
Abstract
Recent structural observations of kinesin-1, the founding member of the kinesin group of motor proteins, have led to substantial gains in our understanding of this molecular machine. Kinesin-1, similar to many kinesin family members, assembles to form homodimers that use alternating ATPase cycles of the catalytic motor domains, or “heads”, to proceed unidirectionally along its partner filament (the microtubule) via a hand-over-hand mechanism. Cryo-electron microscopy has now revealed 8-Å resolution, 3D reconstructions of kinesin-1•microtubule complexes for all three of this motor’s principal nucleotide-state intermediates (ADP-bound, no-nucleotide, and ATP analog), the first time filament co-complexes of any cytoskeletal motor have been visualized at this level of detail. These reconstructions comprehensively describe nucleotide-dependent changes in a monomeric head domain at the secondary structure level, and this information has been combined with atomic-resolution crystallography data to synthesize an atomic-level "seesaw" mechanism describing how microtubules activate kinesin’s ATP-sensing machinery. The new structural information revises or replaces key details of earlier models of kinesin’s ATPase cycle that were based principally on crystal structures of free kinesin, and demonstrates that high-resolution characterization of the kinesin–microtubule complex is essential for understanding the structural basis of the cycle. I discuss the broader implications of the seesaw mechanism within the cycle of a fully functional kinesin dimer and show how the seesaw can account for two types of "gating" that keep the ATPase cycles of the two heads out of sync during processive movement.
Similar content being viewed by others

Introduction
More than many enzymes, the molecular motor proteins invite descriptive, mechanical terms such as "piston", "driveshaft", and "cam" to explain their mechanism of action. The kinesin molecular motor is commonly understood to deliver a kind of "power stroke" with its "force-delivering element", the neck linker, to engage in a "hand-over-hand", "walking" movement. The power stroke is itself proposed to be actuated by a "piston-like" or "seesaw-like" movement of an element known as the "relay helix" (Vale and Milligan 2000). These mechanical analogies, however, are necessarily incomplete, not least because the hydrolysis of ATP to ADP/Pi products, which is a purely chemical step, lies at the heart of the mechanism. Indeed, a major motivation for the study of molecular motors is to understand how chemical energy is converted to directed movement, in a "chemo-mechanical" cycle. As such, exploration of kinesin’s mechanism provides one avenue to better understanding the overall energetics of protein folding and conformational change.
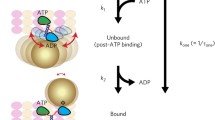
More than two decades of intensive study have established that the kinesin motor domain undergoes the following biochemical transitions during its ATPase cycle (Fig. 1): (1) weak attachment to the microtubule in the motor’s ADP-bound form; (2) release of ADP to form a tightly attached motor–microtubule complex; (3) binding of ATP, which triggers a significant conformational rearrangement of the motor domain, associated with a displacement of the cargo attachment point (the "neck linker", see below) in the direction of travel; and (4) hydrolysis of ATP and release of phosphate, which leads to concurrent detachment of the motor domain from the microtubule (Gilbert and Johnson 1994; Ma and Taylor 1995).

Overview of kinesin and microtubule architecture. a. Schematic depiction of dimeric kinesin during processive movement. The crystallized conformation of dimeric kinesin-1 (Kozielski et al. 1997) is overlaid on a low-resolution density map of the microtubule with fitted atomic coordinates of α- and β-tubulin from the 2D crystal structure of α-β tubulin (Nogales et al. 1998). The microtubule polarity is indicated with an arrow running towards the plus end. ADP is depicted with yellow spheres. b–d Schematic depiction of structural states encountered by the actively hydrolyzing head in a kinesin dimer as it progresses through ADP, no-nucleotide, and ATP-bound states while attached to the microtubule. The ADP attaches weakly to the microtubule, while no-nucleotide and ATP states are tightly attached states (Rosenfeld et al. 1996). Following the model of Rice et al. (1999), the neck linker is depicted in a disordered conformation in the first two nucleotide states, and is docked along the head towards the microtubule plus end in the ATP state. e, f Overview of the atomic structure of kinesin-1 obtained by X-ray crystallography and fitted into a high-resolution cryo-EM map together with the atomic structure of tubulin (Sindelar and Downing 2010). Key structural elements are labeled
Deriving the structural basis for the above steps is clearly critical for better understanding of the motor mechanism. However, a principle difficulty in structural analysis of the kinesin system, as with the other two known cytoskeletal motors (myosin and dynein), has been the inability to obtain atomic-resolution structures of the motor while complexed to the partner filament. To date, the only atomic-resolution structural data available for kinesin comes from X-ray crystal structures of the molecule by itself (Marx et al. 2009) or in complex with a regulatory partner (Vinogradova et al. 2009). However, kinesin’s enzymatic properties change markedly in the absence of microtubules, where for example the ATPase rate drops to a basal level ∼1,000-fold reduced over the motile, microtubule-attached state (Hackney 1988). Such crystal structures, while highly informative, are not necessarily accurate depictions of the enzyme during the actual force-generating process, during which the microtubule is a required substrate. This shortcoming is highlighted by the observation that most crystal structures of kinesin—with one recent exception (Parke et al. 2010)—do not exhibit a hydrolytically competent conformation of the ATP binding site even in the presence of non-hydrolyzable ATP analogs (Kikkawa et al. 2001; Nitta et al. 2004).
The current inability of X-ray crystallography to characterize the kinesin–microtubule complex has motivated the use of an alternative structural method, high-resolution cryo-electron microscopy (cryo-EM), to fill the gap (Kikkawa 2008). Most cryo-EM studies on kinesin published to date, however, are limited to ≥10-Å resolution. This has meant that atomic-level structure in the motor–microtubule complex must be inferred from a combination of crystallography and the EM data. Unfortunately, at ≥10-Å resolution the structural analysis of cryo-EM data has been principally limited to rigid-body fitting of known crystal structures into the resulting 3D maps. As the resolution of a reconstruction drops below 10 Å, however, cryo-EM begins to resolve secondary structure details unambiguously, which can allow subdomain conformations in various crystal structures to be matched to features in the maps. Recent studies have now begun to achieve this goal for the kinesins (Sindelar and Downing 2007; Bodey et al. 2009; Sindelar and Downing 2010).
Here I discuss how a recently introduced "seesaw" model (Sindelar and Downing 2010), derived from cryo-EM reconstructions of kinesin-1 that now consistently achieve 8-Å resolution in all three of the motor’s principal nucleotide states (ADP, no-nucleotide, and ATP analog-bound), may be used to advance our understanding of conventional kinesin-1. I place particular emphasis on how the motor co-modulates its interactions with the microtubule and the nucleotide together within a single catalytic domain. As I discuss, the seesaw model provides an intuitive, atomic-level description of the fundamental biophysical forces that drive kinesin’s ATP-sensing machinery, although the functional changes associated with ADP release, a key piece of the motile mechanism, still remain largely unexplained. Based on the "seesaw" mechanism, I introduce a novel model detailing the coordination between two dimerized "walking" motor domains.
Modulating neck linker conformation and microtubule affinity: two proposed roles for the switch II helix
The atomic architecture of kinesin reveals that this motor possesses Walker-type nucleotide-sensing “switch” motifs common to a broad swath of ATPase and GTPase enzymes (Sablin et al. 1996). An important observation relating to the functioning of these motifs was that the switch II sensor loop is immediately N-terminal to the "switch II helix" that forms a major part of kinesin’s microtubule binding interface (see Table 1 and Fig. 2 for definitions of the switch regions and other key structural elements). Moreover, the switch II helix extends across the full width of the motor domain (Fig. 1f), reaching from the nucleotide cleft across to the opposite side where the C terminus of the catalytic domain emerges and (in conventional kinesins) attaches to cargo, via a putative force-generating element called the neck linker. This arrangement therefore suggested an elegant structural scheme for motor function whereby ATP binding in the motor would simultaneously modulate the microtubule binding affinity and also lead to cargo translocation, with both actions controlled via switch-loop-modulated changes in the geometry of the switch II helix (Kull et al. 1996; Sablin et al. 1996; Vale and Milligan 2000). Each of these of these putative functional roles for the switch II helix, modulation of microtubule attachment affinity and modulation of the conformation of the neck linker, constitutes a prediction which can be tested by structural and biophysical methods.

The seesaw model for activation of kinesin by microtubules. a Schematic showing the correspondence of the crystal structure with elements in the seesaw cartoon. Selected side chain atoms are rendered with van der Waals spheres (fulcrum F82, Y84 are in gray), while the nucleotide is depicted by a ball-and-stick diagram. b Cartoon depiction of the seesaw. Positions of various structural elements are labeled. c Depiction of uncoupled seesaw motion in the absence of microtubules. Disordered neck linker is represented by a dashed magenta line; disordered loop L11 is represented by a dashed red line. While this panel depicts kinesin’s ADP state, the ATP state is expected to explore a similar set of conformations, in the absence of microtubules. d Depiction of microtubule-activated "seesaw" ATP sensing mechanism. The notch in the microtubule surface represents a conserved contact with residue N255 in the switch II helix. Grayed out bottom panels represent conformations disfavored by the seesaw coupling mechanism, so that a "pre-stroke" state is uniquely selected in the absence of ATP, and a "post-stroke" state is uniquely selected in the presence of ATP. Striped areas in the bottom panels represent unfavorable interactions generated either by (bottom left) steric overlap between I254 and the switch loops lining the nucleotide site or (bottom right) hydrophobic void formed by displacement of I254 out of the switch pocket
Perhaps the most direct test of the switch II helix’s proposed functional roles has been via high-resolution cryo-EM studies of the kinesin–microtubule complex. A number of such studies have reported nucleotide-induced swiveling of the kinesin core domain relative to the switch II helix, which appears to remain in a fixed position relative to the microtubule surface (Kikkawa et al. 2001; Kikkawa and Hirokawa 2006; Sindelar and Downing 2010). This swiveling has been interpreted to support a role for the switch II helix in modulating microtubule affinity (Kikkawa et al. 2001), aptly described as a “twist-off” scheme (Schliwa and Woehlke 2001). In the twist-off scheme, the presence or absence of the γ-phosphate moiety would trigger changes in the shape of the motor’s microtubule binding interface via swiveling of the switch II helix.
These and other studies have also reported nucleotide-dependent conformational transitions of the neck linker such that this element is mostly disordered in non-ATP-bound state of the motor, but docks along the motor domain in the ATP state (see Fig. 1). This docking transition appears closely coupled to the orientation of the switch II helix on the motor domain, in support of a role for the switch II helix in docking the neck linker (Vale and Milligan 2000; Kikkawa et al. 2001). Coupling between the switch II helix and the neck linker conformation is further supported by crystallographic observations of "ADP-like" and "ATP-like" arrangements of these elements, such that the C-terminal end of the switch II helix would displace the docked neck linker conformation in the former arrangement, but open up a complementary hydrophobic pocket that facilitates docking in the latter conformation, providing a kind of "power stroke" (Vale and Milligan 2000). Thus, a significant body of structural evidence supports a role for the switch II helix both in the modulation of microtubule attachment affinity as well as modulation of neck linker conformation, via nucleotide-controlled transitions between "ADP-like" and "ATP-like" orientations of the switch II helix.
Two key questions relating to proposed roles of the switch II helix
The above described scheme leaves at least two key problems relating to motor function. The first is how microtubules activate kinesin’s ATP-sensing functions: microtubules are required to accelerate catalysis and also to enable ATP-induced docking of the neck linker. A second problem is why ADP triggers motor detachment from the microtubule, while the no-nucleotide motor state remains very tightly attached. For example, the “twist-off” scheme does not distinguish between these two nucleotide states at all (Kikkawa et al. 2001). The presence of such issues, within the switch II helix scheme, leaves open the possibility that a different mechanism entirely may underlie the motility properties of some or all kinesin motors (Hirose et al. 2006).
Here it should be emphasized that at least three different nucleotide states of kinesin have been identified, each having quite distinct functional properties (Fig. 1b–d). Kinesin begins its motility cycle in the ADP-bound state where the motor can only weakly attach to the microtubule, and docking of the neck linker is disfavored; following ADP release, nucleotide-free kinesin attaches tightly to the microtubule, but the neck linker remains undocked; finally, ATP binds and triggers docking of the neck linker (Rice et al. 1999; Asenjo et al. 2006), while the motor remains tightly attached to the microtubule. Subsequent hydrolysis and phosphate release events reset the cycle to the ADP state. To date, atomic-level structural schemes for kinesin, including the “seesaw” mechanism described below, have largely focused on conformational changes triggered by the presence or absence of the γ-phosphate moiety of ATP. In this simplified, γ-phosphate-oriented framework, ADP-bound and no-nucleotide states would both be loosely categorized as “ADP-like” structural states, while binding of ATP or related transition-state analogs would generate an “ATP-like” structural state (Vale and Milligan 2000). Importantly, the key step of ADP release is not addressed within the γ-phosphate-oriented framework (see the later sections of this review dealing with this issue). On the other hand, as I now discuss, this framework has proven particularly useful in analyzing the mechanics of docking by the neck linker.
Seesaw model can account for microtubule activation of the motor’s ATP sensing functions
To determine how microtubules activate kinesin’s nucleotide-sensing functions, including ATP hydrolysis and ATP-triggered neck linker docking, the conformation of microtubule-attached kinesin (as reported by sub-nanometer cryo-EM reconstructions) must be compared to various atomic-resolution crystal structures. Such comparisons suggested that microtubule attachment functionally activates kinesin through a direct contact involving the absolutely conserved N255 residue within the switch II helix, which apparently mediates the formation of several additional coils at the helix’s N terminus from a region of the protein sequence often manifesting in crystal structures as a disordered loop (L11) (Sindelar and Downing 2007). These additional coils, universally seen in sub-nanometer cryo-EM reconstructions of kinesin-1, are the key element in an elegant "seesaw" mechanism that is proposed to explain why microtubules are required in order for ATP-bound kinesin to proceed with neck linker docking and associated catalysis of the hydrolytic step (Sindelar and Downing 2010).
Seesaw mechanism explains how microtubule attachment can regulate hydrolytic cleavage and neck linker docking
A rather specific molecular mechanism for ATP-induced docking by the neck linker could be derived from the kinesin-1 data, owing to exceptionally high clarity of features in the sub-nanometer cryo-EM maps. This clarity, which indicated that sufficient data had been collected to largely eliminate structural noise up to ∼8–9 Å resolution, permitted the synthesis of high-precision atomic models for many of the key elements visualized in the maps, using a library of crystallized subdomain conformations from various crystal structures (Sindelar and Downing 2010). The resulting analysis showed that kinesin’s central β-sheet pivots relative to the switch II helix around a "fulcrum" of highly conserved, bulky hydrophobic residues positioned roughly at the center of the sheet–helix interface. This movement leads to coupling between the nucleotide binding site and the neck linker because tilting the resulting "seesaw" towards one side or the other will open a hydrophobic cavity between the sheet and the helix on the opposite side (Fig. 2a). Thus, by selectively occupying or reshaping these cavities, events at the nucleotide cleft may exert a strong energetic influence on the neck linker and vice versa.
Detached from microtubules, however, the left end of the switch II helix shows strong evidence of disorder, as reflected in numerous crystal structures where density for this element is absent or displays a large "kink" in the helix (Vinogradova et al. 2008). The resulting absence of specific interactions between the core domain and the switch II helix, on the left side of the seesaw, indicates that under detached conditions the balance between docked and undocked states may be specified principally by elements located on the right side of the seesaw. The resulting absence of structural regulation deriving from the nucleotide binding site would account for kinesin’s apparent nucleotide-independent equilibrium between docked and non-docked neck linker conformations, when detached from the microtubule (Sindelar et al. 2002), as depicted in Fig. 2b.
Once kinesin attaches to the microtubule, however, cryo-EM data indicate that a microtubule contact stabilizes the formation of several coils on the N terminus of the switch II helix (the "switch II helix extension"), as indicated in Fig. 2c (Sindelar and Downing 2007; Kikkawa 2008; Bodey et al. 2009; Sindelar and Downing 2010). Stabilization of the helix extension thereby introduces a hydrophobic interface between kinesin’s central β-sheet and the switch II helix on the nucleotide-binding side of the seesaw (left side of molecule as depicted in Fig. 2). The upper side of this interface, contributed by kinesin’s central β-sheet, is formed by the switch I and switch II nucleotide sensing loops. The lower side of the interface is formed by the extremely conserved I254 side chain from the switch II helix extension (note that the switch II sensor loop is connected to the switch II helix by L11, a flexible loop whose length and sequence is not highly conserved). Crystal structures of myosin and kinesin, which share a very similar switch loop architecture, indicate that binding of ATP analogs in the active site stabilizes a "closed" conformation where the switch I and II sensor loops retract toward the nucleotide to directly interact with the γ-phosphate via an extensive network of hydrogen bonds (Fisher et al. 1995; Minehardt et al. 2001; Parke et al. 2010). In atomic models of kinesin-1 derived from cryo-EM data, ATP-induced retraction of the switch loops opens up an enlarged cavity below the nucleotide cleft and facing the switch II helix, complementary to I254 (Sindelar and Downing 2010) (Fig. 2d, right hand side); this cavity has been termed the "switch pocket". Thus, the seesaw can respond to ATP binding by tilting leftwards such that I254 occupies the switch pocket; this tilting movement is confirmed by high-precision docking alignments of kinesin’s central β-sheet domain into 8-Å resolution EM maps (Sindelar and Downing 2010) and also by a crystal structure of the Eg5 motor complexed with the ATP analog AMPPNP (Parke et al. 2010). In this way, ATP binding can control the seesaw angle by modulating the shape of the switch pocket.
On the opposite side of the motor from the nucleotide cleft (right-hand side in Fig. 2), ATP-triggered leftward tilting by the seesaw opens up a second hydrophobic pocket (this may be thought of as a "docking pocket") into which the neck linker can bind (Fig. 2a and b, right-hand side). An important observation from the kinesin-1 cryo-EM structures was that the C terminus of helix 6 (to which the neck linker attaches) also contributes to the docking interaction (Sindelar and Downing 2010), as has been observed in crystal structure comparisons of "ADP-like" and "ATP-like" kinesin conformations (Khalil et al. 2008). As shown in Fig. 3, the cryo-EM maps indicate that α6 lengthens by at least one turn during the docking transition, which would insert a conserved hydrophobic element from α6 (A321 in human conventional kinesin-1) into the docking pocket, as seen in crystal structures that exhibit a docked neck linker. The neck linker itself also contributes a highly conserved hydrophobic side chain (I325 in human kinesin-1 construct) into the docking pocket in the ATP-bound motor state. The resulting hydrophobic interactions are likely to contribute a sizable component of free energy stabilizing the docked motor conformation, owing to the substantial depth, size, and conserved hydrophobic nature of the docking pocket. The docked neck linker conformation is stabilized by additional interactions with kinesin’s N-terminal "cover strand" (Khalil et al. 2008), although cover-strand interactions do not appear to be directly modulated by the switch II helix. The overall view of kinesin’s microtubule-attached docking transition that emerges is that tight coupling between ATP binding and neck linker docking occurs through seesaw-mediated changes in the hydrophobic pockets present on either side of the motor domain. As described above, however, when kinesin is detached from the microtubule the left-hand side of the seesaw would no longer be regulated by bound nucleotide, accounting for the observed nonproductive behavior of the motor in the absence of microtubules.

Experimental observation of elongation by helix α6 following ATP analog binding, in 8 Å cryo-EM maps. a Cryo-EM density map of microtubule-attached, no-nucleotide kinesin, showing a shortened “sausage” of density for α6 that corresponds to a non-helical conformation of the C terminus as seen in crystal structures of "ADP-like" kinesin. b Density map of microtubule-attached, ADP•Al•Fx kinesin showing elongation of α6, corresponding to occupation of the docking pocket by the neck linker and the C terminus of α6. Density corresponding to the docked conformation of the neck linker is also evident
Discrepancies with earlier crystal structure-based models
It should be noted that the structural states found in the seesaw model of kinesin’s power stroke differ substantially from those found in the recently published scheme of Nitta et al. (2008) and Hirokawa et al. (2009). For example, the Nitta et al. scheme predicts that kinesin’s no-nucleotide state (which they label as an “ATP/ADP exchange state”) exhibits most of the features (including a docked neck linker) that the seesaw scheme presented here would attribute exclusively to the ATP state, for the microtubule-attached motor. Because the Nitta et al. scheme thus requires the switch II helix to assume an “ATP-like” orientation in the absence of ATP, however, this scheme appears to be fundamentally incompatible with the seesaw mechanism as presented here. Another striking difference between the two schemes is the absence of an ordered switch II helix extension in many points in the ATPase cycle proposed by Nitta et al., whereas the seesaw mechanism predicts that the switch II helix extension is universally present, for the microtubule-attached case.
A probable explanation for these discrepancies is that the Nitta et al. structural models are derived exclusively from crystal structures of kinesin in the absence of microtubules. The scheme presented by Nitta et al. may therefore be more relevant to kinesin’s basal ATPase reaction, although it is also possible to interpret the conformations assigned by Nitta et al. to specific nucleotide states as fluctuations that are accessible to many or all nucleotide states, owing to the large variations in the orientation of the switch II helix that are possible when ADP- or ATP analog-bound kinesin is detached from microtubules (Sindelar et al. 2002).
Structures representing kinesin’s microtubule-attached, ATP analog-bound state inform the free energy of hydrolysis
The isotopic measurements of Hackney (2005) demonstrated that, on microtubules, the hydrolysis reaction in kinesin displays some degree of reversibility. These measurements allowed an upper limit of ∼20 kJ/mol to be placed on the free energy liberated during the microtubule-attached motor’s ATP cleavage step, compared with ∼55 kJ/mol net available free energy at physiological concentrations of ADP, ATP, and phosphate. The conclusion from this work was that kinesin redistributes the majority of the available hydrolysis free energy away from the cleavage step, such that >30 kJ/mol of favorable energy accompanies ATP binding during productive motor stepping. This observation was significant because it suggested that a large amount of free energy would be available to drive ATP-triggered docking by the neck linker. Docking of the neck linker, in turn, may serve a crucial role in the motility mechanism by either its “power-stroke” like character, its role in “gating”, or both (see below).
Consideration of the ATP state structures available for kinesin on the microtubule (Sindelar and Downing 2010) may boost this free energy estimate still further. As pointed out by Hackney, his analysis of isotope exchange experiments operates under the assumption that the catalytic water is in free exchange with the solvent medium. The experimental logic used is that the more times a reversal occurs in the nucleotide site, the more labeled oxygen is introduced in the phosphate product when it is finally released and subsequently analyzed. However, the tightly sealed nature of the nucleotide cleft in the presumed catalytically active kinesin ATP structure (Parke et al. 2010; Sindelar and Downing 2010) indicates that catalytic water might not be in free exchange with the solvent medium. If this were the case, then the isotope experiments may have substantially underestimated the amount of reversal that takes place because only two labeled waters are found within the nucleotide-binding cavity where they would be available for exchange. Geometry constraints within the closed nucleotide cleft could still further limit the exchange reaction if the two waters cannot readily swap positions during the catalytic process. In such a circumstance, the maximum observed isotope exchange could be limited to two or even fewer out of four available phosphate oxygen groups, even if many rounds of repeated hydrolysis and synthesis occurred. Any opening of the nucleotide cleft that was sufficient to admit new labeled waters during catalysis would seem just as likely to release phosphate as to admit additional waters, further limiting the possibility of productive solvent exchange. It therefore seems possible that Hackney’s observation of a relatively limited isotope effect may in fact reflect a much larger rate of hydrolysis reversal than he obtained under assumptions of free solvent exchange. If so, then the free energy available from kinesin’s ATP binding step could represent an even larger fraction of the available hydrolysis energy, further strengthening his conclusion. The seesaw mechanism provides an ideal means of harnessing the free energy of ATP binding and transmitting this energy (in a microtubule-dependent manner) across the motor domain to the neck linker.
In this context, however, it should be noted that even in the absence of ATP analogs, EPR measurements have detected evidence of substantial neck linker docking for kinesin’s microtubule-attached state (Rice et al. 2003; Larson et al. 2010). This latter observation led to the suggestion that ATP binding contributes <1 kcal/mol of free energy towards the docking transition. While it has been argued that kinesin could in principle generate productive motility even with such poor coupling (Rice et al. 2003; Schnitzer et al. 2000), this small free energy magnitude seems inconsistent with the tight coupling indicated by the seesaw mechanism. An alternative interpretation of the EPR results is possible, however, that would support strong ATP/neck linker coupling: that the docking reported in kinesin’s non-ATP states reflects an alternative, ordered conformation of the neck linker, perhaps oriented towards the microtubule minus end. This latter interpretation is supported by cryo-EM experiments that indicate partial ordering of the neck linker in ADP-bound and no-nucleotide states, but having a distinctly different orientation than ATP analog docked state (Rice et al. 1999). The dual questions of (1) how much free energy drives neck linker docking and (2) how different ways the neck linker can dock are clearly central to the motility mechanism and deserve future attention.
Revised forward ATP gating mechanism for processive dimer: "seesaw gating" vs. "pure strain gating"
For two heads of a kinesin dimer to avoid premature detachment during processive movement along a microtubule, it is necessary for the heads to keep their ATPase cycles out of phase. One particularly counterproductive sequence of events that should be avoided follows attachment of the leading head and accompanying release of ADP from this head (relatively fast steps in the kinesin cycle). At this point in the cycle, the trailing head remains tightly attached for ∼1/50 s until coupled phosphate release and microtubule detachment occur (this latter event is the primary rate-limiting step in the cycle of microtubule-attached kinesin; Ma and Taylor 1995). During this 1/50 s period, it would be detrimental to motility for ATP to bind the leading head and so trigger a power stroke because the resulting strain might lead to detachment of the leading head rather than the trailing head (Rosenfeld et al. 2003). Indeed, kinetics (Rosenfeld et al. 2003) and optical trapping experiments (Guydosh and Block 2006) have indicated that a form of "gating" occurs in kinesin, such that when the trailing kinesin head is strongly attached, ATP is prevented from binding to the leading head. A series of elegant mutagenesis experiments, where the neck linkers of kinesin dimers were artificially lengthened (Hackney et al. 2003; Shastry and Hancock 2010) and/or an assisting load was applied (Yildiz et al. 2008), have implicated strain between the two kinesin heads (transmitted through the neck linkers) as a likely factor in such this type of ATP gating. However, the detailed molecular mechanism for this process remains uncertain.
The seesaw scheme can account for such an "ATP gating" mechanism as follows (Fig. 4a). Strain generated by a strongly attached trailing head would prevent forward extension of the neck linker and accompanying occupation of the docking pocket by kinesin’s α6/neck linker assembly. Consequently, if the seesaw within the leading head were to assume the ATP-bound conformation having an open docking pocket, no compensating hydrophobic interactions could be made by the α6/neck linker assembly in this head. On the other hand, if the seesaw within leading head maintains a closed docking pocket, formation of the switch pocket on the opposite side of the seesaw (required for the switch loops to assume a strong hydrogen bond network with the γ-phosphate of ATP) will not be favored owing to displacement of I254 (located on the switch II helix extension) away from the seesaw interface. In this latter circumstance, switch pocket formation would create an unfavorable hydrophobic cavity on the left-hand side of the seesaw. Thus, if the lead head is under rearwards strain but simultaneously is bound to ATP, both leftwards as well as rightwards seesaw orientations in this head are disfavored by unoccupied hydrophobic cavities so that ATP binding itself would be disfavored. This mechanism thus provides a means by which the trailing head can gate ATP binding in the lead head.

Model for gating of ATP binding in the leading head, controlled by attachment of the trailing head. a Cartoon schematic indicating how the trailing head in a processively moving kinesin dimer generates rearwards strain on the neck linker that would “gate” ATP binding in the lead head. Rearwards strain prevents the neck linker/α6 assembly from binding into the hydrophobic docking pocket, thus leading to unfavorable energy if the seesaw tilts leftward to accommodate ATP binding (grayed out bottom panel). On the other hand, if the seesaw does not tilt in response to ATP binding, the switch loops cannot deform to interact with γ-phosphate without generating the switch pocket, which will be vacant (grayed out top panel). b Depiction of structure models showing the absence of support for a “twist-off” mechanism of gating between trailing and lead motor domains. Tilting caused by ATP binding in the forward head generates almost no displacement by the neck linker attachment point. Crystal structures were fitted into a composite density map, where the density for the trailing head is from an 8-Å resolution map of the ADP•Al•Fx state of kinesin-1 and density for the leading head is from the corresponding no-nucleotide map (Sindelar and Downing 2010). Axis of rotation identified for core domain movement during the transition from no-nucleotide to ADP•Al•Fx motor states is indicated by the green rod. c Comparison of predicted location of I325 at the α6/neck linker junction, before and after occupation of the docking pocket in the leading head by this residue accompanying neck linker docking. The resulting 5-Å displacement is consistent with a gating mechanism whereby rearwards strain prevents I325 from occupying the docking pocket
A number of distinct but related structural schemes have been proposed to account for this ATP gating function in kinesin. One mode of gating, which I will refer to as “pure strain gating”, would prohibit tilting in the leading head simply via geometric strain originating from the attached trailing head (Sablin and Fletterick 2004; Kikkawa 2008), similar to how a stay rope prevents the mast of a sailing ship from toppling over. This scheme contrasts with the above seesaw gating scheme where strain disfavors tilting of the lead head more indirectly, by preventing the neck linker of the leading head from occupying its docking pocket. As shown in Fig. 4b, however, ATP analog-induced tilting observed in the kinesin-1 system does not significantly increase the predicted strain between leading and trailing heads (∂x < 0.2 Å), thus indicating that this simple type of pure strain gating would not be effective.
A variation of pure strain gating has also been proposed whereby ATP-induced tilting might enhance inter-head strain by introducing steric interference between the rearwards-directed neck linker (in the leading head) and a small "β-domain" protruding from the minus end side of the motor domain (Sablin and Fletterick 2004; Kikkawa 2008). However, the tilting movement found in the kinesin-1 EM data fails to generate obvious interference between the neck linker and the β-domain (Fig. 4b). The “pure strain gating” models were originally based on data from lower-resolution EM maps of KIF1A (∼16–20 Å) where secondary structure of the motor was not resolved, thus giving rise to substantially greater uncertainty in quantification of motor domain movement (Kikkawa et al. 2001). Indeed, subsequent reprocessing of same KIF1A data to ∼10–12 Å resolution (Kikkawa and Hirokawa 2006) led to a substantial revision in the predicted axis of rotation; the result, for ATP analog-triggered tilting, was very similar to what was seen in high-resolution kinesin-1 reconstructions (Sindelar and Downing 2010). Thus, the analysis presented in Fig. 4 tends to disfavor “pure strain gating” type models for KIF1A as well as kinesin-1.
The role of ADP is critical to motor function
The functional role of ADP in kinesin’s cycle may be equally critical to the motor mechanism as ATP. Numerous studies on a wide range of motile kinesin families have shown that the ternary complex of microtubule-attached, ADP-bound kinesin is relatively weak, with dissociation constants of 10–20 μM. In comparison, kinesin’s microtubule attachment affinity is 1 μM or lower in other observed nucleotide states (Rosenfeld et al. 1996; Ma and Taylor 1997). This instability in the ADP state is critically involved in defining both the start and finish of the hydrolysis cycle of the microtubule-attached motor, as was highlighted by the landmark observation that a dimer of ADP•kinesin heads releases only one ADP when it encounters microtubule (Hackney 1994). Remarkably, the dissociation pathway of the three-way ADP•kinesin•microtubule complex has been shown to depend on external strain, such that motor detachment is more likely when an external load is applied to the neck linker from the leading direction, while ADP release is more likely when the load is applied from the trailing direction (Uemura et al. 2002; Uemura and Ishiwata 2003).
These characteristics are consistent with the following ADP-gating scheme for processive movement by kinesin dimers (Hancock and Howard 1999). At the stage in the cycle where one head is ATP-bound and a second, ADP-bound head is searching for the next binding site, a productive forward attachment step by the ADP head would likely involve considerable rearward-directed strain on this head’s neck linker, while the reverse is true for a backward attachment step. Kinesin’s asymmetric behavior under strain with respect to ADP, therefore, naturally favors the forward step (see the final section for an illustration of this mechanism). The role of ADP, particularly with respect to gating, is thus critical to understanding kinesin’s motility mechanism.
Seesaw leads to a picture of how phosphate release can trigger kinesin detachment under forward strain
The seesaw model accounts for load-dependent release of the microtubule-attached kinesin•ADP motor domain in the following way (Fig. 5). In the absence of ATP, the switch pocket collapses and prevents the seesaw from tilting leftward without introducing severe steric overlap between the switch loops and I254 from the switch II helix extension (Fig. 5a). Forward strain on the neck linker, however, favors its docked conformation, which in turn requires leftward tilting of the seesaw. The only way to accommodate the resulting steric overlap between I254 and the switch loops is for the helix extension to revert to its loop structure (seen in crystal structures), which would abolish the interaction seen between residue N255 in the extension and the microtubule surface (Sindelar and Downing 2007), thus destabilizing the kinesin–microtubule interaction. Thus, under forward strain the seesaw model suggests a pathway for microtubule detachment of kinesin•ADP that involves melting of the switch II helix extension, thus accounting for a critical regulatory step in kinesin’s ADP-gated behavior (Fig. 5b).

Cartoon schematic indicating how forward strain on the neck linker could interact with the seesaw to promote detachment in the absence of ATP. The forward strain would favor docking of the neck linker into the docking pocket, thus promoting leftward tilting of the seesaw. However, if ATP is not bound, collapse of the switch pocket would lead to steric interference with I254 in the switch II helix extension in the leftward-tilted case. This action therefore promotes melting of the extension and accompanying weakening of the kinesin–microtubule interaction
The functional relevance of this proposed detachment pathway is best understood in the context of a functioning kinesin dimer (see below for a schematic of the dimer cycle). Kinetic measurements have indicated that, for kinesin dimers, phosphate release in the trailing head is timed within measurement error of microtubule detachment by this head (Rosenfeld et al. 2003). Experiments have suggested that phosphate release may precede detachment by the trailing head (Klumpp et al. 2004), but definitive proof for this order of events is lacking, thus raising the question of whether kinesin detaches as kinesin•ADP•Pi or as kinesin•ADP, and what the structural basis for this decision might be.
Consideration of the seesaw model, in conjunction with the available crystal structure model of kinesin’s microtubule-attached ATP state as modeled by AMPPNP (Parke et al. 2010), leads to the following pathway for kinesin detachment related to phosphate release. Following catalytic cleavage of the β–γ phosphate bond but before phosphate release, the predicted geometry of the cleaved γ-phosphate group appears poised to maintain a large part of the extensive hydrogen bond network that holds the switch loops "closed" (results not shown). This geometry, together with observation of hydrolysis reversal in microtubule-attached kinesin•ADP•Pi (Hackney 2005), indicates that kinesin may maintain its closed switch loop conformation for a substantial interval following ATP cleavage, thus remaining in an "ATP-like" conformation. Kinesin should accordingly remain tightly attached to the microtubule during this period since expected conformational changes would be localized to the nucleotide cleft until phosphate escapes (perhaps due to opening of the switch I "lid" following protonation of the conserved switch I–switch II salt bridge between R203 and E236; Parke et al. 2010). Release of phosphate, however, would permit the switch loops to relax into their ADP conformation, leading to collapse of the switch pocket and subsequent motor detachment under forward strain as outlined above. This line of reasoning (motivated by the seesaw model) would suggest that phosphate release precedes detachment of the motor domain, at least for the trailing head in a processive dimer.
The more general case of ADP-stimulated microtubule detachment remains mysterious
While the seesaw model could thus explain ADP’s destabilizing role in gating by forward strain, the more general phenomenon of instability in the ADP–kinesin–microtubule complex is not so easily accounted for. One explanation for ADP-induced motor detachment involves a "twist-off" scheme (Kikkawa et al. 2001), mentioned above. In the twist-off scheme, ATP-like orientations of the switch II helix on the motor core are hypothesized to protrude from kinesin’s microtubule binding face to form a complementary binding surface for the microtubule (leading to tight attachment), while "ADP-like" orientations of the helix would abolish the complementarity and lead to microtubule detachment. This scheme, however, is inconsistent with high-resolution cryo-EM observations of the no-nucleotide state of kinesin-1, which tightly attaches to the microtubule but was discovered in the EM study to exhibit an "ADP-like" switch II helix orientation (Sindelar and Downing 2007).
In order to reconcile this model with the ADP-like no-nucleotide conformation observed in the kinesin-1 EM study, (Nitta et al. 2008) suggested that the EM study reflected an early, presumably weakly attached intermediate of kinesin during the ADP release process (that is, before ADP is actually released). Contrary to this proposal, however, EM comparisons of ADP-bound and no-nucleotide states of kinesin-1 indicated that density corresponding to bound ADP was specifically absent from the nucleotide cleft under the no-nucleotide experimental conditions, while ADP density was significantly present in the ADP state (Sindelar and Downing 2010).
The cryo-EM maps of (Sindelar and Downing 2010) have since been refined from ∼9-Å to ∼8-Å resolution (C.V.S., unpublished data), and the corresponding increase in map quality further supports the interpretation that the nucleotide cleft is fully vacated under these experimental conditions (Fig. 6). Both no-nucleotide as well as ADP-bound motor states, as visualized in these EM maps, exhibit "ADP-like" orientations of the switch II helix. These observations indicate that "ADP-like" orientations of the switch II helix can occur in both tightly microtubule-attached (no-nucleotide) as well as weakly attached (ADP-bound) states of the motor. The conformation of the switch II helix itself also appears largely the same in these two maps; the density indicates that the helix maintains structure in its N-terminal "extension" coils (Sindelar and Downing 2010). Cryo-EM results with conventional kinesin are thus quite difficult to reconcile with any version of the ADP "twist-off" scheme. An alternative scheme, suggested by Minehardt et al. (2001), might be that microtubule attachment affects the switch I loop through a pathway involving the microtubule-binding loop L8, which connects to the switch I loop via the helix α3. A mutation in L8 is found to stall kinesin in a tightly attached ADP state following hydrolysis, which could lend support to this suggestion (Klumpp et al. 2003).

Illustration of 8-Å resolution ADP and no-nucleotide density maps for microtubule-attached kinesin-1, showing a significant conformational change of the switch I loop (gold) and the P-loop (circled in blue) between these two nucleotide states: in the no-nucleotide state, the switch I loop intrudes into the nucleotide cleft accompanied by loss of ADP density and also loss of P-loop density. ADP is rendered as a ball-and-stick diagram
The related mystery of microtubule-stimulated ADP release
An alternative to studying ADP-stimulated kinesin detachment from the microtubule is to investigate the mechanism of microtubule-stimulated ADP release since these processes are closely linked by thermodynamics. One proposal is that microtubule-triggered "opening" of kinesin’s nucleotide cleft would stimulate ADP release (Hirose et al. 2006; Kikkawa and Hirokawa 2006), although this is in some respects puzzling due to the fact that most known crystal structures of kinesin exhibit an "open" nucleotide cleft but nonetheless have extremely high affinity for ADP (Kull et al. 1996). Indeed, sub-nanometer cryo-EM reconstructions of the kinesin•microtubule complex before and after ADP release suggest a process rather different from "opening" (Sindelar and Downing 2007, 2010). As shown in Fig. 6, the switch I loop in the cryo-EM reconstruction of kinesin-1’s ADP state is remote from the nucleotide density and does not appear to be completely ordered (thus the cleft would be considered "open"). In contrast, the no-nucleotide state exhibits density corresponding to the switch I loop intruding into the nucleotide cleft, accompanied by the apparent loss of density corresponding to the nucleotide-coordinating P-loop. The significance of these observations was highlighted by the ability to resolve well-defined density for the P-loop and switch I in all other sub-nanometer maps characterized for kinesin-1. Coordinated displacement of the P-loop by switch I during ADP release, as implied by these observations, is consistent with an "ejection" role for the switch I loop in which conformational changes induced by microtubule attachment propagate to this loop and lead to dissociation of ADP (Sindelar and Downing 2007).
The role of microtubules remains quite unclear, however, for this putative "ejection" scheme. Microtubule attachment reduces the ADP affinity of free kinesin by approximately three orders of magnitude, from the low nanomolar range to 10–20 μM (Hackney 1988). It is therefore surprising that the position and shape of kinesin’s secondary structure elements appear largely unchanged following ADP release in maps of the microtubule–kinesin-1 complex (Sindelar and Downing 2010). Moreover, the orientation of the core domain relative to the switch II helix remains unchanged within the ability to discriminate in these maps. Indeed, the above described changes in switch I and the P-loop are essentially the only reliably resolved differences between ADP and no-nucleotide states in the kinesin-1 cryo-EM maps, despite the 8-Å resolution now uniformly achieved for all nucleotide states in this construct. Moreover, the overall motor architecture seen in these kinesin-1 maps appears very similar to crystallized conformations such as that seen in the KIF1A•ADP complex, where ADP is bound very tightly (Kikkawa et al. 2001). Thus, comparison of kinesin-1 EM maps to each other or to ADP-bound kinesin crystal structures does not yield an obvious explanation for the observed instability of the kinesin•ADP•microtubule complex.
It may be concluded that, whatever the mechanism by which the kinesin•ADP•microtubule complex is destabilized, the process must involve relatively subtle conformational rearrangements that are not evident in EM maps having resolutions in the 8 Å range. It should be noted that at least two cryo-EM studies of other kinesin family members have concluded that other types of structural changes not seen in the kinesin-1 studies may occur in the no-nucleotide microtubule co-complex (Hirose et al. 2006; Peters et al. 2010). However, these other studies reported resolutions no higher than 10 Å, such that secondary structure elements were not always unambiguously resolved. It is therefore of considerable interest to obtain higher-resolution cryo-EM maps describing microtubule complexes of other kinesin variants in order to further test and elaborate on the various proposed schemes pertaining to the role of ADP.
A working model of dimeric kinesin based on the seesaw
Elements in the preceding discussion may be assembled into a working model describing processive movement by a kinesin dimer, as shown in Fig. 7 which highlights a series of key structural intermediates that may occur in the cycle. In state 1, the trailing head contains bound ATP and/or ADP•Pi within the tightly closed nucleotide-binding cavity of kinesin’s actively hydrolyzing conformation. Neck linker docking in the trailing head biases the partner head to search toward the leading position, where it can attach to the microtubule in an ADP-bound state. In state 2, attachment of the lead head, in concert with rearwards strain on its neck linker, promotes subtle rearrangements in the motor domain (possibly involving L8) that in turn promote the "nucleotide-ejecting" conformation of the switch I loop, causing loss of ADP in this head. Subsequent ATP binding in the lead head, in state 2, is inhibited by rearward strain on the lead head’s neck linker, according to the gating scheme described in Fig. 4. In state 3, catalytic cleavage and phosphate release in the trailing head lead to collapse of the switch pocket, but forward strain on the neck linker of this head prevents compensatory seesaw tilting to relieve the resulting steric overlap between the switch pocket and the I254 from the switch II helix extension. Thus, in state 3, collapse of the switch pocket forces melting of the switch II helix extension, leading to detachment of the trailing head from the microtubule. Subsequently, ATP binding in the leading head resets the system to state 1, but with the kinesin dimer displaced 8 nm towards the microtubule plus end and identity of the leading and trailing heads swapped.

Cartoon schematic indicating how the seesaw mechanism leads to gating between heads of dimeric kinesin. See text for detailed description
The largest differences between the scheme presented here and earlier proposals for kinesin dimer operation relate to integration of the "seesaw" mechanism. The seesaw mechanism serves a key function in the current dimer scheme by generating coupling between the formation of two distinct hydrophobic pockets on either side of kinesin, the switch pocket and the docking pocket. The switch pocket and the docking pocket, in turn, both exhibit obvious and direct energetic coupling to key motor control elements (ATP and the neck linker, respectively). These features appear to form a tightly coupled structural pathway that would be well suited to drive important ADP and ATP gating functions as described here, in addition to the "power stroke" function previously linked to the seesaw (Sindelar and Downing 2010). Incidentally, strain-gating via the neck linkers as described here and elsewhere would be relevant not only to dimer function, but more generally to any system involving multiple kinesins working to cooperatively generate transport, so long as the individual kinesin heads were connected by a sufficiently rigid linkage—for example, two kinesin heads connected via a DNA spacer (Miyazono et al. 2010), or even a glass surface coated with monomeric head domains as found in microtubule gliding assays (Vale et al. 1985).
"Power stroke" or "ratchet"?
A long-standing question of interest has been whether ATP-triggered docking of kinesin’s neck linker is favorable enough to constitute a "power stroke" that would drive the cargo forward via what is essentially an elastic deformation. It is commonly pointed out, for example, that thermal energy is likely to drive large fluctuations of the unbound head in an actively moving kinesin dimer, due in part to the relatively weak observed stability of the docked neck linker conformation even in the presence of ATP (Rice et al. 2003). In addition, docking of the neck linker by itself would not even generate half of the 8 nm displacement that must be contributed by each catalytic domain during a complete turnover (Rice et al. 1999). Thus, a "power stroke" by the neck linker is clearly not adequate by itself to account for the prominent 8-nm steps that are the hallmark of kinesin motility.
Contrasting to such a "power stroke" scheme of kinesin motility are “thermal ratchet” type schemes in which the stepping process would driven solely by strain-gating that prevents the freely diffusing head from taking a step in the wrong direction. Such "ratchet" models have been explored theoretically and are proposed to account for kinesin’s stepping behavior, although it remains unclear whether discrepancies between these models and experimental observations can be fully rationalized (Fox and Choi 2001). It has also been noted that characteristics of “power stroke” and “ratchet” models are not entirely distinct (Block 2007).
While a detailed discussion of this topic is beyond the scope of the current review, it is interesting to note that in the seesaw model presented here, the neck linker could be viewed as having dual roles. The strain-gating function of the neck linker, as has been described above, arises from the way that externally imposed forces would promote or hinder binding of the neck linker into the docking pocket. This strain modulation in turn would affect the affinity of various elements (nucleotide/motor/microtubule) for each other at appropriate points in the cycle. At the same time, the free energy of neck linker docking remains strongly coupled, in the seesaw model, to the free energy of ATP binding. While ATP-driven neck linker docking would clearly be subject to large thermal fluctuations, the docking interactions nevertheless seem likely to kinetically accelerate forward movement by the diffusing head, at the same time slowing backward movement. Such a kinetic effect could be seen as a type of “power stroke”. It remains a challenge for the future to quantify the role of strain-gating and power strokes in the kinesin system and other motors, using increasingly detailed physical models.
Summary
The seesaw model provides a lens through which to interpret kinesin’s control pathway that links the nucleotide binding site to the neck linker, and how this pathway relates to gating of partner heads, in terms of fundamental biophysical forces including steric, hydrophobic, and hydrogen bond interactions. While many aspects of the scheme presented here remain speculative, the scheme nevertheless provides a framework for further scrutiny of kinesin’s various experimentally determined features. In contrast to kinesin, the other cytoskeletal motors dynein and myosin currently lack high-resolution structural descriptions of the motor–filament complexes: the best reported resolution for a 3D cryo-EM map of actomyosin is 14 Å (Holmes et al. 2003), while dynein–microtubule reconstructions are currently limited to >20 Å even in the best case (Mizuno et al. 2007). Considering what is now known about microtubule-induced rearrangements in kinesin, it seems probable that continued study of these other cytoskeletal motors will harbor future surprises.
This review has focused on the mechanism of conventional kinesin I, the founding member of this motor family. Other variants of kinesin exist with strikingly different functional features, including reversed directionality as well as microtubule depolymerization. The seesaw mechanism likely to plays a key role in most or all of these other systems as well, owing to the extremely high degree of sequence and/or structure conservation in the amino acid sequences defining all of the seesaw elements listed in Table 1 (see http://www.cellbio.duke.edu/kinesin/). While the near-universal sequence conservation in the switch sensor loops across all kinesin families has long been noted, the conserved nature of other important seesaw elements (including I254/N255 in the switch II helix extension as well as the fulcrum residues) has received less attention. It is particularly striking that, despite the absence of a conventionally defined “neck linker” in minus-end and depolymerizing kinesin motors, these other motors maintain sequence conservation in the docking pocket as well as the complementary hydrophobic insertion elements seen at positions 322, 325 in kinesin’s neck linker. “ATP-like” conformations of two minus-end motors have been reported, and both exhibit docked conformations of a “neck mimic” element analogous to the neck linker (Vinogradova et al. 2004; Heuston et al. 2010). While an analogous “ATP-like” crystallized conformation from the depolymerizing kinesin family has not yet been seen, it is quite tempting to predict that a docked neck mimic will be found there as well. More broadly, it may be postulated that the entire seesaw assembly described here, including the neck linker/neck mimic, acts as the fundamental “engine” onto which additional modular components may be attached to yield different functional results.
On a final note, it must be said that even in the kinesin variants most similar to conventional kinesin I, important functional differences have been observed. For example, the Eg5 member of the kinesin-5 family was found to exhibit strikingly different nucleotide dependence of neck linker docking compared with kinesin-1, and even appears to dock the neck linker prior to ATP binding (Rosenfeld et al. 2005; Larson et al. 2010). Future, higher-resolution structural studies of these and other kinesin family members are likely to add much to our understanding of these fascinating molecular machines.
References
Asenjo AB, Weinberg Y, Sosa H (2006) Nucleotide binding and hydrolysis induces a disorder-order transition in the kinesin neck-linker region. Nat Struct Mol Biol 13:648–654
Block SM (2007) Kinesin motor mechanics: binding, stepping, tracking, gating, and limping. Biophys J 92:2986–2995
Bodey AJ, Kikkawa M, Moores CA (2009) 9-Angström structure of a microtubule-bound mitotic motor. J Mol Biol 388:218–224
Fisher AJ, Smith CA, Thoden J, Smith R, Sutoh K, Holden HM, Rayment I (1995) Structural studies of myosin:nucleotide complexes: a revised model for the molecular basis of muscle contraction. Biophys J 68:19S–26S, discussion 27S–28S
Fox RF, Choi MH (2001) Rectified Brownian motion and kinesin motion along microtubules. Phys Rev E Stat Nonlin Soft Matter Phys 63:051901
Gilbert SP, Johnson KA (1994) Pre-steady-state kinetics of the microtubule-kinesin ATPase. Biochemistry 33:1951–1960
Guydosh NR, Block SM (2006) Backsteps induced by nucleotide analogs suggest the front head of kinesin is gated by strain. Proc Natl Acad Sci USA 103:8054–8059
Hackney DD (1988) Kinesin ATPase: rate-limiting ADP release. Proc Natl Acad Sci USA 85:6314–6318
Hackney DD (1994) Evidence for alternating head catalysis by kinesin during microtubule-stimulated ATP hydrolysis. Proc Natl Acad Sci USA 91:6865–6869
Hackney DD (2005) The tethered motor domain of a kinesin–microtubule complex catalyzes reversible synthesis of bound ATP. Proc Natl Acad Sci USA 102:18338–18343
Hackney DD, Stock MF, Moore J, Patterson RA (2003) Modulation of kinesin half-site ADP release and kinetic processivity by a spacer between the head groups. Biochemistry 42:12011–12018
Hancock WO, Howard J (1999) Kinesin’s processivity results from mechanical and chemical coordination between the ATP hydrolysis cycles of the two motor domains. Proc Natl Acad Sci USA 96:13147–13152
Heuston E, Bronner CE, Kull FJ, Endow SA (2010) A kinesin motor in a force-producing conformation. BMC Struct Biol 10:19
Hirokawa N, Pfister KK, Yorifuji H, Wagner MC, Brady ST, Bloom GS (1989) Submolecular domains of bovine brain kinesin identified by electron microscopy and monoclonal antibody decoration. Cell 56:867–878
Hirokawa N, Nitta R, Okada Y (2009) The mechanisms of kinesin motor motility: lessons from the monomeric motor KIF1A. Nat Rev Mol Cell Biol 10:877–884
Hirose K, Akimaru E, Akiba T, Endow SA, Amos LA (2006) Large conformational changes in a kinesin motor catalyzed by interaction with microtubules. Mol Cell 23:913–923
Holmes KC, Angert I, Kull FJ, Jahn W, Schröder RR (2003) Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature 425:423–427
Huang TG, Suhan J, Hackney DD (1994) Drosophila kinesin motor domain extending to amino acid position 392 is dimeric when expressed in Escherichia coli. J Biol Chem 269:16502–16507
Khalil AS, Appleyard DC, Labno AK, Georges A, Karplus M, Belcher AM, Hwang W, Lang MJ (2008) Kinesin’s cover-neck bundle folds forward to generate force. Proc Natl Acad Sci USA 105:19247–19252
Kikkawa M (2008) The role of microtubules in processive kinesin movement. Trends Cell Biol 18:128–135
Kikkawa M, Hirokawa N (2006) High-resolution cryo-EM maps show the nucleotide binding pocket of KIF1A in open and closed conformations. EMBO J 25:4187–4194
Kikkawa M, Sablin EP, Okada Y, Yajima H, Fletterick RJ, Hirokawa N (2001) Switch-based mechanism of kinesin motors. Nature 411:439–445
Klumpp LM, Brendza KM, Rosenberg JM, Hoenger A, Gilbert SP (2003) Motor domain mutation traps kinesin as a microtubule rigor complex. Biochemistry 42:2595–2606
Klumpp LM, Hoenger A, Gilbert SP (2004) Kinesin’s second step. Proc Natl Acad Sci USA 101:3444–3449
Kozielski F, Sack S, Marx A, Thormählen M, Schönbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E (1997) The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell 91:985–994
Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD (1996) Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature 380:550–555
Larson AG, Naber N, Cooke R, Pate E, Rice SE (2010) The conserved L5 loop establishes the pre-powerstroke conformation of the Kinesin-5 motor, eg5. Biophys J 98:2619–2627
Ma YZ, Taylor EW (1995) Mechanism of microtubule kinesin ATPase. Biochemistry 34:13242–13251
Ma YZ, Taylor EW (1997) Kinetic mechanism of a monomeric kinesin construct. J Biol Chem 272:717–723
Marx A, Hoenger A, Mandelkow E (2009) Structures of kinesin motor proteins. Cell Motil Cytoskeleton 66:958–966
Minehardt TJ, Cooke R, Pate E, Kollman PA (2001) Molecular dynamics study of the energetic, mechanistic, and structural implications of a closed phosphate tube in ncd. Biophys J 80:1151–1168
Miyazono Y, Hayashi M, Karagiannis P, Harada Y, Tadakuma H (2010) Strain through the neck linker ensures processive runs: a DNA–kinesin hybrid nanomachine study. EMBO J 29:93–106
Mizuno N, Narita A, Kon T, Sutoh K, Kikkawa M (2007) Three-dimensional structure of cytoplasmic dynein bound to microtubules. Proc Natl Acad Sci USA 104:20832–20837
Nitta R, Kikkawa M, Okada Y, Hirokawa N (2004) KIF1A alternately uses two loops to bind microtubules. Science 305:678–683
Nitta R, Okada Y, Hirokawa N (2008) Structural model for strain-dependent microtubule activation of Mg-ADP release from kinesin. Nat Struct Mol Biol 15:1067–1075
Nogales E, Wolf SG, Downing KH (1998) Structure of the alpha beta tubulin dimer by electron crystallography. Nature 391:199–203
Parke CL, Wojcik EJ, Kim S, Worthylake DK (2010) ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J Biol Chem 285:5859–5867
Peters C, Brejc K, Belmont L, Bodey AJ, Lee Y, Yu M, Guo J, Sakowicz R, Hartman J, Moores CA (2010) Insight into the molecular mechanism of the multitasking kinesin-8 motor. EMBO J 29:3437–3447
Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M et al (1999) A structural change in the kinesin motor protein that drives motility. Nature 402:778–784
Rice S, Cui Y, Sindelar C, Naber N, Matuska M, Vale R, Cooke R (2003) Thermodynamic properties of the kinesin neck-region docking to the catalytic core. Biophys J 84:1844–1854
Rosenfeld SS, Rener B, Correia JJ, Mayo MS, Cheung HC (1996) Equilibrium studies of kinesin–nucleotide intermediates. J Biol Chem 271:9473–9482
Rosenfeld SS, Fordyce PM, Jefferson GM, King PH, Block SM (2003) Stepping and stretching. How kinesin uses internal strain to walk processively. J Biol Chem 278:18550–18556
Rosenfeld SS, Xing J, Jefferson GM, King PH (2005) Docking and rolling, a model of how the mitotic motor Eg5 works. J Biol Chem 280:35684–35695
Sablin EP, Fletterick RJ (2004) Coordination between motor domains in processive kinesins. J Biol Chem 279:15707–15710
Sablin EP, Kull FJ, Cooke R, Vale RD, Fletterick RJ (1996) Crystal structure of the motor domain of the kinesin-related motor ncd. Nature 380:555–559
Schliwa M, Woehlke G (2001) Molecular motors. Switching on kinesin. Nature 411:424–425
Schnitzer MJ, Visscher K, Block SM (2000) Force production by single kinesin motors. Nat Cell Biol 2:718–723
Shastry S, Hancock WO (2010) Neck linker length determines the degree of processivity in kinesin-1 and kinesin-2 motors. Curr Biol 20:939–943
Sindelar CV, Downing KH (2007) The beginning of kinesin’s force-generating cycle visualized at 9-A resolution. J Cell Biol 177:377–385
Sindelar CV, Downing KH (2010) An atomic-level mechanism for activation of the kinesin molecular motors. Proc Natl Acad Sci USA 107:4111–4116
Sindelar CV, Budny MJ, Rice S, Naber N, Fletterick R, Cooke R (2002) Two conformations in the human kinesin power stroke defined by X-ray crystallography and EPR spectroscopy. Nat Struct Biol 9:844–848
Uemura S, Ishiwata S (2003) Loading direction regulates the affinity of ADP for kinesin. Nat Struct Biol 10:308–311
Uemura S, Kawaguchi K, Yajima J, Edamatsu M, Toyoshima YY, Ishiwata S (2002) Kinesin–microtubule binding depends on both nucleotide state and loading direction. Proc Natl Acad Sci USA 99:5977–5981
Vale RD, Fletterick RJ (1997) The design plan of kinesin motors. Annu Rev Cell Dev Biol 13:745–777
Vale RD, Milligan RA (2000) The way things move: looking under the hood of molecular motor proteins. Science 288:88–95
Vale RD, Reese TS, Sheetz MP (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42:39–50
Vinogradova MV, Reddy VS, Reddy ASN, Sablin EP, Fletterick RJ (2004) Crystal structure of kinesin regulated by Ca(2+)-calmodulin. J Biol Chem 279:23504–23509
Vinogradova MV, Malanina GG, Reddy VS, Reddy ASN, Fletterick RJ (2008) Structural dynamics of the microtubule binding and regulatory elements in the kinesin-like calmodulin binding protein. J Struct Biol 163:76–83
Vinogradova MV, Malanina GG, Reddy ASN, Fletterick RJ (2009) Structure of the complex of a mitotic kinesin with its calcium binding regulator. Proc Natl Acad Sci USA 106:8175–8179
Yang JT, Laymon RA, Goldstein LS (1989) A three-domain structure of kinesin heavy chain revealed by DNA sequence and microtubule binding analyses. Cell 56:879–889
Yildiz A, Tomishige M, Gennerich A, Vale RD (2008) Intramolecular strain coordinates kinesin stepping behavior along microtubules. Cell 134:1030–1041
Acknowledgments
Research described here was supported by National Institutes of Health grants GM46033 and GM51487 and by the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. I am also grateful to Enrique De La Cruz for critical comments on the manuscript, and I particularly thank the reviewers for numerous insightful comments and suggestions.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sindelar, C.V. A seesaw model for intermolecular gating in the kinesin motor protein. Biophys Rev 3, 85–100 (2011). https://doi.org/10.1007/s12551-011-0049-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-011-0049-4