
Abstract
The mitochondrial calcium-activated potassium channel (mitoKCa) and the mitochondrial ATP-sensitive potassium channel (mitoKATP) are both involved in cardiac preconditioning. Here, we examined whether these two channels are also involved in ischemic or pharmacological postconditioning. Using Langendorff perfusion, rat hearts were made hypoxic for 45 min and then reoxygenated for 30 min. Ischemic postconditioning (IPT) was achieved through application of 3 cycles of 10 s of reperfusion and 10 s of ischemia before reoxygenation, with and without paxilline (Pax; a mitoKCa blocker) or 5-hydroxydecanoate (5-HD; a mitoKATP blocker). Pharmacological postconditioning was carried out for 5 min at the onset of reoxygenation using NS1619 (a mitoKCa opener) or diazoxide (Dia; a mitoKATP opener). Pax and 5-HD abolished IPT-induced cardioprotection from reoxygenation injury, whereas administration of NS1619 or Dia significantly improved cardiac contractile activity and reduced aspartate aminotransferase (an index of myocyte injury) release following reoxygenation. In addition, isolated rat myocytes were loaded with tetramethylrhodamine methyl ester (TMRE; fluorescent mitochondrial membrane potential indicator) and 2′,7′-dichlorofluorescein [DCFH; fluorescent reactive oxygen species (ROS) indicator] or Fluo-4-acetoxymethyl ester (Fluo-4-AM; fluorescent calcium indicator). When TMRE-loaded myocytes were laser illuminated, the DCFH and Fluo-4 fluorescence increased, and TMRE fluorescence decreased. These effects were significantly inhibited by NS1619 and Dia. We therefore conclude that IPT may protect the heart through activation of mitoKATP and mitoKCa channels, and that opening of these channels at the onset of reoxygenation protects the heart from reoxygenation injury, most likely by reducing excess generation of ROS and the resultant Ca2+ overload.
Similar content being viewed by others


Introduction
Preconditioning is a well-characterized phenomenon in which a brief period of ischemia or addition of a pharmacological agent can protect the myocardium from infarction induced by a subsequent and prolonged ischemic attack [1–5]. More recently, it was reported that postconditioning [6–9]—i.e., repetitive cycles of reperfusion and ischemia or addition of pharmacological agents at the onset of reperfusion—enables substantial salvage of the myocardium and offers a degree of protection similar to that obtained with preconditioning. Cardioprotection through preconditioning and postconditioning can be induced by a variety of stimuli [10, 11], but the opening of mitochondrial ATP-sensitive potassium channels (mitoKATP), which are involved in the regulation of mitochondrial function, appears to be a common step in mediating the cardioprotective effects of ischemic and pharmacological preconditioning and postconditioning [3, 9, 12, 13]. Although the mechanism remains unclear, it is thought that the activity of this channel may suppress the opening of the mitochondrial permeability transition pore (mPTP).
In addition, recent studies have shown that activation of another potassium channel, the mitochondrial calcium-activated potassium channel (mitoKCa), is also involved in preconditioning [14, 15]. The opening of mitoKCa channels induces both early and delayed preconditioning in adult mice [16], reduces generation of reactive oxygen species (ROS), and improves mitochondrial bioenergetics after ischemia–reperfusion in guinea pig hearts [4]. However, it is uncertain whether activation of mitoKCa channels is involved in ischemic postconditioning (IPT) or pharmacological postconditioning.
Events thought to be involved in reperfusion/reoxygenation injury include robust generation of ROS [17–19], Ca2+ overload [20, 21] and the opening of mPTP [22]. Our aim was to test the hypothesis that the effects of IPT are associated with activation of mitoKCa and mitoKATP channels, and that the activation of these channels at the onset of reoxygenation protects hearts from injury by reducing ROS generation and the accumulation of cytosolic Ca2+. To investigate this issue, we examined the effects of the mitoKCa channel opener NS1619 (1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-trifluoromethyl-2H-benzimidazol-2-one) and the mitoKATP channel opener diazoxide (Dia) on cardiac function and on ROS generation, mPTP opening, and cytosolic Ca2+ levels in isolated cardiac myocytes.
Materials and methods
This investigation conformed to the Guide for the Care and Use of Laboratory Animals [US National Institute of Health publication, DHEW publication No. (NIH) 85-23, 1996] and was approved by the Juntendo University Faculty of Medicine (Tokyo, Japan) animal experimentation committee.
Langendorff perfusion
Male Sprague–Dawley (SD) rats weighing 285–340 g (7–8 weeks) were used. After anesthetizing the rats with pentobarbital sodium (55 mg kg−1, i.p.), they were decapitated and their hearts were quickly excised for Langendorff perfusion [23]. Each heart was perfused with modified Krebs–Henseleit solution (containing in mM: NaCl 116.0, NaHCO3 25.0, MgSO4 1.2, KCl 4.7, KH2PO4 1.2, CaCl2 2.0, glucose 5.5, pH 7.4) in a retrograde direction at a constant flow rate of 13 ml min−1 without recirculation. The perfusate was warmed to 38 °C and oxygenated with a 95 % O2–5 % CO2 gas mixture to maintain the partial pressure of O2 above 400 mmHg. During the period of hypoxia, glucose in the perfusate was replaced with equimolar sucrose, and the solution was saturated with a 95 % N2–5 % CO2 gas mixture to maintain the partial pressure of O2 at approximately 20 mmHg (hypoxic solution). In addition, a latex balloon was inserted through the mitral annulus into the left ventricular cavity, and distilled water (0.1–0.2 ml) was injected into the balloon until it was inflated to just above the level required to produce a visible (1–2 mmHg) elevation in left ventricular end-diastolic pressure (LVEDP). Left ventricular developed pressure (LVDP), LVEDP, heart rate (HR), and coronary perfusion pressure were monitored throughout the experiments. The extent of the irreversible myocardial damage was assessed by measuring the amount of aspartate aminotransferase (AST) released into the coronary effluent. The AST activity in 10-μl aliquots of coronary effluent was estimated using a dry chemical method with a commercially available kit (Fuji Film, Tokyo, Japan). The released AST activity was normalized to IU g−1 min−1 (dry tissue).
Perfusion protocols
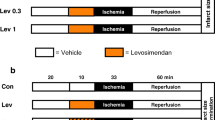
After stabilization for 20 min, the hearts were subjected to 45 min of hypoxia followed by 30 min of reoxygenation (Fig. 1). IPT was attained by 3 cycles of 10 s of reperfusion and 10 s of ischemia before reoxygenation (by stopping coronary perfusion) in the absence or presence of paxilline (Pax, mitoKCa blocker; 1 μM) or 5-hydroxydecanoate (5-HD, mitoKATP blocker; 100 μM). Pharmacological postconditioning was performed for 5 min at the onset of reoxygenation using NS1619 (mitoKCa opener; 30 μM) or Dia (mitoKATP opener; 100 μM).

Protocols for the Langendorff experiment. Hearts were stabilized for at least 20 min, after which they were subjected to 45 min of hypoxic perfusion followed by 30 min of reoxygenation. CT control, IPT ischemic postconditioning, I–R ischemia–reperfusion, Pax paxilline (mitoKCa channel blocker, 1 μM), 5-HD 5-hydroxydecanoate (mitoKATP channel blocker, 100 μM), NS1619 (mitoKCa channel opener, 30 μM), Dia diazoxide (mitoKATP channel opener, 100 μM), n number of hearts
Isolation of ventricular myocytes
Ventricular myocytes were isolated from adult male SD rats (285–340 g). After anesthetizing the rats with pentobarbital sodium (55 mg/kg, i.p.), the hearts were rapidly excised and perfused using a Langendorff apparatus, first with normal Tyrode solution (in mM: NaCl 135, KCl 5.4, CaCl2 1.8, MgCl2 1.0, HEPES 10, glucose 10, pH 7.4) for 5 min, then with Ca2+-free Tyrode for 5 min, and finally with collagenase (0.45 mM; S-1) solution for 5 min. All solutions were saturated with 100 % O2 and warmed to 37 °C. After digestion, the ventricles were cut into small pieces and dissociated in nominally Ca2+-free Tyrode solution in a shaking water bath. The resultant suspension was filtered through nylon mesh (ϕ 200 μm) to obtain the dissociated myocytes. The Ca2+ concentration was then gradually increased to 2.0 mM, and the ventricular myocytes were stored in normal Tyrode solution at room temperature (22–25 °C).
Induction of mPTP opening in ventricular myocytes
Intermittent laser illumination of cardiomyocytes loaded with a mitochondria-specific fluorescent dye, tetramethylrhodamine ethyl ester (TMRE), generates ROS within the mitochondria that provoke the mPTP [24–27]. Oxidative stress generated upon hypoxia/reoxygenation of myocardium also involves the excess production of ROS within the mitochondria, and this model should therefore simulate the events associated with hypoxia/reoxygenation-induced cell injury.
In the present study, isolated myocytes were loaded with TMRE (2 μM) for 10 min, washed, and then loaded with the fluorescent Ca2+ indicator Fluo-4 acetoxymethyl ester (Fluo-4-AM, 10 μM) or the ROS-specific indicator 2′,7′-dichlorofluorescein (DCFH; 10 μM) for 30 min. After washout of the fluorescent dyes, the isolated ventricular myocytes were stored at room temperature in normal Tyrode solution. To elicit mPTP opening, TMRE-loaded myocytes were intermittently illuminated (for 4 s with 20-s intervals) at 543 nm for 15 min using a 5-mW He/Ne laser system (Laser-Fertigung, Hamburg, Germany). mPTP opening was detected as a reduction in 574 nm fluorescent emission.
The combination of a 488-nm 200-mW argon laser (Laser-Fertigung) and 520 nm long pass emission filters were used to visualize the Fluo-4 and DCFH fluorescences. In the experiment, the dye-loaded myocytes were perfused with oxygenated Krebs–Henseleit solution, and NS1619 (30 μM) or Dia (100 μM) was administered throughout the laser illumination. In addition, changes in cell length induced by the intermittent laser illumination were measured and expressed as the ratio of final/initial cell length.
To measure time-dependent fluorescence changes, data were acquired every 20 s. Confocal imaging was carried out using a Zeiss laser-scanning microscope (LSM 510 META v.3.2; Carl Zeiss, Germany) equipped with a Zeiss Plan Apochromat 40× immersion objective.
Chemicals
Pax, NS1619, 5-HD and Dia were purchased from Biomol (PA, USA). Collagenase was from Nitta Gelatin (Osaka, Japan). TMRE and Fluo-4-AM were from Molecular Probes (Eugene, OR, USA). DCFH was from Wako Pure Chemicals (Osaka, Japan).
Statistical analysis
Data are presented as mean ± SEM. Differences between means were analyzed using Student’s t test or ANOVA and the Bonferroni method, as deemed appropriate. Values of P < 0.05 were considered significant.
Results
Blocking of mitoKCa or mitoKATP channel inhibits IPT-induced cardioprotection
Figure 2 shows the effects of Pax, a mitoKCa channel blocker and 5-HD, a mitoKATP channel blocker on IPT-induced cardioprotection. The changes in HR are shown in Fig. 2a. Recovery of HR at 10 min into reoxygenation was significantly greater (P < 0.05) in the IPT group (275 ± 13 bpm, mean ± SEM) compared to the CT group (214 ± 21 bpm), the IPT + Pax group (191 ± 33 bpm), and the IPT + 5HD group (191 ± 34 bpm). After 30 min of reoxygenation, the HR had recovered to the basal level in all four groups, and there was no significant difference among them.

Effects of mitoKCa and mitoKATP channel inhibition on IPT-induced cardioprotection. Filled circles, filled squares, open triangles and filled triangles indicate the control (CT), IPT, IPT + Pax, and IPT + 5-HD groups, respectively. a Heart rate, b pressure-rate product (PRP), c released AST. *P < 0.05 versus CT; # P < 0.05 versus IPT
The changes in cardiac contractile activity are shown in Fig. 2b, where it is expressed as a pressure-rate product (PRP = HR × LVDP) and normalized as a percentage of the basal PRP (N). Upon reoxygenation, recovery of PRP was significantly faster and greater in the IPT group (57.9 ± 2.0 %, R30, P < 0.05) than in the CT group (47.2 ± 1.8 %, R30). In the IPT + Pax group (51.9 ± 2.9 %, R30) and in the IPT+5-HD (39.4 ± 3.3 %, R30), the changes in PRP upon reoxygenation were similar to those in the CT group, and recovery levels were significantly lower (P < 0.05) than in the IPT group.
Irreversible cardiac damage was estimated based on the amount of AST released into the coronary effluent (Fig. 2c). After the first 2 min of reoxygenation, the AST release was significantly lower (P < 0.05) in the IPT group (1.2 ± 0.2 IU g−1 min−1) than in the CT group (2.4 ± 0.2 IU g−1 min−1), IPT + Pax group (2.0 ± 0.2 IU g−1 min−1), and IPT + 5-HD (2.6 ± 0.3 IU g−1 min−1) group.
Pax (n = 6) or 5-HD (n = 6) were also given for 5 min without any other treatment at the onset of reoxygenation. There were no significant differences in HR, PRP, and AST release among the CT, Pax, and 5-HD groups (data not shown).
Pharmacological postconditioning with a mitoKCa or mitoKATP channel opener mitigates hypoxia-reoxygenation injury
Our observation that blocking of mitoKCa and mitoKATP channels inhibited the cardioprotective effects of IPT prompted us to assess the efficacy of pharmacological postconditioning with a mitoKCa or mitoKATP channel opener, NS1619 or Dia, respectively (Fig. 3). We found that HR recovered to a similar level in the three groups tested (Fig. 3a). On the other hand, NS1619 improved the recovery of PRP. At the end of the reoxygenation, PRP had recovered to 59.8 ± 6.0 % in the NS1619 group, which was significantly higher (P < 0.05) than in the CT group (46.6 ± 2.8 %, R30) (Fig. 3b). Dia had a similarly beneficial effect on PRP, so that the recovery of PRP (56.8 ± 2.6 %, R30) was significantly better (P < 0.05) in the Dia group than the CT group (Fig. 3b). Finally, after the first 2 min of reoxygenation, significantly less (P < 0.05) AST were released in the NS1619 (1.0 ± 0.3 IU g−1 min−1) and Dia (1.4 ± 0.1 IU g−1 min−1) groups than in the CT group (2.9 ± 0.5 IU g−1 min−1) (Fig. 3c).

Effects of pharmacological postconditioning using a mitoKATP or mitoKCa channel opener. Filled circles, open inverted triangles and filled inverted triangles indicate the control (CT), NS1619 and diazoxide (Dia) groups, respectively. a Heart rate, b pressure-rate product (PRP), c released AST. *P < 0.05 versus CT
MitoKCa and mitoKATP channel openers inhibit ROS production and Ca2+ accumulation in cardiac myocytes
We next assessed the ability of NS1619 and Dia to protect against laser-induced cardiac myocyte damage. Figure 4a shows representative sequential confocal images of TMRE/DCFH-loaded (left panel) and TMRE/Fluo-4-loaded (right panel) cardiomyocytes before and after 15 min of intermittent laser illumination. Increases and decreases in the levels of intracellular ROS and Ca2+ are reflected by corresponding changes in the fluorescence intensity of DCFH (yellow) and Fluo-4 (green), respectively, while mPTP openings are indicated by reductions in TMRE fluorescence (red). After 15 min of intermittent laser illumination, DCFH intensity had increased to 1.69 ± 0.19-fold (mean ± SEM, expressed as fold changes of intensity before laser illumination) in the CT group (Fig. 4b). This increase in the relative DCFH intensity was significantly (P < 0.05) diminished to 1.13 ± 0.12-fold in the NS1619 group and to 1.26 ± 0.10-fold in the Dia group (Fig. 4b), indicating a reduction in ROS generation.

Effects of mitoKCa and mitoKCa channel openers on mitochondrial membrane potential, ROS production, cytosolic Ca2+ concentration and cell length. a Representative sequential confocal images of TMRE (red)/DCFH (yellow)-loaded and TMRE/Fluo-4 (green)-loaded myocytes before and after 15 min of intermittent laser illumination. b Changes in ROS production (DCFH fluorescence). c Changes in mitochondrial membrane potential (TMRE fluorescence). d Changes in the cytosolic Ca2+ concentration (Fluo-4 fluorescence). e Changes in cell length (final/initial of cell length). N before laser illumination, CT control, Dia diazoxide, n number of cells. *P < 0.05 versus CT (color figure online)
In untreated cells, TMRE fluorescence remained unchanged through a 15-min observation period (data not shown). By contrast, the relative TMRE intensity had declined to 0.48 ± 0.07-fold after 15 min of intermittent laser illumination in the CT group (Fig. 4c), indicating a loss of mitochondrial membrane potential due to the opening of mPTP. NS1619 and Dia significantly mitigated this reduction in the TMRE fluorescence, such that the relative TMRE intensity was 0.86 ± 0.05-fold in the NS1619 group and 0.84 ± 0.12-fold in the Dia group, both of which were significantly (P < 0.05) higher than in the CT group (Fig. 4c).
Fluo-4 fluorescence increased during the intermittent laser illumination, indicating a rise in cytosolic Ca2+ concentration, and NS1619 and Dia attenuated the laser-induced increase in the Fluo-4 intensity. The relative Fluo-4 intensity was 1.28 ± 0.14-fold in the NS1619 group and 1.15 ± 0.17-fold in the Dia group, which was significantly (P < 0.05) lower than in the CT group (1.75 ± 0.17-fold) (Fig. 4d).
Intermittent laser illumination also induced cell shortening (final/initial cell length in the CT group: 0.52 ± 0.07-fold), and those were significantly (P < 0.05) mitigated by NS1619 (0.90 ± 0.07-fold) and by Dia (0.80 ± 0.08-fold) (Fig. 4e).
Discussion
In this study, we found that (1) the recovery of LVDP and PRP upon reoxygenation was facilitated by IPT, and that the effect of IPT was inhibited by blockade of the mitoKCa or mitoKATP channels; (2) pharmacological postconditioning using a mitoKCa or mitoKATP channel openers improved recovery of PRP and inhibited AST release upon reoxygenation; and (3) pharmacological postconditioning inhibited the ROS production, cytosolic Ca2+ accumulation, and depolarization of mitochondrial membrane potential caused by laser-induced mPTP opening in isolated myocytes.
The recovery of cardiac contractile activity upon reoxygenation was improved by IPT, and that beneficial effect was antagonized by mitoKCa channel blockade and mitoKATP channel blockade (Fig. 2), which indicates IPT is mediated, at least in part, by activation of mitoKCa channels and mitoKATP channels. MitoKATP channels has been reported to play a role in mediating IPT-induced cardioprotection [7, 9], and our finding that 5-HD administered at the onset of reoxygenation inhibited IPT-induced cardioprotection is consistent with those earlier reports (Fig. 2). In the present study, we also demonstrated IPT-mediated cardioprotection is abolished by not only mitoKATP channel blockade but also by mitoKCa channel blockade. This is the first study which examined two types of mitochondrial K+ channels on IPT induced cardiac protection in one experiment. Conversely, direct activation of mitoKCa channel with the opener NS1619 (Fig. 3) or direct activation of mitoKATP channel with the opener Dia (Fig. 3) at the onset of reoxygenation have cardioprotective effects like that of IPT. Taken together, these findings suggest that activation of mitochondrial K+ channels is a key cardioprotective event, and that IPT activates both mitoKCa and mitoKATP channels.
There have been a number of studies implicating mitoKATP channels in cardioprotection [28–30], and more recently the involvement of mitoKCa channels in preconditioning [1, 14, 15] or postconditioning [31] have also been suggested. For example, it was recently shown that treatment with the mitoKCa channel opener NS1619 can induce cardioprotection [14, 15, 31, 32]. Similarly, application of NS11021, a novel large-conductance Ca2+-activated K+ channel activator, prior to or at the onset of reperfusion protects the heart from ischemia–reperfusion injuries [1]. On the other hand, it has also been reported that NS1619 failed to exert a postconditioning effect in an earlier study [16]. This discrepancy may be due to the differences in the experimental model between studies in the previous study and in the present study.
IPT reportedly protects the heart by inhibiting ROS generation [19]. We therefore examined the effect of mitoKCa and mitoKATP channel activation on ROS generation induced by intermittent laser illumination. Previous reports have shown that both ROS generation and intracellular Ca2+ overload play crucial roles in the induction of cell death during reperfusion/reoxygenation [33, 34]. A burst of ROS increases Ca2+ influx through L-type Ca2+ channels, increasing the cytosolic Ca2+ concentration [35, 36]. In addition, ROS reportedly increase the activity of the Na+/H+ exchanger, which further increases cytosolic Ca2+ by stimulating reverse mode Na+/Ca2+ exchange [37].
We found that, in the CT group, intermittent laser illumination of TMRE-loaded myocytes consistently resulted in mPTP opening, as indicated by the loss of TMRE fluorescence (Fig. 4a, c), which is well known to reflect collapse of the mitochondrial membrane potential [24–27]. This model is a widely used and reliable means of reproductively inducing a loss of mitochondrial membrane potential, which has been identified unequivocally as a reflection of the mPTP opening, and therefore simulates the events associated with reoxygenation-induced cell injury. In fact, mitochondrial Ca2+ overload and ROS generation are the major triggers of mPTP opening [14, 38]. Evidence suggests that IPT may mitigate mPTP opening, and that activation of the salvage kinase cascade and mitochondrial KATP/ROS/PKC pathway may be crucial for cardioprotection [8, 38]. Our observation that NS1619 and Dia each inhibited the ROS generation and accumulation of cytosolic Ca2+ (Fig. 4b, d) induced by intermittent laser illumination indicates that ROS generation and cytosolic Ca2+ overload can be mitigated by activation of mitoKCa and mitoKATP channels. In addition, NS1619 and Dia delayed the onset of cell shortening by the intermittent laser illumination, and thus delayed the cell death (Fig. 4e). We therefore suggest that activation of mitoKCa and mitoKATP channels, which mitigates ROS generation, cytosolic Ca2+ overload, and depolarization of mitochondrial membrane potential, is an important component of IPT- and drug-induced cardioprotection.
Although neither NS1619 nor Pax are mitoKCa channel specific, since large-conductance Ca2+-activated K+ channels are absent from the myocardial sarcolemmal membrane [15], it would be reasonable to assume that they affected the mitoKCa channel. However, we cannot completely exclude the possibility that the protection of NS1619 is due to mechanisms other than the activation of mitoKCa channels. Possible upstream mechanisms of mitoKCa channel activation remains to be elucidated. Recently, it has also been reported that NS1619 could inhibit the L-type Ca2+ channel, which may protect myocardium from hypoxia-reoxygenation injury by reducing Ca2+ influx [39].
In the present study, we found neither Pax nor 5-HD affect the protection of IPT when it is given at the onset of reoxygenation. Pax [16, 32] and 5-HD [7, 9] are often used in Langendorff perfusion studies investigating preconditioning signaling mechanisms. It has been reported that Pax does not act on mitoKATP channels [32], and that 5-HD alone does not alter hypoxia/reoxygenation-induced changes in mitochondrial membrane potential and mPTP opening [7]. Therefore, we can exclude the possibilities that Pax or 5-HD is potentially harmful to the myocardium.
In summary, this is the first study demonstrating that the IPT protects the heart through activation of not only the mitoKATP but also the mitoKCa channels. At the onset of reoxygenation, activation of these channels can inhibit the opening of mPTP caused by ROS production and cytosolic Ca2+ overload, thereby preventing hypoxia-reoxygenation injury.
References
Bentzen BH, Osadchii O, Jespersen T, Hansen RS, Olesen SP, Grunnet M (2009) Activation of big conductance Ca2+-activated K+ channels (BK) protects the heart against ischemia–reperfusion injury. Pflugers Arch 457:979–988
Kloner RA, Jennings RB (2001) Consequences of brief ischemia: stunning, preconditioning, and their clinical implications: part 2. Circulation 104:3158–3167
Murphy E, Steenbergen C (2007) Preconditioning: the mitochondrial connection. Annu Rev Physiol 69:51–67
Stowe DF, Aldakkak M, Camara AK, Riess ML, Heinen A, Varadarajan SG, Jiang MT (2006) Cardiac mitochondrial preconditioning by Big Ca2+-sensitive K+ channel opening requires superoxide radical generation. Am J Physiol Heart Circ Physiol 290:H434–H440
Yellon DM, Downey JM (2003) Preconditioning the myocardium: from cellular physiology to clinical cardiology. Physiol Rev 83:1113–1151
Kin H, Zhao ZQ, Sun HY, Wang NP, Corvera JS, Halkos ME, Kerendi F, Guyton RA, Vinten-Johansen J (2004) Postconditioning attenuates myocardial ischemia–reperfusion injury by inhibiting events in the early minutes of reperfusion. Cardiovasc Res 62:74–85
Mykytenko J, Reeves JG, Kin H, Wang NP, Zatta AJ, Jiang R, Guyton RA, Vinten-Johansen J, Zhao ZQ (2008) Persistent beneficial effect of postconditioning against infarct size: role of mitochondrial KATP channels during reperfusion. Basic Res Cardiol 103:472–484
Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, Losano G, Pagliaro P (2005) Post-conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol 101:168–179
Penna C, Rastaldo R, Mancardi D, Raimondo S, Cappello S, Gattullo D, Losano G, Pagliaro P (2006) Post-conditioning induced cardioprotection requires signaling through a redox-sensitive mechanism, mitochondrial ATP-sensitive K+ channel and protein kinase C activation. Basic Res Cardiol 101:180–189
Ferdinandy P, Schulz R, Baxter GF (2007) Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev 59:418–458
Hausenloy DJ, Tsang A, Yellon DM (2005) The reperfusion injury salvage kinase pathway: a common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med 15:69–75
Cohen MV, Yang XM, Liu GS, Heusch G, Downey JM (2001) Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial KATP channels. Circ Res 89:273–278
Penna C, Mancardi D, Rastaldo R, Losano G, Pagliaro P (2007) Intermittent activation of bradykinin B2 receptors and mitochondrial KATP channels trigger cardiac postconditioning through redox signaling. Cardiovasc Res 75:168–177
Shintani Y, Node K, Asanuma H et al (2004) Opening of Ca2+-activated K+ channels is involved in ischemic preconditioning in canine hearts. J Mol Cell Cardiol 37:1213–1218
Xu W, Liu Y, Wang S, McDonald T, Van Eyk JE, Sidor A, O’Rourke B (2002) Cytoprotective role of Ca2+-activated K+ channels in the cardiac inner mitochondrial membrane. Science 298:1029–1033
Wang X, Yin C, Xi L, Kukreja RC (2004) Opening of Ca2+-activated K+ channels triggers early and delayed preconditioning against I/R injury independent of NOS in mice. Am J Physiol Heart Circ Physiol 287:H2070–H2077
Grill HP, Zweier JL, Kuppusamy P, Weisfeldt ML, Flaherty JT (1992) Direct measurement of myocardial free radical generation in an in vivo model: effects of postischemic reperfusion and treatment with human recombinant superoxide dismutase. J Am Coll Cardiol 20:1604–1611
Hearse DJ (1991) Reperfusion-induced injury: a possible role for oxidant stress and its manipulation. Cardiovasc Drugs Ther 5:225–235
Sun HY, Wang NP, Kerendi F, Halkos M, Kin H, Guyton RA, Vinten-Johansen J, Zhao ZQ (2005) Hypoxic postconditioning reduces cardiomyocyte loss by inhibiting ROS generation and intracellular Ca2+ overload. Am J Physiol Heart Circ Physiol 288:H1900–H1908
Mukherjee SB, Das M, Sudhandiran G, Shaha C (2002) Increase in cytosolic Ca2+ levels through the activation of non-selective cation channels induced by oxidative stress causes mitochondrial depolarization leading to apoptosis-like death in Leishmania donovani promastigotes. J Biol Chem 277:24717–24727
Toda T, Kadono T, Hoshiai M, Eguchi Y, Nakazawa S, Nakazawa H, Higashijima N, Ishida H (2007) Na+/H+ exchanger inhibitor cariporide attenuates the mitochondrial Ca2+ overload and PTP opening. Am J Physiol Heart Circ Physiol 293:H3517–H3523
Hausenloy DJ, Yellon DM, Mani-Babu S, Duchen MR (2004) Preconditioning protects by inhibiting the mitochondrial permeability transition. Am J Physiol Heart Circ Physiol 287:H841–H849
Watanabe M, Okada T (2003) Lysophosphatidylcholine-induced myocardial damage is inhibited by pretreatment with poloxamer 188 in isolated rat heart. Mol Cell Biochem 248:209–215
Ruiz-Meana M, Abellán A, Miró-Casas E, Garcia-Dorado D (2007) Opening of mitochondrial permeability transition pore induces hypercontracture in Ca2+ overloaded cardiac myocytes. Basic Res Cardiol 102:542–552
Duchen MR (2000) Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium 28:339–348
Halestrap AP, Clarke SJ, Javadov SA (2004) Mitochondrial permeability transition pore opening during myocardial reperfusion—a target for cardioprotection. Cardiovasc Res 61:372–385
Hüser J, Rechenmacher CE, Blatter LA (1998) Imaging the permeability pore transition in single mitochondria. Biophys J 74:2129–2137
Fryer RM, Hsu AK, Nagase H, Gross GJ (2000) Opioid-induced cardioprotection against myocardial infarction and arrhythmias: mitochondrial versus sarcolemmal ATP-sensitive potassium channels. J Pharmacol Exp Ther 294:451–457
Garlid KD, Dos Santos P, Xie ZJ, Costa AD, Paucek P (2003) Mitochondrial potassium transport: the role of the mitochondrial ATP-sensitive K+ channel in cardiac function and cardioprotection. Biochim Biophys Acta 1606:1–21
Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, Lodge NJ, Smith MA, Grover GJ (1997) Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Possible mechanism of cardioprotection. Circ Res 81:1072–1082
Huhn R, Heinen A, Weber NC, Schlack W, Preckel B, Hollmann MW (2010) Ischaemic and morphine-induced post-conditioning: impact of mKCa channels. Br J Anaesth 105:589–595
Cao CM, Xia Q, Gao Q, Chen M, Wong TM (2005) Calcium-activated potassium channel triggers cardioprotection of ischemic preconditioning. J Pharmacol Exp Ther 312:644–650
Akao M, O’Rourke B, Teshima Y, Seharaseyon J, Marbán E (2003) Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ Res 92:186–194
Herzog WR, Vogel RA, Schlossberg ML, Edenbaum LR, Scott HJ, Serebruany VL (1997) Short-term low dose intracoronary diltiazem administered at the onset of reperfusion reduces myocardial infarct size. Int J Cardiol 59:21–27
Bagchi D, Wetscher GJ, Bagchi M, Hinder PR, Perdikis G, Stohs SJ, Hinder RA, Das DK (1997) Interrelationship between cellular calcium homeostasis and free radical generation in myocardial reperfusion injury. Chem Biol Interact 104:65–85
Gen W, Tani M, Takeshita J, Ebihara Y, Tamaki K (2001) Mechanisms of Ca2+ overload induced by extracellular H2O2 in quiescent isolated rat cardiomyocytes. Basic Res Cardiol 96:623–629
Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA (2002) H2O2-induced Ca2+ overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol Heart Circ Physiol 283:H598–H605
Sivaraman V, Mudalagiri NR, Di Salvo C, Kolvekar S, Hayward M, Yap J, Keogh B, Hausenloy DJ, Yellon DM (2007) Postconditioning protects human atrial muscle through the activation of the RISK pathway. Basic Res Cardiol 102:453–459
Park WS, Kang SH, Son YK, Kim N, Ko JH, Kim HK, Ko EA, Kim CD, Han J (2007) The mitochondrial Ca2+-activated K+ channel activator, NS 1619 inhibits L-type Ca2+ channels in rat ventricular myocytes. Biochem Biophys Res Commun 362:31–36
Acknowledgments
This study was partially supported by a High Technology Research Center Grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Jin and J. Wu are joint first authors.
About this article
Cite this article
Jin, C., Wu, J., Watanabe, M. et al. Mitochondrial K+ channels are involved in ischemic postconditioning in rat hearts. J Physiol Sci 62, 325–332 (2012). https://doi.org/10.1007/s12576-012-0206-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-012-0206-y