiology 2008; 109: 872-8.
Valenzuela C, Delpon E, Tamkun MM, Tamargo J, Snyders DJ. Stereoselective block of a human cardiac potassium channel (Kv1.5) by bupivacaine enantiomers. Biophys J 1995; 69: 418-27.
Gonzalez T, Arias C, Caballero R, et al. Effects of levobupivacaine, ropivacaine and bupivacaine on HERG channels: stereoselective bupivacaine block. Br J Pharmacol 2002; 137: 1269-79.
Rivera-Acevedo RE, Pless SA, Ahern CA, Schwarz SKW. The quaternary lidocaine derivative, QX-314, exerts biphasic effects on transient receptor potential vanilloid subtype 1 channels in vitro. Anesthesiology 2011 (in press).
Garattini S. Toxic effects of chemicals: difficulties in extrapolating data from animals to man. Crit Rev Toxicol 1985; 16: 1-29.
Acknowledgements
The authors sincerely thank Drs. Ernest Puil and Richard A. Wall (professors emeriti) for their valuable discussion, suggestions, and comments. We also thank Jimmy T. C. Wang (graduate student) for his technical assistance (all, Department of Anesthesiology, Pharmacology & Therapeutics, The University of British Columbia).
Financial support
Supported in part by the Jean Templeton Hugill Anesthesia Research Foundation and a Canadian Anesthesiologists’ Society Research Award, as well as a Canadian Anesthesiologists’ Society/Abbott Laboratories Ltd Career Scientist Award in Anesthesia granted to Dr. Schwarz. Dr. Schwarz is recipient of a Leaders Opportunity Fund award from the Canada Foundation for Innovation.
Conflicts of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was presented in part at the 2009 Annual Meeting of the Canadian Anesthesiologists’ Society Annual Meeting (Vancouver, BC, Canada), where it won the Award for Best Paper in Regional Anesthesia and Acute Pain.
Rights and permissions
About this article
Cite this article
Cheung, H.M.C., Lee, S.M., MacLeod, B.A. et al. A comparison of the systemic toxicity of lidocaine versus its quaternary derivative QX-314 in mice. Can J Anesth/J Can Anesth 58, 443–450 (2011). https://doi.org/10.1007/s12630-011-9479-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12630-011-9479-5