
Abstract
The goal of our study was to evaluate the origin of the increased O2 consumption in electrically stimulated left ventricular slices of isoproterenol-induced hypertrophied rat hearts with normal left ventricular pressure. O2 consumption per minute (mVO2) of mechanically unloaded left ventricular slices was measured in the absence and presence of 1-Hz field stimulation. Basal metabolic mVO2, i.e., mVO2 without electrical stimulation, was significantly smaller, but mVO2 for the total Ca2+ handling in excitation–contraction coupling (E–C coupling mVO2), i.e., delta mVO2 (=mVO2 with stimulation − mVO2 without stimulation), was significantly larger in the hypertrophied heart. Furthermore, the fraction of E–C coupling mVO2 was markedly altered in the hypertrophied heart. Namely, mVO2 consumed by sarcoplasmic reticulum Ca2+-ATPase (SERCA2) was depressed by 40%; mVO2 consumed by the Na+/K+-ATPase (NKA)-Na+/Ca2+ exchange (NCX) coupling was increased by 100%. The depressed mVO2 consumption by SERCA2 was supported by lower protein expressions of phosphorylated-Ser16 phospholamban and SERCA2. The increase in NKA–NCX coupling mVO2 was supported by marked augmentation of NCX current. However, the increase in NCX current was not due to the increase in NCX1 protein expression, but was attributable to attenuation of the intrinsic inactivation mechanisms. The present results demonstrated that the altered origin of the increased E–C coupling mVO2 in hypertrophy was derived from decreased SERCA2 activity (1ATP: 2Ca2+) and increased NCX activity coupled to NKA activity (1ATP: Ca2+). Taken together, we conclude that the energetically less efficient Ca2+ extrusion pathway evenly contributes to Ca2+ handling in E–C coupling in the present hypertrophy model.
Similar content being viewed by others


Introduction
Cardiac hypertrophy is a major risk factor for the development of heart failure and sudden cardiac death. Therefore, to reduce mortality from cardiovascular diseases, elucidation of the mechanisms involved in cardiac hypertrophy is pivotal. It is recognized that activation of the sympathetic nervous system is associated with cardiac hypertrophy and heart failure in humans [1, 2]. In addition, chronic treatment of hearts with an α, β-stimulant, norepinephrine, or a β-stimulant, isoproterenol, induces cardiac hypertrophy accompanied with enhanced fibrosis among cardiac interstitial cells in experimental animal models [3–6]. So far, various mechanisms for this remodeling have been proposed [5–9]. We have recently reported that 3-day subcutaneous infusion of isoproterenol to rats induced cardiac hypertrophy with unchanged left ventricular (LV) systolic and diastolic function that was reversible to the control levels after the cessation of infusion [10, 11]. In this model, a significant decrease of mechanical work due to the decrease of LVV (LV volume) was observed. Furthermore, this short-term model did not show any marked differences in the LV mechanical work capability compared to that induced by a 2-week isoproterenol infusion [11]. The expression levels of cardiac sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2), phospholamban (PLB) and phosphorylated Ser16 (phospho-ser16) PLB (p-PLB) in this model were also decreased, suggesting that the Ca2+-handling of the cardiac myocyte was already altered [10, 11]. However, functional changes of molecules associated with the Ca2+ handling are not completely understood.
Previously, we demonstrated that the oxygen consumption per minute (mVO2) of the mechanically unloaded slices without electrical field stimulation is related to basal metabolism, and the increments in mVO2 from the basal metabolic mVO2 with 1-Hz electrical field stimulation is related to total Ca2+ handling in excitation–contraction (E–C) coupling (E–C coupling mVO2) [12–15]. Previous reports have reported that freely shortening myocardial slices under mechanically unloaded conditions do not require a significant additional VO2 for crossbridge cycling [16, 17]. This finding is further supported by our results showing that 5 mmol/l 2,3-butanedione monoxime (BDM: a specific cross-bridge cycling inhibitor) does not affect the rat LV myocardial slice VO2, although 5 mmol/l BDM markedly reduces the slice motility [14, 15]. On the other hand, we observed an increase in the slice VO2 by barium because of the enhanced cross-bridge cycling [12]. This increase in VO2 was abolished by 5 mmol/l BDM, but not affected by cyclopiazonic acid (CPA; SR Ca2+-ATPase inhibitor). Therefore, the measured increment in VO2 by electrical stimulation under the mechanically unloaded condition corresponds to the VO2 for non-mechanical activities of myocardium [14, 15].
The aim of the present study was to measure mVO2 in the LV slice preparation of the hypertrophic heart to provide key information about causes of the altered Ca2+ handling in E–C coupling from the viewpoint of myocardial energetics. If a severe imbalance between larger O2 demand and smaller O2 supply elicited by energetically less efficient Ca2+ handling continues in the hypertrophic heart, it leads to heart failure for various reasons, including Ca2+ overload.
Some parts of these data have been reported previously in abstract form [18].
Methods
Experimental procedures followed the regulations of and were approved by the Animal Care and Use Committee of Nara Medical University.
Animals and drug infusion
Male Wistar rats weighing 250–370 g (8–10 weeks) were used in the experiments. Delivery of drug was achieved by implanting an osmotic minipump (model 1003D, Alzet, Durect Corp, Cupertino, CA) subcutaneously in the neck under pentobarbital (50 mg/kg i.p.) anesthesia. Either isoproterenol (2.4 mg kg−1 day−1 for 3 days) or vehicle (0.1% ascorbic acid in saline 2.4 μl/day for 3 days) was infused subcutaneously [3].
Animals and left ventricular myocardial slice preparation
Male Wistar rats that underwent isoproterenol or saline infusion for 3 days were anesthetized with pentobarbital sodium (50 mg/kg, i.p.). The heart and its slices were prepared as described before [12–15]. The whole heart was excised under perfusion with Tyrode solution oxygenated with 100% O2 at 13°C for 5 min. The composition of Tyrode solution (in mM) was 136.0 NaCl, 5.4 KCl, 1.0 MgCl2, 0.3 NaH2PO4, 0.9 CaCl2, 10.0 glucose and 5.0 HEPES, with pH adjusted to 7.4 with NaOH at 30°C. After the perfusion, both atria, the four valves including the connective tissue, the aorta and the pulmonary artery were removed from the heart. The heart was longitudinally cut into two or three pieces, and each of the pieces was cut into 300-μm-thick slices in parallel with the epicardium with a microslicer DTK-3000 (Dosaka EM, Kyoto, Japan). The slices with the thickness (300 μm) we chose seem to be sufficiently oxygenated by diffusion, according to the published data and calculation [14, 15, 19–21]. This cut was chosen so that most fibers were parallel-sectioned to fiber orientation according to the known fiber architecture of the heart wall. We obtained 12–20 slices (average single-side surface area: approximately 30 mm2) from each LV. The slices were stored in Tyrode solution oxygenated with 100% O2 at 18°C for 30 min, and subsequently at 25°C at least for 30 min. Finally, the mVO2 of slices was measured between 1.5 and 2.5 h after making slices. Consequently, acute effects of isoproterenol were removed before starting experiments.
Assessment of mVO2 of LV slices
One or two sets of six to ten LV slices from each heart were used to measure mVO2 in the absence and presence of electrical field stimulation. Stimulation consisted of 1-Hz rectangular pulses, 0.5 ms in duration and current of 0.4–0.8 mA (voltage: 8–16 V). The direction of stimulation was alternated by a polarity change. We did not find the bubbles of oxygen and hydrogen on the platinum electrodes (distance: 0.5 cm) under a microscope. mVO2 of each set of slices under mechanically unloaded conditions was measured polarographically with an oxygen electrode (model 1T-125, Instech Labs; Plymouth Meeting, PA) and current amplifier (model 102, Instech Labs) as described previously [12–15, 22]. This oximetric system was calibrated at three different O2 concentrations ([O2], in mg/l): 0 (5% Na2SO3 solution), 7.3 (Tyrode solution saturated with air) and 35.1 (Tyrode solution saturated with 100% O2) at 30°C in each experiment. We first measured the background mVO2 (0.19 ± 0.07 mg O2 min−1; n = 257 experiments) without slices in the Tyrode-filled chamber at the beginning and end of each measurement after sufficient saturation with 100% oxygen. Volume of Tyrode solution in the chamber was measured at the end of the three-repeated measurements. Wet weight of a set of slices used was 73.1 ± 11.3 mg for saline-infused hearts (CONT) and 79.6 ± 11.6 mg for isoproterenol-infused hypertrophied hearts (HYP).
We obtained mVO2 of slices by subtracting the background mVO2 (A mg O2 min−1) from the measured mVO2 (B mg O2 min−1) in the presence of LV slices. The LV slice mVO2 (μl O2 min−1 g−1) was calculated as [(B − A)/slice wet weight (in g)] × volume of Tyrode solution in the chamber (in l) × [22.4 l/32 g] × 1 g. The time series of oxygen concentration were recorded by a desktop computer with a 12-bit A/D converter [AD12-16(PCI)E; Contec] at a sampling rate of 2 Hz [22]. Slope of A or B was determined by regression analysis in 120-s periods during non-stimulation (2 min) and in the last 80-s periods during stimulation (3 min).
Experimental procedure
Tyrode solutions were oxygenated with 100% O2 and preheated to 30°C in a water bath. Slices without any mechanical load were placed into the oximetric chamber filled with the prepared Tyrode solution, which was continuously oxygenated. The chamber was warmed with a Microwarm Plate DC-MP10DM (Kitazato, Tokyo, Japan). After the slices were superfused for 10 min in the tightly closed chamber, mVO2 of the quiescent slices in the absence of stimulation [−St mVO2] was obtained for 120 s of non-stimulation period (2 min), and mVO2 of the activated and contracting slices in the presence of stimulation [+St mVO2] was obtained for the last 80 s of the stimulation period (3 min). The slices in the chamber were then washed and oxygenated for 10 min. mVO2 of mechanically unloaded slices without stimulation [−St mVO2] corresponded to basal metabolic mVO2. The increment in mVO2 by stimulation, i.e., delta mVO2 (=[+St mVO2] − [−St mVO2]) corresponded to E–C coupling mVO2 [13–15].
We used the following protocols 1–4 in LV slices from CONT and HYP (Fig. 1). The second measurement value was used to calculate an average expressed by raw data (Figs. 2, 4) or % of first measurement value in normal Tyrode solution (1st NT) (Fig. 3).

Protocols for experiments in saline-infused normal hearts (CONT) and isoproterenol-infused hypertrophic hearts (HYP). Dob: dobutamine. CPA: cyclopiazonic acid. KBR: KB-R7943. CE: 5-(and -6)-carboxyeosin diacetate. −St mVO 2 myocardial oxygen consumption per minute in the absence of stimulation. +St mVO 2 mVO2 in the presence of stimulation. Abbreviations used in this figure were also used in Figs. 2, 3, 4

Comparison of mean basal metabolic mVO2 and excitation–contraction (E–C) coupling mVO2 between LV slices from HYP (n = 12 sets from 6 hearts) and LV slices from CONT (n = 10 sets from 5 hearts). *P < 0.05 vs. CONT. **P < 0.001 vs. CONT. Upper insets indicated the method for preparing the LV slice

Dependences of E–C coupling mVO2 on [Ca2+]o (0.9–7.2 mM) in CONT (a) and HYP (b) LV slices and on Dob (0–10 μM) in CONT (c) and HYP (d) LV slices. # P < 0.05 vs. each 0.9 mM [Ca2+]o and 0 μM Dob. 1st NT 1st measurement value in normal Tyrode solution. Numerical value in each bar indicates set number of slices. Each mean ± SD value was expressed as % of 1st NT
Protocol 1
To obtain basal metabolic and E–C coupling mVO2, after 25-min superfusion of the slices with normal Tyrode solution (0.9 mM Ca2+), the first measurement was performed as described above. Subsequently, the second and third measurements were performed in normal Tyrode solution. Thus, the measurements were repeated three times to ascertain the reproducibility of measurements.
Protocol 2
After the first measurement in normal Tyrode solution, to examine dependencies of basal metabolic and E–C coupling mVO2 on Ca2+ concentrations within 2.7, 4.5 and 7.2 mM, the second measurement of the slices was performed in Tyrode solution containing each concentration of Ca2+. The third measurement of the slices was performed in normal Tyrode solution as in protocols 3 and 4.
Protocol 3
After the first measurement in normal Tyrode solution, to examine dependencies of basal metabolic and E–C coupling mVO2 on dobutamine (Dob; 1, 10 μM), the second measurement of the slices was performed in Tyrode solution containing Dob (1, 10 μM).
Protocol 4
After the first measurement in normal Tyrode solution, to examine the effects of cyclopiazonic acid (CPA; 30 μM) (Sigma; St. Louis, MO) used as a SERCA2 inhibitor [13, 14], KB-R7943 (KBR; 10 μM) (a gift from Kanebo; Tokyo, Japan) used as a bidirectional-mode NCX inhibitor [23, 24], and 5-(and -6)-carboxyeosin diacetate (CE; 20 μM) (Molecular Probes, Eugene, OR) used as a sarcolemmal Ca2+-ATPase inhibitor, though less selective [25, 26] on E–C coupling mVO2, the second measurement of the slices was performed. CPA and KBR and CE were dissolved in dimethyl sulfoxide (DMSO), and the solution was further diluted with normal Tyrode solution just before use (1.0–1.3% of DMSO final concentration). This concentration of DMSO did not affect basal metabolic and E–C coupling mVO2 [13, 14].
From our previous results, we revealed that KBR at 10 μM almost completely blocked reverse-mode NCX for initial reperfusion for 5 min and forward-mode NCX during the following 10-min perfusion upon ischemic reperfusion [23]. Furthermore, KBR (10 μM) was always added after treatment with CPA to minimize any non-specific effects. In this protocol, field stimulation was performed for the former 5 min during the 10-min superfusion with CPA, CPA + KBR, or CPA + KBR + CE (Fig. 1).
Histological analysis
LV was fixed with 3.7% paraformaldehyde in phosphate-buffered saline, embedded in paraffin, and cut into 6-μm slices, which were stained with hematoxylin-eosin for morphological analysis or with Masson’s trichrome (MTC) staining for detection of fibrosis.
Polyacrylamide gel electrophoresis and Western blots for SERCA2, PLB, NCX1, PLM, NKA and p-PLB
Membrane proteins were isolated from the left ventricular wall of each frozen heart. The frozen hearts were homogenized and centrifuged at 1,000×g for 10 min. The supernatants were centrifuged at 100,000×g for 60 min at 4°C. The 100,000×g pellets were cellular membrane fractions and used for immunoblotting of SERCA2, phospholamban (PLB), Na+/Ca2+ exchanger-1 (NCX1), phospholemman (PLM), Na+/K+-ATPase α-1 (NKA) and phosphorylated-Ser16 PLB (p-PLB).
The same amounts of membrane proteins (10 μg/lane) were separated on SDS-polyacrylamide gels (10% for SERCA2 and NKA, 15% for PLB, p-PLB and PLM, and 7.5% for NCX1) in a minigel apparatus (Mini-PROTEAN II, Bio-Rad) and transferred to polyvinyliodene difluoride membranes. The membranes were blocked (4% Block Ace, Dainippon Pharmaceutical Co., Osaka) and then incubated with anti-SERCA2 antibody (1:1,000 dilution, Affinity Bio Reagents), anti-PLB antibody (1:2,000 dilution, Upstate Biotechnology), anti-p-PLB (Ser16) antibody (1:1,000 dilution, Upstate Biotechnology), anti-NCX1 antibody (1:200 dilution, a generous gift from Dr. Iwamoto, Fukuoka University), anti-PLM antibody (1:100 dilution, ABGENT), or anti-NKA antibody (1:10,000 dilution, Upstate Biotechnology). The antigens were detected by the luminescence method (ECL Western blotting detection kit, Amersham) with peroxidase-linked anti-mouse IgG (1:2,000 dilution) or peroxidase-linked anti-rabbit IgG (1:2,000 or 1:5,000 dilution). The amounts of membrane proteins were determined to obtain the linear response of ECL-immunoblot. After immunoblotting, the film was scanned with a scanner, and the intensity of the bands was calculated by NIH image analysis.
Myocytes study
Cell isolation
The rats were deeply anesthetized by intraperitoneal injection of pentobarbital sodium (>0.1 mg/g body weight). LV myocytes were dissociated from the hearts as described in previous studies [27]. The dissociated myocytes were kept in a HEPES-buffered DMEM solution (ICN Biomedicals) and used for the patch clamp and fluorescence studies within 8 h. All procedures were approved by the Animal Research Committee of the Graduate School of Medicine, Kyoto University.
Solutions
The standard pipette solution contained (in mM) 30 CsOH, 40 aspartate, 20 TEACl, 40 EGTA, 5 MgATP, 33.8 CaCl2, 1.19 MgCl2, 10 HEPES and 50 NaOH (pH = 7.2 with CsOH). Free Ca2+ concentration was calculated to be 0.8 μM. Composition of bath solution for recording NCX current (I NCX) was 145 NaCl, 2 BaCl2, 3 MgCl2, 5 HEPES, 0.2 EGTA (or 0 when adding 2 CaCl2), 0.05 mM ouabain and 0.002 nicardipine (pH = 7.4 with NaOH), and that for NKA current (I NKA) was 144 NaCl, 0.5 MgCl2, 1.8 CaCl2, 5 NiCl2, 4 BaCl2 and 10 HEPES (pH = 7.4 with NaOH). When measuring the inactivation of I NCX, the pipette solution contained 20 CsOH, 20 TEACl, 50 EGTA, 2 MgATP, 21.3 CaCl2, 0.71 MgCl2, 50 HEPES and 100 NaOH (pH = 7.2 with CsOH), and the bath solution contained 145 LiCl, 2 BaCl2, 2 MgCl2, 5 HEPES, 0.5 EGTA (or 0 when adding 2 CaCl2), 0.05 mM ouabain and 0.002 nicardipine (pH = 7.4 with NMDG), according to a previous study [28].
Electrophysiology
The myocytes were voltage clamped using the whole cell method with an Axopatch 200B amplifier (Axon Instruments). Holding potential was −40 mV. Current–voltage (I–V) relationships were measured by applying ramp pulses and normalized by membrane capacitance. I NCX was induced by applying 2 mM Ca2+ for 5 or 30 s at intervals of 30 or 120 s and determined as the difference current in the same manner as described in our previous studies [27, 28]. I NKA was induced similarly by applying 10 mM K+ [29, 30]. The cell size was evaluated by measuring cell area using a cooled CCD camera (ORCA-ER, Hamamatsu Photonics).
[Na+]i fluorescence measurements
[Na+]i was monitored by dual-wavelength ratio imaging of a Na+-sensitive dye, sodium-binding benzofuran isophthalate (SBFI, Invitrogen). Isolated myocytes were loaded with 10 μM SBFI-AM for 90 min at 21°C in the HEPES-buffered D-MEM solution containing non-ionic surfactant Pluronic F-127 (0.05% w/v). After washing out the external dye, we allowed SBFI AM to de-esterify for 20 min before proceeding with [Na+]i measurements. The loaded myocytes, which were settled onto the glass bottom of a recording chamber mounted on an inverted microscope (Eclipse TE2000, Nikon, with a ×20 objective), were superfused with the NT at 30°C and electrically stimulated at 1 Hz. Fluorescent image pairs of single cells (excitation at 340 and 380 nm; emission of >510 nm) were acquired using the cooled CCD camera. At the end of each experiment, a calibration was performed according to the methods by Despa et al. [31]. In brief, myocytes were exposed to various [Na+]o, 0 to 30 mM, in the presence of 10-μM gramicidin D and 100-μM strophanthidin. The fluorescence ratio plotted against [Na+]o showed a linear relationship. According to this calibration curve, each [Na+]i value was calculated.
Statistics
All data were presented as mean ± SD. Differences between two mean values were evaluated by paired or non-paired Student’s t-test. Multiple comparisons were performed by one-way ANOVA and Bonferroni’s t-test or Dunnett’s t-test, or two-way ANOVA. In all statistical tests, P values less than 0.05 were considered statistically significant.
Results
We confirmed first a formation of HYP in rats treated with the isoproterenol infusion. The thickness of the LV free wall was significantly increased, and each LV and right ventricle (RV) dry and wet weight ratio to body weight also significantly increased from CONT (Table 1). Cell size, measured with area of myocyte, was also significantly larger in HYP than the one of CONT. These data are substantially the same as those previously reported by us [10].
Collagen accumulation
Distinct collagen accumulation was confirmed in the HYP slices used in the present study, as shown by previous papers including ours [9, 10]. Our data also indicated that collagen contents measured by hydroxyproline methods in HYP (1.69 ± 0.21% collagen/LV dry weight; n = 6) were significantly (P < 0.05) larger than those in CONT (1.33 ± 0.23% collagen/LV dry weight; n = 5), though the difference was small.
Composition of myocardial slice mVO2
Basal metabolic mVO2 without stimulation in the HYP slices (19.7 ± 1.9 μlO2 min−1 g−1; n = 12 sets from 6 hearts) was significantly smaller than that of the CONT slices (23.5 ± 1.6 μlO2 min−1 g−1; n = 10 sets from 5 hearts) (Fig. 2). The basal metabolic VO2 of the whole heart measured by arresting it with KCl (0.03 mM) was 24.3 μlO2 min−1 g−1 (resting VO2; 8.1 mW g−1 or 2.43 mlO2 min−1 100 g−1) [32, 33]. These data are in good agreement with the present results and those from the Alpert and Gibbs laboratories in regard to the activation or tension-independent heat [34].
Delta mVO2 in the HYP slices was significantly, but moderately larger than that of the CONT slices (14.5 ± 3.7 vs. 11.7 ± 2.3 μlO2 min−1 g−1) (Fig. 2), suggesting that the energy expenditure for total Ca2+ handling in E–C coupling is accelerated in the HYP slices.
E–C coupling mVO2: dependency on [Ca2+]o, but no dependency on Dob in HYP slices
E–C coupling mVO2 was further examined at various [Ca2+]o in 8 groups (4 CONT and 4 HYP) of 6–14 sets of slices from 3 to 7 hearts. There were no significant increases in mean basal metabolic mVO2 values in each set of the CONT and HYP slices at increased [Ca2+]o from 0.9 mM [Ca2+]o to 7.2 mM [Ca2+]o (data not shown). E–C coupling mVO2 was also confirmed in these experiments: 11.5 ± 3.3 μlO2 min−1 g−1 in the CONT slices and 14.7 ± 5.0 μlO2 min−1 g−1 in the HYP slices at 0.9 mM [Ca2+]o. E–C coupling mVO2 increased up to 147% of 1st NT in the CONT slices and up to 134% of 1st NT in the HYP slices in a [Ca2+]o-dependent manner, respectively (Fig. 3a, b). There were no significant differences in the responses to [Ca2+]o between CONT and HYP slices. These data indicated that overall [Ca2+]o-dependence of energy expenditure for Ca2+ handling is not markedly affected in HYP slices.
In contrast, dobutamine, a β-adrenergic receptor stimulant, did not increase E–C coupling mVO2 at 1–10 μM in the HYP slices, while it dose-dependently increased E–C coupling mVO2 in the CONT slices (Fig. 3c, d). These data suggested a possibility for a downregulation of β-adrenergic receptors in isoproterenol-induced hypertrophied hearts for only 3 days.
Origin of E–C coupling mVO2
To explore mechanisms of the alteration of E–C coupling mVO2 in the HYP heart, we studied the contributions of SERCA and NCX to the E–C coupling mVO2 as shown in Fig. 4. To obtain reliable values, Tyrode solution containing CPA, CPA + KBR, or CPA + KBR + CE was applied to LV slices.

Effects of CPA, CPA + KBR and CPA + KBR + CE on E–C coupling mVO2 in CONT (a) (n = 10–12 sets from 5–6 hearts) and HYP (b) slices (n = 8–14 sets from 4–7 hearts). Each bar shown in a and b was not series data. Ordinate axis: E–C coupling mVO2 values by each second measurement. c Origin of E–C coupling mVO2 in CONT and HYP slices shown in a and b. SERCA2: sarcoplasmic reticulum Ca2+-ATPase. NCX: Na+/Ca2+ exchanger. NHE: Na+/H+ exchanger *P < 0.01 vs. None. # P < 0.01 vs. CPA. ∞ P < 0.05 vs. None in CONT slices. † P < 0.01 vs. CPA in CONT slices. § P < 0.05 vs. CPA + KBR in CONT slices
Inhibition of SERCA by CPA (30 μM) significantly (P < 0.05) decreased the mean E–C coupling mVO2 both in the CONT (n = 7 sets) and HYP slices (n = 8 sets) compared to each no treatment (none in Fig. 4a, b). However, the remaining mean delta mVO2 in HYP slices (53.7 ± 15.4% of 1st NT) was significantly (P < 0.01) larger than the one in the CONT slices (26.8 ± 7.7% of 1st NT). KBR at 1 μM can selectively inhibit reverse-mode NCX [24], but KBR at 10 μM can inhibit bidirectional-mode NCX as described in “Materials and methods” [23]. The following suppression of NCX by a combination of 30 μM CPA and 10 μM KBR further decreased mean E–C coupling mVO2 almost to zero in CONT slices (n = 10 sets) (Fig. 4a) and to 13.2 ± 8.2% of 1st NT in the HYP slices (n = 6 sets) (Fig. 4b). In the presence of CPA, [Ca2+]i must have increased, and thus forward-mode NCX must have been activated. Therefore, this decreased mean E–C coupling mVO2 by 10 μM KBR in the presence of CPA was consumed by Na+/K+-ATPase (NKA) coupled to NCX [23, 32]. An addition of 20 μM CE, a blocker of PMCA, which has a lower selectivity, did not further decrease mean E–C coupling mVO2 in the HYP slices (n = 9 sets) (Fig. 4b). These data are summarized in Fig. 4c. Mean E–C coupling mVO2 consumed by SERCA2 was smaller in HYP slices (46%) than in CONT slices (77%). On the contrary, mean E–C coupling mVO2 consumed by Na+/K+-ATPase (NKA) coupled to NCX [23, 32] was larger in the HYP slices (40%) than in the CONT slices (20%) (Fig. 4c). These data demonstrated that SERCA2 is downregulated and the NKA–NCX coupling is upregulated in the HYP heart at the functional level. The third small component of E–C coupling mVO2 was not determined yet. However, in longer-term isoproterenol-induced cardiac hypertrophy, functional evidence for enhanced Na+/H+ exchange-1 (NHE-1) activity has been reported [35]. Accordingly, the NKA–NHE-1 coupling may contribute to the third small component of E–C coupling mVO2.
Comparison of immunoblottings of SERCA2, PLB, p-PLB, NCX1, PLM and NKA between CONT and HYP
Expression of proteins representative in E–C coupling was studied with Western blot analysis in Fig. 5. Summarized data of SERCA2, PLB, p-PLB, NCX1, PLM and NKA expression in HYP (n = 5–6 hearts) in comparison with CONT (n = 5 hearts) indicated that protein levels of SERCA2, PLB and p-PLB were significantly lower in HYP than CONT. Protein level of NCX1, however, was not significantly different between CONT and HYP. Protein level of PLM was significantly higher, and the one of NKA was significantly lower in HYP than CONT (Fig. 5).

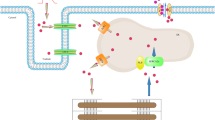
Comparison of immunoblottings of cardiac sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2), phospholamban (PLB), phosphorylated-Ser16 PLB (p-PLB), Na+/Ca2+ exchanger-1 (NCX1), Phospholemman (PLM) and Na+/K+-ATPase (NKA) between CONT and HYP. Upper inset Schematic illustration for Ca2+ handling in a cardiac myocyte of hypertrophic rat heart. a The ratio of handled Ca2+/ATP at SR. b The ratio of handled Ca2+/ATP at sarcolemma. Left trace each representative immunoblotting of SERCA2, PLB pentamer, p-PLB, NCX1, PLM and NKA in CONT and HYP slices. Lower graph indicates the quantitative protein levels measured from the immunoblottings. The levels of SERCA2, PLB, p-PLB and NKA were significantly lower, while the level of PLM was significantly higher in HYP (n = 5–6) than CONT (n = 5). *P < 0.05 vs. values in CONT (unpaired t-test)
The decrease in the expressions of p-PLB and SERCA2 is probably the major cause of the decrease of E–C coupling mVO2 consumed by SERCA2. However, the unaltered expression of NCX and slightly decreased NKA expression are apparently inconsistent with the increase in E–C coupling mVO2 consumed by the NKA–NCX coupling. NCX and NKA functions were further assessed by directly measuring the transporter-mediated current in the following experiments.
Functional study of NCX and NKA
The steady state current–voltage relationship of NCX current (I NCX) was measured in voltage-clamped ventricular myocytes. I NCX density was approximately two-fold larger in myocytes dissociated from HYP than the one of myocytes from CONT at all the membrane voltage examined (Fig. 6a). The intrinsic inactivation mechanisms of NCX1 [28, 36, 37] was assessed in a manner similar to a previous study [28] in Fig 6b. Slower inactivation of I NCX was observed in myocytes from HYP, suggesting the attenuation of inactivation mechanisms. The mean time constant of I NCX inactivation in HYP myocytes (n = 7) was significantly larger than that in CONT myocytes (n = 4) (11.4 ± 1.6 s vs. 8.4 ± 1.2 s) (Fig. 6c). It was likely that the augmentation of I NCX density in the HYP without the increase in protein expression was caused by the decrease in inactive NCX, i.e., the increase in functionally active NCX.

aI–V relationship of NCX current (INCX) measured in myocytes. The current density, which was normalized to the membrane capacitance, was significantly larger in HYP (filled circlesn = 17) than in CONT (open circlesn = 8). b Time-dependent inactivation decay of INCX in a CONT (gray circles) and a HYP (black circles) myocyte stimulated with 2 mM Ca2+ in Na+ free Tyrode solution. c Mean time constants of CONT myocytes (open columnn = 4) and HYP (solid columnn = 7) myocytes. A single exponential function was fit to the current decay. *P < 0.01 vs. CONT myocytes. **P < 0.005 vs. CONT myocytes
Although the protein level of NKA was significantly lower in HYP than CONT (Fig. 5), there were no significant differences in I NKA density between CONT and HYP myocytes (Fig. 7a). Therefore, it is concluded that the increase in E–C coupling mVO2 consumed by the NKA–NCX coupling is mainly caused by the functional upregulation of NCX.

aI–V relationship of NKA current (INKA) measured in CONT (open circlesn = 6) and HYP (filled circlesn = 7) myocytes. b [Na]i measured in CONT (open columnn = 13) and HYP (filled columnn = 8) myocytes
[Na+]i fluorescence measurements
As expected, [Na]i, which was measured with a Na+-sensitive dye (SBFI) during 1-Hz field stimulation, had a similar level in myocytes dissociated from CONT (19.4 ± 3.5 mM; n = 13) to that in HYP (17.8 ± 3.9 mM; n = 8) (Fig. 7b).
Discussion
The most important finding in this study was that delta mVO2 (=+St mVO2 − (−St mVO2)), which is associated with the total Ca2+ handling in E–C coupling [14, 15], moderately but significantly increased in the present HYP (see Fig. 2). The E–C coupling mVO2s were tightly related to SERCA2 and to NKA coupled with NCX. The contribution of SERCA2 was decreased and that of NKA–NCX coupling was increased in HYP. The functional attenuation of SERCA2 was supported by the lower protein level of p-PLB and SERCA2 (see Fig. 5). Probable reasons for this depression of SERCA2 activity in the present HYP would be activation of the MAPK kinase (MKK)6-p38 mitogen-activated protein kinase (MAPK) signaling pathway participating in downregulation of the SERCA2 gene transcription and the consequent activation of the calcineurin/nuclear factor of the activated T cells (NFAT) pathway [38], although decisive reasons were not yet clarified. Furthermore, it is possible that NFAT3 in concert with GATA4 could be involved in downregulation of the expression of the SERCA2 gene during cardiac hypertrophy [38].
The increase in E–C coupling mVO2 consumed by the NKA–NCX coupling was not caused by the upregulation of protein expression of either NKA or NCX. Our study actually demonstrated that the increase in active NCX, which was caused by the attenuation of inactivation, is the major cause of the increase, although the inactivation (I 1 and I 2 inactivation) [28, 36, 37] could not be distinguished by the present protocol. Concomitant changes in other proteins, such as PLM, might also be related to the attenuation, but the underlying mechanisms need to be further clarified. Accordingly, it is plausible that the increase in Na+ influx via forward-mode NCX results in the increased [Na]i, which in turn increases the Na+ efflux via NKA. Consequently, changes in [Na]i would be small. The actually observed increase in E–C coupling mVO2 in hypertrophic hearts supports this speculation, although further studies are needed.
On the other hand, it was found that in isoproterenol-induced hypertrophied left ventricular myocytes of rats by 7-day treatment, the density of the NKA current was reduced by 40%, and the intracellular Na+ content was increased by 55% [39]. These findings may explain the propensity for arrhythmias seen in this model of hypertrophy [39]. In the present study, [Na]i, which was measured with a Na+-sensitive dye (SBFI) during 1-Hz field stimulation, had a similar level in myocytes dissociated from CONT (19.4 ± 3.5 mM; n = 13) to that in HYP (17.8 ± 3.9 mM; n = 8). The difference in our results might have resulted from a different administration method (intraperitoneal injection vs. osmotic pump), dose (5 mg/kg once daily vs. 2.4 mg kg−1 day−1) and term (7 vs. 3 days).
Interestingly, in the failing heart of hypothyroid rats, the O2 consumption for total Ca2+ handling in the E–C coupling significantly decreased, but did not increase [40]. Thus, the remodeling of Ca2+ handling in the E–C coupling may depend on causal factors for heart failure.
Theoretically, energy demand for Ca2+ uptake by SERCA2 (1ATP: 2Ca2+) (recirculation fraction; RF) is one-half of that for Ca2+ extrusion mediated via forward-mode NCX coupled to NKA (1ATP: 1Ca2+) (1-RF) [32, 41]. Accordingly, when the contribution of energetically more efficient SERCA2 for Ca2+ uptake was less prominent and the contribution of energetically less efficient forward-mode NCX for Ca2+ extrusion coupled to NKA was more prominent, myocardial O2 consumption for total Ca2+ handling in the E–C coupling increased like in the hypertrophic model. Thus, the Ca2+ handling mechanism in E–C coupling in the hypertrophic heart is energetically less efficient than in controls. Since the prominent NCX to NKA coupling works in compensation for depressed SERCA2 activity, intracellular Ca2+ levels at the resting and/or excited states would be unaltered in the present short-term hypertrophic heart. Although the relaxation rate in the hypertrophic heart was somewhat slower than in the control hearts [42], our findings that systolic and diastolic functions were unchanged [11, 12] further supported this speculation.
In the previous study on NCX inactivation (28), fast and slow components were found in the NCX current decay during external Ca2+ application. The fast component was attributed to the inactivation process (time constant = 3–5 s) and the slow component to Na+ depletion/Ca2+ accumulation under the plasmalemma (time constant = several tens of seconds). In our current study (Fig. 6b, c), the contribution of Na+ depletion/Ca2+ accumulation was probably small because the amplitude of external Ca2+-induced NCX current was about 50–30% of the one in the previous study [28]. In fact, we could not detect the slower component in either CONT or HYP. Although the possibility of intracellular ion changes could not be completely negated, we assumed the decay of NCX current was largely due to the intrinsic inactivation of NCX.
According to the previous studies [28, 36], the majority of NCX was expected to be recovered from the inactive states in the absence of both external Na+ and Ca2+ (experiments in Fig. 6b, c) and to locate in the active state. Applying external Ca2+ initiated Na+–Ca2+ exchange cycle and the inactivation processes. Therefore, the peak amplitude of NCX current under this condition reflects the total number of NCX. Interestingly, no significant difference in the peak NCX current was observed between the two groups: 1.48 ± 0.21 (CONT) vs. 1.20 ± 0.45 (HYP) pA/pF. This might be in line with the result of Western blots (Fig. 5). On the other hand, in the presence of external Na+, NCX was expected to locate in both active and inactive states (experiments in Fig. 6a), and applying Ca2+ did not initiate measureable inactivation. Therefore, the difference in NCX current density found in Fig. 6a suggests the increase in active NCX, i.e., a decrease in inactive NCX.
It has been shown that myocardial substrate metabolism influences LV energetics in in situ hearts [43, 44]. The present experiment was completed in the sufficiently oxygenated Tyrode solution containing 10 mM glucose at a mechanically unloading state, lower temperature (30°C) and low frequency of field stimulation (1 Hz). Thus, the cardiac energy substrate is thought to be exogenous glucose. The myocardium becomes a net lactate producer only when there is accelerated glycolysis in the face of impaired oxidation of pyruvate, such as occurs with ischemia [44]. Actually, in this study, we detected no lactate in the perfusion solution of the LV slice throughout the experiments. Therefore, the contribution of anaerobic glycolysis is negligible in this study.
To evaluate LV function in in situ rat hearts, the systolic pressure-volume area (PVA) has been proposed as a good mechanoenergetic index [45, 46]. PVA, which is a total mechanical energy per beat, has been shown to linearly relate with myocardial O2 consumption per beat in in vitro cross-circulated (blood-perfused) rat hearts [23, 40, 47]. PVA-independent myocardial O2 consumption at PVA = 0 corresponds to energy requirements for Ca2+ handling in E–C coupling and basal metabolism [15, 23, 32]. Using this cross-circulated whole heart preparation of the same HYP model as the present one, we found that PVA-independent myocardial O2 consumption was unchanged compared to CONT (our unpublished observation). This result is consistent with the present one showing a nearly equal sum of E–C coupling and basal mVO2 values between CONT and HYP slices (see Fig. 2). The results indicated that the present oximetric system using rat LV slices enables us to evaluate LV energetics even in an HYP model under mechanically unloaded conditions (PVA = 0).
Mean mVO2 without stimulation in the CONT slices corresponded to basal metabolic mVO2 obtained by KCl arrest in blood-perfused whole heart preparations [23, 40, 47], supporting the reliability of the basal metabolic mVO2 value obtained in the CONT slices. The basal metabolic mVO2 significantly decreased in the HYP slices. The mechanisms associated with basal metabolic mVO2 have not yet been revealed. However, modulation of NKA activity is not probably the cause, because NKA current density was not decreased in myocytes from the present HYP, although the slightly decreased NKA and the increased PLM protein levels suggested a possibility for depression of NKA activity [48].
Changes of energy reserve in cardiac myocytes are also unlikely because the mean PCr/ATP (β-ATP) ratio of LV slices of the same HYP model without stimulation was unchanged compared to CONT (our unpublished observation). It is plausible that altered mitochondrial energy metabolism, altered mechanically unloading condition by contractile states, differences in slice composition (increased collagen accumulation) or any of a number of factors contributes to the decreased basal metabolic mVO2.
In conclusion, the E–C coupling mVO2 was moderately increased in the Iso-induced hypertrophied hearts. Decreased SERCA2 activity and increased NCX activity coupled with NKA contribute to the increase in E–C coupling mVO2. Therefore, the Ca2+ handling mechanism in E–C coupling in the hypertrophied heart is energetically less efficient. This mechanoenergetic imbalance may have a deleterious effect on the progress of heart failure.
Abbreviations
- BDM:
-
2,3-Butanedione monoxime
- BW:
-
Body weight
- CE:
-
5-(and -6)-Carboxyeosin diacetate
- CONT:
-
Saline-infused normal hearts
- CPA:
-
Cyclopiazonic acid
- DMSO:
-
Dimethyl sulfoxide
- Dob:
-
Dobutamine
- DW:
-
Dry weight
- E–C coupling:
-
Excitation–contraction coupling
- 1st NT:
-
1st Measurement value in normal Tyrode solution
- HYP:
-
Isoproterenol-infused hypertrophic hearts
- KBR:
-
KB-R7943
- LV:
-
Left ventricle
- MAPK:
-
Mitogen-activated protein kinase
- MKK:
-
MAPK kinase
- MTC:
-
Masson’s trichrome
- mVO2 :
-
O2 consumption per minute
- NCX:
-
Na+/Ca2+ exchanger
- NFAT:
-
Nuclear factor of activated T cells
- NHE:
-
Na+/H+ exchanger
- NKA:
-
Na+/K+-ATPase
- NT:
-
Normal Tyrode solution
- PKA:
-
cAMP-dependent protein kinase
- PLB:
-
Phospholamban
- p-PLB:
-
Phosphorylated-Ser16 PLB
- PLM:
-
Phospholemman
- PMCA:
-
Plasma membrane Ca2+-ATPase
- PVAmLVV :
-
Left ventricular systolic pressure-volume area at midrange left ventricular volume
- RF:
-
Recirculation fraction
- RV:
-
Right ventricle
- SBFI:
-
Sodium-binding benzofuran isophthalate
- SERCA2:
-
Sarcoplasmic reticulum Ca2+-ATPase
- St (−) mVO2 :
-
mVO2 of the quiescent slices in the absence of stimulation
- St (+) mVO2 :
-
mVO2 of the activated and contracting slices in the presence of stimulation
- WW:
-
Wet weight
References
Floras JS (2003) Sympathetic activation in human heart failure: diverse mechanisms, therapeutic opportunities. Acta Physiol Scand 177:391–398. doi:10.1046/j.1365-201X.2003.01087.x
Lohse MJ, Engelhardt S, Eschenhagen T (2003) What is the role of beta-adrenergic signaling in heart failure? Circ Res 93:896–906. doi:10.1161/01.RES.0000102042.83024.CA
Boluyt MO, Long X, Eschenhagen T, Mende U, Schmitz W, Crow MT, Lakatta EG (1995) Isoproterenol infusion induces alterations in expression of hypertrophy-associated genes in rat heart. Am J Physiol 269:H638–H647
Scheuer J (1999) Catecholamines in cardiac hypertrophy. Am J Cardiol 83:70H–74H. doi:10.1016/S0002-9149(99)00264-7
Sugden PH (1999) Signaling in myocardial hypertrophy: life after calcineurin? Circ Res 84:633–646
Suzuki J, Ohno I, Nawata J, Miura S, Ikeda J, Shirato K (1999) Overexpression of insulin-like growth factor-I in hearts of rats with isoproterenol-induced cardiac hypertrophy. J Cardiovasc Pharmacol 34:635–644. doi:10.1097/00005344-199911000-00003
Leenen FH, White R, Yuan B (2001) Isoproterenol-induced cardiac hypertrophy: role of circulatory versus cardiac renin-angiotensin system. Am J Physiol 281:H2410–H2416
Morisco C, Zebrowski DC, Vatner DE, Vatner SF, Sadoshima J (2001) Beta-adrenergic cardiac hypertrophy is mediated primarily by the beta (1)-subtype in the rat heart. J Mol Cell Cardiol 33:561–573. doi:10.1006/jmcc.2000.1332
Zhang GX, Ohmori K, Nagai Y, Fujisawa Y, Nishiyama A, Abe Y, Kimura S (2007) Role of AT1 receptor in isoproterenol-induced cardiac hypertrophy and oxidative stress in mice. J Mol Cell Cardiol 42:804–811. doi:10.1016/j.yjmcc.2007.01.012
Kitagawa Y, Yamashita D, Ito H, Takaki M (2004) The reversible effects of isoproterenol-induced hypertrophy on in situ left ventricular function in rat hearts. Am J Physiol 287(1):H277–H285
Takeshita D, Shimizu J, Kitagawa Y, Yamashita D, Tohne K, Nakajima-Takenaka C, Ito H, Takaki M (2008) Isoproterenol-induced hypertrophied rat hearts: does short-term treatment correspond to long-term treatment? J Physiol Sci 58(3):1–10. doi:10.2170/physiolsci.RP004508
Kohzuki H, Misawa H, Sakata S, Ohga Y, Suga H, Takaki M (1999) Energy expenditure by Ba2+ contracture in rat ventricular slices derives from cross-bridge cycling. Am J Physiol 277:H74–H79
Kohzuki H, Misawa H, Sakata S, Ohga Y, Takaki M (2001) Sustained high O2 use for Ca2+ handling in rat ventricular slices under decreased free shortening after ryanodine. Am J Physiol 281:H566–H572
Takaki M, Kohzuki H, Kawatani Y, Yoshida A, Ishidate H, Suga H (1998) Sarcoplasmic reticulum Ca2+ pump blockade decreases O2 use of unloaded contracting rat heart slices: thapsigargin and cyclopiazonic acid. J Mol Cell Cardiol 30:649–659. doi:10.1006/jmcc.1997.0630
Yasuhara S, Takaki M, Kikuta A, Ito H, Suga H (1996) Myocardial VO2 of mechanically unloaded contraction of rat ventricular slices measured by a new approach. Am J Physiol 270:H1063–H1070
Duwel CMB, Westerhof N (1988) Feline left ventricular oxygen consumption is not affected by volume expansion, ejection or redevelopment of pressure during relaxation. Pflügers Arch 412:409–416. doi:10.1007/BF01907560
Holroyd SM, Gibbs CL (1992) Is there a shortening-heat component in mammalian cardiac muscle contraction? Am J Physiol 262:H200–H208
Ito H, Shimizu J, Matsuoka S, Kim B, Takeuchi A, Nakajima-Takenaka C, Yamashita D, Sakata S, Takaki M (2008) Role of NCX in increased O2 consumption in hypertrophied rat hearts. J Physiol Sci 58(Suppl):S184
Delbridge LM, Loiselle DS (1981) An ultrastructural investigation into the size dependency of contractility of isolated cardiac muscle. Cardiovasc Res 15:21–27. doi:10.1093/cvr/15.1.21
Gibbs CL (1978) Cardiac energetics. Physiol Rev 58:174–254
Loiselle DS (1979) The effects of temperature on the energetics of rat papillary muscle. Pflügers Arch 379:173–180. doi:10.1007/BF00586944
Yamashita D, Kohzuki H, Kitagawa Y, Nakashima T, Kikuta A, Takaki M (2004) Oxygen consumption of mechanically unloaded contractions of mouse left ventricular myocardial slices. Am J Physiol 287(1):H54–H62
Hagihara H, Yoshikawa Y, Ohga Y, Takenaka C, Murata K, Taniguchi S, Takaki M (2005) Na+/Ca2+ exchange inhibition protects the rat heart from ischemic-reperfusion injury by blocking energy-wasting processes. Am J Physiol 288:H1699–H1707
Iwamoto T, Watano T, Shigekawa M (1996) A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem 271:22391–22397. doi:10.1074/jbc.271.37.22391
Choi HS, Eisner DA (1999) The role of sarcolemmal Ca2+-ATPase in the regulation of resting calcium concentration in rat ventricular myocytes. J Physiol 515:109–118. doi:10.1111/j.1469-7793.1999.109ad.x
Choi HS, Eisner DA (1999) The effects of inhibition of the sarcolemmal Ca2+-ATPase on systolic calcium fluxes and intracellular calcium concentration in rat ventricular myocytes. Pflügers Arch 437:966–971. doi:10.1007/s004240050868
Lin X, Jo H, Sakakibara Y, Tambara K, Kim B, Komeda M, Matsuoka S (2006) β-Adrenergic stimulation does not activate Na+/Ca2+ exchange current in guinea pig, mouse, and rat ventricular myocytes. Am J Physiol 290:C601–C608. doi:10.1152/ajpcell.00452.2005
Matsuoka S, Hilgemann DW (1994) Inactivation of outward Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J Physiol 476:443–458
Erlenkamp S, Glitsch HG, Kockskämper J (2002) Dual regulation of cardiac Na+-K+ pumps and CFTR Cl- channels by protein kinases A and C. Pflüger Arch 444(1–2):251–262. doi:10.1007/s00424-002-0802-4
Fujioka Y, Matsuoka S, Ban T, Noma A (1998) Interaction of the Na+-K+ pump and Na+-Ca2+ exchange via [Na]i in a restricted space of guinea-pig ventricular cells. J Physiol 509:457–470. doi:10.1111/j.1469-7793.1998.457bn.x
Despa S, Islam MA, Pogwizd SM, Bers DM (2002) Intracellular [Na+] and Na+ pump rate in rat and rabbit ventricular myocytes. J Physiol 539:133–143. doi:10.1113/jphysiol.2001.012940
Takaki M (2004) Left ventricular mechanoenergetics in small animals. Jpn J Physiol 54:175–207. doi:10.2170/jjphysiol.54.175
Tsuji T, Ohga Y, Yoshikawa Y, Sakata S, Abe T, Tabayashi N, Kobayashi S, Kohzuki H, Yoshida K, Suga H, Kitamura S, Taniguchi S, Takaki M (2001) Rat cardiac contractile dysfunction induced by Ca2+ overload: possible link to the proteolysis of fodrin. Am J Physiol 281:H1286–H1294
Gibbs CL, Loiselle DS (2001) Cardiac basal metabolism. Jpn J Physiol 51:399–426. doi:10.2170/jjphysiol.51.399
Ennis IL, Escudero EM, Console GM, Camihort G, Dumm CG, Seidler RW, Camilion de Hurtado MC, Cingolani HE (2003) Regression of isoproterenol-induced cardiac hypertrophy by Na+/H+ exchange inhibition. Hypertension 41:1324–1329. doi:10.1161/01.HYP.0000071180.12012.6E
Fujioka Y, Hiroe K, Matsuoka S (2000) Regulation kinetics of Na+-Ca2+ exchange current in guinea-pig ventricular myocytes. J Physiol 529:611–623. doi:10.1111/j.1469-7793.2000.00611.x
Matsuoka S, Nicoll DA, Reilly RF, Hilgemann DW, Philipson KD (1993) Initial localization of regulatory regions of the cardiac sarcolemmal Na+-Ca2+ exchanger. Proc Natl Acad Sci USA 90:3870–3874. doi:10.1073/pnas.90.9.3870
Zarain-Herzberg A (2006) Regulation of the sarcoplasmic reticulum Ca2+ ATPase expression in the hypertrophic and failing heart. Can J Physiol Pharmacol 84:509–521
Mészáros J, Khananshvili D, Hart G (2001) Mechanisms underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. Am J Physiol 281:H903–H914
Ohga Y, Sakata S, Takenaka C, Abe T, Tsuji T, Taniguchi S, Takaki M (2002) Cardiac dysfunction in terms of left ventricular mechanical work and energetics in hypothyroid rats. Am J Physiol 283:H631–H641
Cooper PJ, Ward M-L, Hanley PJ, Denyer GR, Loiselle DS (2001) Metabolic consequences of a species difference in Gibbs free energy of Na+/Ca2+ exchange: rat versus guinea pig. Am J Physiol 280:R1221–R1229
Nakajima-Takenaka C, Shimizu J, Tohne K, Sakata S, Takaki M (2007) Left ventricular lusitropic dysfunction of isoproterenol-induced hypertrophied rat heart. J Physiol Sci 57(Suppl):S209
Korvald C, Elvenes OP, Myrmel T (2000) Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol 278:H1345–H1351
Stanley WC, Recchina FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129. doi:10.1152/physrev.00006.2004
Kuzumoto N, Kitagawa Y, Uemura K, Ueyama T, Yoshida K, Furuya H, Takaki M (2004) A brief regional ischemic-reperfusion enhances propofol-induced depression in left ventricular function of in situ rat hearts. Anesthesiology 101:879–887. doi:10.1097/00000542-200410000-00012
Tachibana H, Takaki M, Lee S, Ito H, Yamaguchi H, Suga H (1997) New mechanoenergetic evaluation of left ventricular contractility in in situ rat hearts. Am J Physiol 272:H2671–H2678
Yoshikawa Y, Hagihara H, Ohga Y, Nakajima-Takenaka C, Murata K, Taniguchi S, Takaki M (2005) Calpain inhibitor-1 protects the rat heart from ischemic-reperfusion injury: analysis by mechanical work and energetics. Am J Physiol 288:H1690–H1698
Zhang X-Q, Moorman JR, Ahlers BA, Carl LL, Lake DE, Song J, Mounsey JP, Tucker AL, Chan YM, Rothblum LI, Stahl RC, Carey DJ, Cheung JY (2006) Phospholemman overexpression inhibits Na+-K+-ATPase in adult rat cardiac myocytes: relevance to decreased Na+ pump activity in postinfarction myocytes. J Appl Physiol 100:212–220. doi:10.1152/japplphysiol.00757.2005
Acknowledgments
This work was supported in part by Grants-in-aid for Scientific Research 17390231, 186501251, 20300162 and 19700385 from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. The authors thank Dr. T. Iwamoto, Fukuoka University, Japan, for the generous gift of anti-NCX1 antibody.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Shimizu, J., Yamashita, D., Misawa, H. et al. Increased O2 consumption in excitation–contraction coupling in hypertrophied rat heart slices related to increased Na+–Ca2+ exchange activity. J Physiol Sci 59, 63–74 (2009). https://doi.org/10.1007/s12576-008-0006-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-008-0006-6