
Abstract
Background
Hereditary renal tubular disease can cause hypercalciuria, acid-base imbalance, hypokalemia, hypomagnesemia, rickets, kidney stones, etc. If these diseases are not diagnosed or treated in time, they can cause kidney damage and electrolyte disturbances, which can be detrimental to the maturation and development of the child. Glomerular involvement in renal tubular disease patients has only been considered recently.
Methods
We screened 71 papers (including experimental research, clinical research, etc.) about Dent’s disease, Gitelman syndrome, and cystinosis from PubMed, and made reference.
Results
Glomerular disease was initially underestimated among the clinical signs of renal tubular disease or was treated merely as a consequence of the tubular damage. Renal tubular diseases affect glomerular podocytes through certain mechanisms resulting in functional damage, morphological changes, and glomerular lesions.
Conclusions
This article focuses on the progress of changes in glomerular podocyte function in Dent disease, Gitelman syndrome, and cystinosis for the purposes of facilitating clinically accurate diagnosis and scientific treatment and improving prognosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The renal tubule is an important part of the kidney that determines the body’s water-electrolyte and acid-base balance, reabsorbs nutrients, and concentrates or dilutes the urine. Renal tubular disease can cause hypercalciuria, acid-base imbalance, hypokalemia, hypomagnesemia, rickets, kidney stones, etc. If these diseases are not diagnosed and treated in time, they can hinder the maturation and development of the child and cause long-term recurrent kidney stones or electrolyte imbalances, exacerbating kidney impairment. Kidney tubular diseases in children are typically genetic, including Gitelman syndrome, Dent disease, and cystinosis (CTNS).
Podocytes are important intrinsic cells of the glomerulus that receive various pathological stimuli. Architecturally, the glomerulus or renal corpuscle consists of aglomerular tuft and Bowman’s capsule. The basic unit of the glomerular tuft is a single capillary. The glomerular basement membrane (GBM) provides the primary structural scaffold for the glomerular tuft. Endothelial and smooth muscle-like mesangial cells providing capillary support are located inside the GBM, whereas podocytes are attached to the outer part of the GBM [1]. Podocytes constitute the molecular and charge barrier of the glomerular filtration membrane, withstanding the force per unit area in the glomeruli. Podocyte foot processes elaborate into a highly branched interdigitating network with foot processes of neighboring podocytes. The slit diaphragm bridges the filtration slits between opposing podocyte foot processes [2], thereby establishing the final barrier to urinary protein loss [3]. The podocyte adjusts and maintains homeostasis, although excessive stress can lead to maladjustment, accompanied by complex biological changes, including loss of integrity and abnormal metabolism (the results are foot processes effacement reflected by the simplification of the foot processes structure and loss of the normal interdigitating pattern and proteinuria [4]). The latest developments in the interaction between the renal tubule and podocytes and the functional changes of podocytes in renal tubular diseases are summarized below.
Podocyte changes in Dent disease
Dent disease is a rare X-linked recessive renal disorder that is found almost exclusively in males, manifested as renal tubular disease, hypercalciuria, and renal tubular proteinuria. Dent disease is characterized by low-molecular-weight (LMW) proteinuria, hypercalciuria, kidney stones, variable manifestations of proximal tubular dysfunction, and progressive renal failure, which ultimately leads to chronic kidney disease in adulthood [5, 6]. Dent disease may vary in clinical presentation with proteinuria alone or in combination with nephrocalcinosis or nephrolithiasis, with or without chronic kidney disease [7]. Dent disease can start in early childhood, usually before the age of ten [8, 9]. Asymptomatic cases are occasionally diagnosed in adult age, while 30–80% of patients aged between 30 and 50 years old will progress to end-stage renal disease [10,11,12]. In approximately 65% of patients, mutations of chloride voltage-gated channel 5 gene (CLCN5) are responsible for Dent disease type 1 [13, 14], while in 10–15% of patients, mutations in the oculocerebrorenal syndrome of Lowe gene (OCRL) cause Dent disease type 2 [15]. The remaining 25% of patients have a Dent disease phenotype, but specific genetic mutations have not been described [9, 16].
In recent years, glomerular involvement in Dent disease has been considered. Since the discovery of CLCN5 and OCRL expression in the glomerular compartment, a new theory has emerged according to which these two proteins’ loss of function lead to primary glomerular cell damage [17, 18]. Glomerular damage accounted for the nephrotic-range proteinuria observed in more than 30% of patients with Dent disease [19]. CLCN5 encodes for electrogenic chloride channel Cl−/H+ antiporter ClC-5 that is primarily expressed in proximal tubular cells, but it is also expressed in epithelial cells of ascending limb of loop of Henle and alpha intercalated cells of collecting duct [20]. Proximal tubular cells also reportedly express CLCN5 in the brush border plasma membrane, where it is needed for LMW protein reabsorption [20]. There is increasing evidence of glomerular protein handling by podocytes [21,22,23]. CLCN5 encoded protein ClC-5, plays a role in the uptake of LMW proteins through receptor-mediated proximal tubule endocytosis. Human podocytes were also demonstrated to be capable of internalizing albumin predominantly through a cubilin-amnionless mediated mechanism. Furthermore, the excess albumin environment induced an increase in CLCN5 expression in these cells [21]. Overexpression of CLCN5 found in biopsies of patients with proteinuria suggests that this condition may play a role in its expression, and podocytes may play a key role in albumin processing therein [17]. Similar to proximal tubular cells, the endocytosis mechanism plays a role in podocytes and in maintaining the glomerular filtration barrier [24]. The importance of endocytosis in podocyte homeostasis is confirmed [21, 25, 26].
Gianesello et al. demonstrated that human podocytes were able to internalize albumin under normal condition, suggesting that these cells are committed to protein uptake [21]. At the proximal renal tubular level, ClC-5 (encoded by CLCN5) and Megalin (encoded by LRP2) are part of a molecular complex involved in the endocytosis and re-uptake of LMW proteins and albumin. Piwon et al. showed that disruption of the mouse CLCN5 gene causes proteinuria by strongly reducing apical proximal tubular endocytosis. Both receptor-mediated and fluid phase endocytosis are affected [27]. In addition to renal tubular dysfunction, CLCN5 mutations may also cause podocyte dysfunction, leading to histological manifestations of focal segmental glomerulosclerosis [28,29,30]. Compared with the control group, CLCN5 knock down human podocytes have a reduced proliferation rate, increased cell migration rate as assessed by the scratch test, and a defect in the endocytosis of transferrin [28]. It is reported that the increased cell migration rate is abnormal and is a sign of podocyte damage [31,32,33,34]. Glomerulosclerosis was a common finding in kidney biopsies from patients with Dent disease type 1 [35]. Bignon et al. [6] provided some evidence that focal segmental glomerulosclerosis or focal global glomerulosclerosis observed in Dent disease might be the result of primary podocyte injury independent of tubular injury.
Mutations in the OCRL gene cause both Dent disease type 2 and Lowe syndrome [36], suggesting a genotype-phenotype correlation [37]. The condition of patients with Dent disease type 2 is mild, and renal impairment is milder than in patients with Lowe syndrome [12]. Renal calculi in patients with Dent disease type 2 are considerably less than in patients with Dent disease type 1 [38]. The OCRL gene is expressed in all human cells except cells of hematopoietic origin, it is widely expressed in the kidney, including the glomerulus and most tubular segments [39, 40]. The OCRL gene was very recently reported to be more widely expressed in human glomeruli than CLCN5—the former in podocytes, mesangial cells and endothelial cells, the latter in podocytes and parietal epithelial cell (PECs) [18]. Whereas the OCRL is expressed in podocytes, mesangial cells and endothelial cells, CLCN5 is expressed in podocytes and PECs [18]. OCRL is primarily expressed in the trans-Golgi network, early endosomes, and lysosomes (in HeLa, normal rat kidney NRK, and COS-7 cells, fibroblasts, zebrafish embryos) [41,42,43]. It was suggested that OCRL is involved in regulating endocytic trafficking, actin cytoskeleton dynamics, and slit diaphragm maintenance. Mutations of the OCRL gene could disrupt these mechanisms, thereby inducing glomerular damage [18]. Given that mutations in CLCN5 and OCRL produce very similar kidney defects in human patients [44], one might expect that CLC-5 and OCRL cooperate in a similar or shared cellular process. OCRL is located at various positions in the sequence encoding the endocytosis pathway and is thought to play a role by coupling the endocytotic membrane with dephosphorylation of inositol 5-phosphatase [41, 45]. Preston et al. [18] showed that OCRL is expressed in podocytes in vivo and is able to interact with CD2AP, an important protein whose function is to maintain the slit dia-phragm between adjacent podocyte foot processes. Their results raise the possibility that defective OCRL can directly cause a glomerulopathy. The functions of OCRL and proton-chloride ion-exchange transporter 5 are concentrated on a shared mechanism, and their damage has a significant effect on proximal tubule endocytosis [46]. Podocyte foot process effacement was discovered in patients with Dent disease, which suggested that glomerulosclerosis in these patients might be the result of a combination of primary podocyte injury and a reaction secondary to tubulointerstitial lesions (tubulointerstitial injury was commonly present and associated with the proportion of globally sclerotic glomeruli) [35].
Podocyte changes in Gitelman syndrome
Gitelman syndrome, also known as familial hypokalemia-hypomagnesemia, is an autosomal recessive salt loss renal tubular disease characterized by hypomagnesemia, hypocalciuria, and hyperaldosteronism, the cause of hypokalemia and metabolic alkalosis [47]. Gitelman syndrome is typically caused by mutations in the SLC12A3 gene encoding the thiazide-sensitive NaCl cotransporter or the CLCNKB gene encoding the chloride channel ClC-Kb [48]. Most cases are caused by mutations in the SLC12A3 gene, and more than 140 different SLC12A3 mutations have been identified in patients with Gitelman syndrome. In most cases, symptoms do not appear before the age of six, and the disease is usually diagnosed during adolescence or adulthood.
According to reports, renal biopsy in patients with Gitelman syndrome showed an enlarged proximal tubule and thickened mesangium under a light microscope [49]. Similar observations were made in renal biopsies from SLC12A3 knockout mice, showing thickened GBM in a wide range of segments, with the thickness of these irregular GBMs accompanied by podocyte foot processes. The disappearance and occasional formation of pseudocysts in podocytes confirm the potential association of glomerular defects with Gitelman syndrome [49]. The lesions observed in this case and the mouse model suggest that there may be a link between the loss of NaCl cotransporter function and podocyte dysfunction. One hypothesis is that the chronic activation of the renin-angiotensin-aldosterone pathway leads to increased systemic and local levels of angiotensin II (Ang II) and renin, which may in turn cause podocyte damage.
Mechanical stress of podocytes stimulates local Ang II synthesis by non-angiotension converting enzyme pathways that presumably involve chymase [50]. Ang II induces transforming growth factor-β1 (TGF-β1) in the various renal cells [51, 52]. TGF-β is well-known among growth factors for its potent and widespread actions. Almost every cell in the body has been shown to make some form of TGF-β, and almost every cell expresses receptors for TGF-β. TGF-β plays an important role in podocyte isolation [53,54,55]. One paper showed that TGF-β1 reduced nephrin expression in conditionally immortalized human podocytes [56]. Ang II has a direct effect on the integrity of the ultrafiltration barrier and reduces the cell surface and extracellular matrix of podocytes. Ang II reduces the synthesis of negatively charged proteoglycans [57, 58]. Complete nephrin (nephropathy protein)-nephrin signaling is important for podocyte survival; thus, Ang II-mediated nephrin inhibition leads to podocyte apoptosis [58]. Ang II stimulates albumin endocytosis in proximal tubule cells via Ang II type 2 receptor–mediated protein kinase B activation. However, an increase in tubular albumin reabsorption activates the tubular renin–angiotensin–aldosterone system, leading to a vicious circle [59]. In another report of Gitelman syndrome, renal biopsy showed severe non-apoptotic podocyte detachment in the glomeruli and thickening of the intimal fibers of the small arteries [60].
Podocyte changes in cystinosis
CTNS is an autosomally recessive lysosomal storage disease caused by a deficiency of cystinosin (a lysosomal membrane cystine transporter). This defect causes cystine to crystallize in the lysosomes of many tissues, particularly in the kidneys and cornea. Renal manifestations of CTNS include Fanconi syndrome, mild proteinuria, and progressive renal failure. CTNS is caused by a pathogenic mutation in the human CTNS gene encoding cystinosin [61]. The kidney is initially affected by widespread proximal tubular dysfunction, which rapidly affects the glomeruli and progresses into end-stage renal failure and multiple organ dysfunction. The accumulation of cystine may involve abnormal nuclear division, with a lack of cytokinesis in the injured podocytes resulting in the appearance of multinucleation [62]. This provides further evidence for the diagnosis of cystine disease. Sharma et al. found that a patient had extensive giant cell transformation of glomerular podocytes, accompanied by focal atrophy and expansion of renal tubules [63].
Studies have shown that the CTNS gene is essential for the function of zebrafish anterior renal podocytes and proximal renal tubules. Anterior kidneys of CTNS-knockout zebrafish show enlarged lysosomes in proximal renal tubular cells, part of podocytes disappear, and slit membrane stenosis [64]. Podocytes can move in the glomerulus and break through the Bowman's capsule and be, rapidly replaced by a stellate cell. This change in podocyte movement is considered to be the basis of the disappearance of the foot process and proteinuria [65]. The number of podocytes in the urine of patients with cystine disease is considerably greater than in the urine of normal subjects. The impaired ability of cells to adhere to substrates may be the cause of mass loss of glomerular podocytes, resulting in damage in this region. Increased movement of podocytes lacking cystinosin is associated with increased phosphorylation of protein kinases [66]. Protein kinase 1 is predominantly expressed in the proximal tubules of the kidney, while protein kinase 2 is mainly expressed in the glomeruli, which protects the glomeruli and prevents podocyte dedifferentiation and death [67].
Podocyte injury mechanism in renal tubular diseases
Mechanical injury
Podocyte detachment in Gitelman syndrome may be related to obstruction of the nephron and decreased protein expression. Mechanical stretching and TGF-β stress may induce podocyte apoptosis or separation from the GBM [68]. In cystine disease, cystine crystals are deposited in podocyte lysosomes, resulting in the appearance of multinucleation, changes in the cytoskeleton, and enhanced podocyte motility, etc.
Gene defects cause podocyte damage
SLC12A3 gene mutation can cause podocyte non-apoptotic detachment. CLCN5-mutated podocytes undergo endocytosis and reduced proliferative ability, accompanied by enhanced migration ability [28]. Furthermore, these mutations cause changes in the podocyte cytoskeleton, damage podocyte adhesion sites, and enhance the mobility of individual podocytes, causing detachment and death.
Pro-inflammatory factors and cytokines
Ang II can induce proteinuria through hemodynamic and non-hemodynamic mechanisms involving vascular endothelial growth factor and TGF-β1 [69]. Contrary to physiological conditions, the pathophysiology of podocyte injury is generally related to increased expression of TGF-β, which plays an important role in podocyte isolation [70]. In response to TGF-β and other TGF-dependent stimuli, mature podocytes undergo dedifferentiation, resulting in the disappearance of foot processes.
Epigenetics
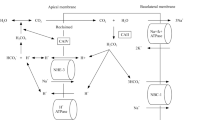
A decrease in sirtuin 1 (Sirt1) expression in the renal tubules causes a decrease in Sirt1 levels in the glomeruli, suggesting that molecular changes in the renal tubules induce phenotypic changes in the glomeruli and podocytes, with the disappearance of more podocytes. Additionally, this reveals the role of nicotinamide mononucleotide as a mediator of interaction between renal tubular cells and podocytes, as nicotinamide mononucleotide derived from renal tubular cells is absorbed by podocytes [71] (Fig. 1).

Podocyte injury mechanism in renal tubular diseases. FSGS focal segmental glomerulosclerosis, FGGS focal global glomerulosclerosis, LMW low-molecular-weight, CLCN5 chloride voltage-gated channel 5 gene, CD2AP CD2-associated protein, GBM glomerular basement membrane, RAAS renin–angiotensin–aldosterone system, CTNS cystinosis, CKD chronic kidney disease, TGF-ß transforming growth factor-β, PF podocyte foot, SD slit diaphragm, Ang II angiotensin II
Conclusions
To study podocyte damage and its mechanism in inherited renal tubular disease, from a clinical perspective, it is useful to explain the phenotype of glomerular lesions in patients with renal tubular disease, to guide clinical treatment and prognosis. Research into the glomerulus-tubule dialogue and mutual feedback pathways should be further investigated. Moreover, further research on the specific roles and molecular mechanisms involved in functioning of glomerulus, renal tubules, etc., should be expanded.
This article reviewed the glomerular podocyte damage caused by the three inherited renal tubular diseases, Gitelman syndrome, Dent disease, and cystine disease. However, other renal tubular diseases may affect glomerular podocyte morphology, abnormal function, and quantity, and affect the phenotype and prognosis of patients. From this perspective, actively exploring podocyte lesions of the renal tubular disease may have important clinical significance.
References
Greka A, Mundel P. Cell biology and pathology of podocytes. Annu Rev Physiol. 2012;74:299–323.
Mundel P, Kriz W. Structure and function of podocytes: an update. Anat Embryol (Berl). 1995;192:385–97.
Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet. 2000;24:333–5.
Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest. 2001;108:1583–7.
Wrong OM, Norden AG, Feest TG. Dent’s disease; a familial proximal renal tubular syndrome with low-molecular-weight proteinuria, hypercalciuria, nephrocalcinosis, metabolic bone disease, progressive renal failure and a marked male predominance. QJM. 1994;87:473–93.
Bignon Y, Alekov A, Frachon N, Lahuna O, Doh-Egueli CJB, Deschênes G, et al. A novel CLCN5 pathogenic mutation supports Dent disease with normal endosomal acidification. Hum Mutat. 2018;39:1139–49.
Thakker RV. Pathogenesis of Dent’s disease and related syndromes of X-linked nephrolithiasis. Kidney Int. 2000;57:787–93.
Devuyst O, Thakker RV. Dent’s disease. Orphanet J Rare Dis. 2010;5:28.
Blanchard A, Curis E, Guyon-Roger T, Kahila D, Treard C, Baudouin V, et al. Observations of a large Dent disease cohort. Kidney Int. 2016;90:430–9.
Claverie-Martin F, Ramos-Trujillo E, Garcia-Nieto V. Dent’s disease: clinical features and molecular basis. Pediatr Nephrol. 2011;26:693–4.
Sekine T, Komoda F, Miura K, Takita J, Shimadzu M, Matsuyama T, et al. Japanese Dent disease has a wider clinical spectrum than Dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant. 2014;29:376–84.
Zaniew M, Bokenkamp A, Kolbuc M, La Scola C, Baronio F, Niemirska A, et al. Long-term renal outcome in children with OCRL mutations: retrospective analysis of a large international cohort. Nephrol Dial Transplant. 2018;33:85–94.
Waldegger S, Jentsch TJ. From tonus to tonicity: physiology of CLC chloride channels. J Am Soc Nephrol. 2000;11:1331–9.
Thakker RV. Chloride channels cough up. Nat Genet. 1997;17:125–7.
Hoopes RR Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, et al. Dent disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–7.
Bockenhauer D, Bokenkamp A, Nuutinen M, Unwin R, Van’t Hoff W, Sirimanna T, et al. Novel OCRL mutations in patients with Dent-2 disease. J Pediatr Genet. 2012;1:15–23.
Ceol M, Tiralongo E, Baelde HJ, Vianello D, Betto G, Marangelli A, et al. Involvement of the tubular ClC-type exchanger ClC-5 in glomeruli of human proteinuric nephropathies. PLoS One. 2012;7:e45605.
Preston R, Naylor RW, Stewart G, Bierzynska A, Saleem MA, Lowe M, et al. A role for OCRL in glomerular function and disease. Pediatr Nephrol. 2020;35:641–8.
Gianesello L, Del Prete D, Anglani F, Calò LA. Genetics and phenotypic heterogeneity of Dent disease: the dark side of the moon. Hum Genet. 2020. https://doi.org/10.1007/s00439-020-02219-2.
Devuyst O, Christie PT, Courtoy PJ, Beauwens R, Thakker RV. Intra-renal and subcellular distribution of the human chloride channel, CLC-5, reveals a pathophysiological basis for Dent’s disease. Hum Mol Genet. 1999;8:247–57.
Gianesello L, Priante G, Ceol M, Radu CM, Saleem MA, Simioni P, et al. Albumin uptake in human podocytes: a possible role for the cubilin-amnionless (CUBAM) complex. Sci Rep. 2017;7:13705.
Kinugasa S, Tojo A, Sakai T, Tsumura H, Takahashi M, Hirata Y, et al. Selective albuminuria via podocyte albumin transport in puromycin nephrotic rats is attenuated by an inhibitor of NADPH oxidase. Kidney Int. 2011;80:1328–38.
Dobrinskikh E, Okamura K, Kopp JB, Doctor RB, Blaine J. Human podocytes perform polarized, caveolae-dependent albumin endocytosis. Am J Physiol Renal Physiol. 2014;306:F941–51.
Inoue K, Ishibe S. Podocyte endocytosis in the regulation of the glomerular filtration barrier. Am J Physiol Renal Physiol. 2015;309:F398-405.
Fyfe JC, Madsen M, Højrup P, Christensen EI, Tanner SM, de la Chapelle A, et al. The functional cobalamin (vitamin B12)-intrinsic factor receptor is a novel complex of cubilin and amnionless. Blood. 2004;103:1573–9.
Coudroy G, Gburek J, Kozyraki R, Madsen M, Trugnan G, Moes SK, et al. Contribution of cubilin and amnionless to processing and membrane targeting of cubilin-amnionless complex. J Am Soc Nephrol. 2005;16:2330–7.
Piwon N, Gunther W, Schwake M, Bösl MR, Jentsch TJ. ClC-5 Cl–channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–73.
Solanki AK, Arif E, Morinelli T, Wilson RC, Hardiman G, Deng P, et al. A novel CLCN5 mutation associated with focal segmental glomerulosclerosis and podocyte injury. Kidney Int Rep. 2018;3:1443–53.
Fervenza FC. A patient with nephrotic-range proteinuria and focal global glomerulosclerosis. Clin J Am Soc Nephrol. 2013;8:1979–87.
Beara-Lasic L, Cogal A, Mara K, Enders F, Mehta RA, Haskic Z, et al. Prevalence of low molecular weight proteinuria and Dent disease 1 CLCN5 mutations in proteinuric cohorts. Pediatr Nephrol. 2020;35:633–40.
Ding F, Li X, Li B, Guo J, Zhang Y, Ding J. Calpain-mediated cleavage of calcineurin in puromycin aminonucleoside-induced podocyte injury. PLoS One. 2016;11:e0155504.
Kumagai T, Baldwin C, Aoudjit L, Nezvitsky L, Robins R, Jiang R, et al. Protein tyrosine phosphatase 1B inhibition protects against podocyte injury and proteinuria. Am J Pathol. 2014;184:2211–24.
Welsh GI, Saleem MA. The podocyte cytoskeleton–key to a functioning glomerulus in health and disease. Nat Rev Nephrol. 2011;8:14–21.
Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med. 2008;14:55–63.
Wang X, Anglani F, Beara-Lasic L, Mehta AJ, Vaughan LE, Hernandez LH, et al. Glomerular pathology in Dent disease and its association with kidney function. Clin J Am Soc Nephrol. 2016;11:2168–76.
Lewis RA, Nussbaum RL, Brewer ED. Lowe syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. 24 July 2001 [Updated 18 Apr 2019].
Hichri H, Rendu J, Monnier N, Coutton C, Dorseuil O, Poussou RV, et al. From Lowe syndrome to Dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat. 2011;32:379–88.
Bökenkamp A, Böckenhauer D, Cheong HI, Hoppe B, Tasic V, Unwin R, et al. Dent-2 disease: a mild variant of Lowe syndrome. J Pediatr. 2009;155:94–9.
Olivos-Glander IM, Jänne PA, Nussbaum RL. The oculocerebrorenal syndrome gene product is a 105-kD protein localized to the Golgi complex. Am J Hum Genet. 1995;57:817–23.
Erb BC, Velazquez H, Gisser M, Shugrue CA, Reilly RF. cDNA cloning and localization of OCRL-1 in rabbit kidney. Am J Physiol. 1997;273:F790–5.
Oltrabella F, Pietka G, Ramirez IBR, Mironov A, Starborg T, Drummond IA, et al. The Lowe syndrome protein OCRL1 is required for endocytosis in the zebrafish pronephric tubule. PLoS Genet. 2015;11:e1005058.
Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell. 2005;16:3467–79.
Ungewickell A, Ward ME, Ungewickell E, Majerus PW. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A. 2004;101:13501–6.
Hoopes RR Jr, Raja KM, Koich A, Hueber P, Reid R, Knohl SJ, et al. Evidence for genetic heterogeneity in Dent’s disease. Kidney Int. 2004;65:1615–20.
Hyvola N, Diao A, Mckenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. 2006;25:3750–61.
Inoue K, Balkin DM, Liu L, Nandez R, Wu Y, Tian X, et al. Kidney tubular ablation of Ocrl/Inpp5b phenocopies Lowe syndrome tubulopathy. J Am Soc Nephrol. 2017;28:1399–407.
Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–35.
Knoers NVAM, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis. 2008;3:22.
Demoulin N, Aydin S, Cosyns JP, Dahan K, Cornet G, Auberger I, et al. Gitelman syndrome and glomerular proteinuria: a link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol Dial Transplant. 2014;29(Suppl 4):iv117–20.
Durvasula RV, Petermann AT, Hiromura K, Blonski M, Pippin J, Mundel P, et al. Activation of a local tissue angiotensin system in podocytes by mechanical strain. Kidney Int. 2004;65:30–9.
Wolf G. Link between angiotensin II and TGF-beta in the kidney. Miner Electrolyte Metab. 1998;24:174–80.
Sharma K, Cook A, Smith M, Valancius C, Inscho EW. TGF-beta impairs renal autoregulation via generation of ROS. Am J Physiol Renal Physiol. 2005;288:F1069–77.
Yamaguchi Y, Iwano M, Suzuki D, Nakatani K, Kimura K, Harada K, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54:653–64.
Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–14.
Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–34.
Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-beta: a model for diabetic podocytopathy. Diabetes. 2011;60:1779–88.
Wolf G, Butzmann U, Wenzel UO. The renin-angiotensin system and progression of renal disease: from hemodynamics to cell biology. Nephron Physiol. 2003;93:P3-13.
Brinkkoetter PT, Holtgrefe S, Van Der Woude FJ, Yard BA. Angiotensin II type 1-receptor mediated changes in heparansulfate proteoglycans in human SV40 transformed podocytes. J Am Soc Nephrol. 2004;15:33–40.
Caruso-Neves C, Kwon SH, Guggino WB. Albumin endocytosis in proximal tubule cells is modulated by angiotensin II through an AT2 receptor-mediated protein kinase B activation. Proc Natl Acad Sci U S A. 2005;102:17513–8.
Takahashi N, Kimura H, Mizuno S, Hara M, Hirayama Y, Kurosawa H, et al. Severe intraglomerular detachment of podocytes in a Gitelman syndrome patient. Clin Exp Nephrol. 2012;16:495–500.
Town M, Jean G, Cherqui S, Attard M, Forestier L, Whitmore SA, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–24.
Chandra M, Stokes MB, Kaskel F. Multinucleated podocytes: a diagnostic clue to cystinosis. Kidney Int. 2010;78:1052.
Sharma A, Gupta R, Sethi SK, Bagga A, Dinda AK. Giant cell transformation of podocytes: a unique histological feature associated with cystinosis. Indian J Nephrol. 2011;21:123–5.
Elmonem MA, Khalil R, Khodaparast L, Khodaparast L, Arcolino FO, Morgan J, et al. Cystinosis (ctns) zebrafish mutant shows pronephric glomerular and tubular dysfunction. Sci Rep. 2017;7:42583.
Peti-Peterdi J, Sipos A. A high-powered view of the filtration barrier. J Am Soc Nephrol. 2010;21:1835–41.
Ivanova EA, Arcolino FO, Elmonem MA, Rastaldi MP, Giardino L, Cornelissen EM, et al. Cystinosin deficiency causes podocyte damage and loss associated with increased cell motility. Kidney Int. 2016;89:1037–48.
Canaud G, Bienaime F, Viau A, Treins C, Baron W, Nguyen C, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19:1288–96.
Schiffer M, Bitzer M, Roberts IS, Kopp JB, ten Dijke P, Mundel P, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest. 2001;108:807–16.
Rüster C, Wolf G. Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol. 2006;17:2985–91.
Wu DT, Bitzer M, Ju W, Mundel P, Böttinger EP. TGF-beta concentration specifies differential signaling profiles of growth arrest/differentiation and apoptosis in podocytes. J Am Soc Nephrol. 2005;16:3211–21.
Hasegawa K, Wakino S, Simic P, Sakamaki Y, Minakuchi H, Fujimura K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19:1496–504.
Funding
No financial or non-financial benefits have been received or will be received from any party related directly or indirectly to the subject of this article.
Author information
Authors and Affiliations
Contributions
HLM wrote the initial draft. MJH reviewed and revised the manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Ethical approval
Not needed.
Conflict of interest
All authors disclosed no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, LM., Mao, JH. Glomerular podocyte dysfunction in inherited renal tubular disease. World J Pediatr 17, 227–233 (2021). https://doi.org/10.1007/s12519-021-00417-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12519-021-00417-0