
Abstract
Background
The safety and tolerability of regadenoson, a pharmacologic stress agent that is excreted primarily by the kidneys, were examined in subjects with chronic kidney disease (CKD).
Methods
This multicenter, double-blind, randomized, placebo-controlled study involved men and women, ≥18 years of age, with stage 3 or 4 [estimated glomerular filtration rate (eGFR) 30-59 mL/minute/1.73 m2 and 15-29 mL/minute/1.73 m2, respectively] CKD and known or suspected coronary artery disease. Subjects were randomized 2:1 to receive one 10-second intravenous injection of regadenoson 0.4 mg or placebo. The primary outcome measure was the frequency of serious adverse events over 24-h post-dose.
Results
The study included 432 subjects with stage 3 (regadenoson n = 287; placebo n = 145) and 72 with stage 4 (regadenoson n = 47; placebo n = 25) CKD. No serious adverse events or deaths were reported over 24-h post-dose. The overall adverse event incidence was higher with regadenoson than placebo (62.6% vs 21.2%; P < .0001). Of the most common adverse events (≥5%) reported by subjects receiving regadenoson, headache (24.9% vs 7.1%), dyspnea (19.2% vs 0.6%), chest discomfort (14.7% vs 0.6%), nausea (14.7% vs 1.2%), flushing (12.0% vs 1.8%), and dizziness (9.6% vs 0.6%) occurred significantly more often (P < .0001) with regadenoson than placebo. There were no trends for clinically meaningful changes in eGFR from baseline to 24-h post-dose in subjects with stage 3 or 4 CKD.
Conclusions
Regadenoson was not associated with any serious or unexpected adverse events in subjects with stage 3 or 4 CKD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Single-photon emission computed tomography (SPECT) stress imaging is widely used in the clinical assessment of patients with known or suspected coronary artery disease (CAD). Although exercise stress is the preferred modality, many patients are unable to exercise sufficiently because of age and physical limitations. Pharmacologic stress agents, such as adenosine and dipyridamole, are commonly used as effective substitutes for exercise stress in the diagnosis and prognosis of CAD in these individuals. Adenosine and dipyridamole are non-selective adenosine agonist stress agents which induce coronary hyperemia directly or indirectly via adenosine 2A (A2A) cell membrane G-protein-coupled receptors found in abundance on arteriolar smooth muscle cells. However, they also act on other adenosine receptor subtypes, such as A1, A2B, and A3 adenosine receptors, causing frequent clinically important side effects (e.g., atrioventricular block, peripheral vasodilation, and bronchoconstriction) as well as other less serious side effects.1-3 Logically, one may assume that a pharmacologic stress agent that selectively targets the A2A receptor would provide adequate coronary vasodilatation for SPECT and lessen or avoid other undesirable side effects.
Regadenoson is a selective A2A receptor agonist that was approved for use as a pharmacologic stressor for SPECT by the US Food and Drug Administration in April 2008.4-11 The affinity of regadenoson for adenosine A2A receptors is 9-fold greater than for adenosine A1 receptors, and it has a very low affinity for A2B and A3 receptors.5 Regadenoson has a triphasic half-life. The half-life of the initial phase is 2-4 minutes and coincides with the period of maximal coronary hyperemia; the second phase is 15-30 minutes and is not associated with a pharmacodynamic effect, as evidenced by resolution of side effects; and the terminal half-life is approximately 2 h. The extended terminal half-life is not considered clinically significant, as accumulation is not a risk following administration of a single intravenous (IV) bolus dose.12
A significant proportion of patients referred for pharmacologic stress SPECT are elderly.13 Many of these individuals are likely to have chronic kidney disease (CKD). This results from the gradual decline in GFR with age of approximately 0.4 mL/minute/year and the prevalence of comorbidities, such as diabetes and hypertension, that are associated with the development of CKD.14-16 Thus, it is important to assess the safety and tolerability of regadenoson in this population. Although regadenoson was safe and well tolerated in 2 pivotal, Phase 3 clinical trials involving over 1,200 patients undergoing SPECT myocardial perfusion imaging (MPI),17,18 individuals with renal impairment were excluded from these trials.
The objective of this study was to examine the safety and tolerability of a single IV dose (0.4 mg/5 mL) of regadenoson in subjects with stage 3 or 4 CKD. A placebo group was included to characterize adverse event rates in an untreated population.
Methods
Study Participants
Participants in this study were men and women outpatients, ≥18 years of age, with stage 3 or 4 CKD at screening. Estimated glomerular filtration rate (eGFR) was calculated from the serum creatinine concentration using the Modification of Diet in Renal Disease (MDRD) equation19,20 and was used to stage each subject’s level of CKD according to the National Kidney Foundation classification.21 Stage 3 CKD was defined as eGFR 30-59 mL/minute/1.73 m2 and stage 4 as eGFR 15-29 mL/minute/1.73 m2. Subjects also had diagnosed CAD or ≥2 of the following CAD risk factors: type 2 diabetes, hypertension, hypercholesterolemia, current or history of cigarette smoking (minimum 10 pack-years exposure), or obesity (body mass index >30 kg/m2). Subjects had no specific indication for cardiac imaging.
Subjects were excluded if they had other clinically significant illnesses (e.g., neurologic, gastrointestinal, renal, hepatic, cardiovascular, metabolic, endocrine, hematologic, or psychiatric disorders), medical conditions, or laboratory abnormalities within 2 weeks prior to screening that, in the investigator’s judgment, might interfere with study assessments; second- or third-degree heart block or sinus node dysfunction in the absence of a functioning pacemaker; symptomatic hypotension; allergy or intolerance to aminophylline or regadenoson and its excipients; or a hemoglobin level ≤9 g/dL. Also excluded were women who were pregnant, lactating, or of childbearing potential and not using contraception, subjects who were participating in another clinical trial or had received an investigational drug in the 30 days prior to screening, and those who had undergone surgery in the previous 3 months. Subjects with asthma or chronic obstructive pulmonary disease were not excluded from the trial. However, no formal criteria were included in the protocol to specifically capture whether subjects had these coexisting conditions.
Study Design and Treatments
This Phase 4, double-blind, randomized, placebo-controlled, parallel-group clinical trial was conducted at 32 sites in the United States (ClinicalTrials.gov number, NCT00863707). The first participant was enrolled in April 2009 and the study was completed (last subject last visit) in December 2009. The study was conducted in compliance with the principles of the International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use and Good Clinical Practice. The protocol was approved by the Institutional Review Board/Independent Ethics Committee of each study center. Each participant signed a written informed consent form prior to any study-related procedures.
Following a screening visit, eligible subjects returned to the clinic within 3 weeks and were randomized to double-blind treatment with either regadenoson 0.4 mg/5 mL or matching placebo using a 2:1 (regadenoson:placebo) computer-generated randomization schedule prepared by the sponsor prior to study initiation. The study drug was administered as a single IV bolus injection over 10 seconds, followed immediately by a 5 mL 0.9% sodium chloride USP (saline) flush. If necessary, aminophylline was given in doses ranging from 50 to 250 mg by slow IV injection (50-100 mg over 30-60 seconds) to attenuate severe and/or persistent adverse reactions. Subjects were monitored in the clinic for 8-h post-dose; clinically stable subjects were then discharged. The follow-up visit was conducted 24 ± 4 h after study drug administration.
Subjects were required to abstain from eating and drinking for 30 minutes before and after dosing, and from smoking for 3 h before to 8 h after dosing. Foods and beverages containing methylxanthines (i.e., caffeine, theobromine, or theophylline) and medications containing theophylline were prohibited from 12 h before study drug administration to the follow-up visit. Dipyridamole was withheld for at least 2 days prior to dosing.
Safety Assessments
Adverse events (coded using Medical Dictionary for Regulatory Activities [MedDRA] version 11.1 terms) were collected from the time of study drug administration to the follow-up visit and were assessed by the investigator for severity and causal relationship to the study drug. An adverse event was defined as any unfavorable and unintended sign (including an abnormal laboratory finding), symptom or disease temporally associated with the use of a study drug, whether or not it was related to the study drug. All adverse events occurring during the study were followed up until resolved or judged to be no longer clinically significant, or until they became chronic to the extent that they could be fully characterized. The primary outcome measure was the frequency of serious adverse events. In accordance with the ICH of Technical Requirements for Registration of Pharmaceuticals for Human Use and Good Clinical Practice, serious adverse events were defined as any adverse event that (i) results in death, persistent, or significant disability or incapacity; (ii) consists of a congenital anomaly or birth defect; (iii) requires inpatient hospitalization or prolongation of hospitalization; (iv) is life-threatening; or (v) is considered medically important.
Vital signs [systolic blood pressure (SBP), diastolic blood pressure (DBP), heart rate (HR), and respiratory rate] and 12-lead electrocardiograms (ECGs) were assessed at screening, at baseline (prior to study drug administration), at 5, 15, and 30 minutes and 1, 2, 3-7, and 8 h after regadenoson or placebo administration and at the follow-up visit, 24 ± 4 h later. The proportion of subjects meeting each of the following prespecified vital sign criteria was also determined:
-
HR increase of >40 beats per minute (bpm)
-
SBP <90 mm Hg
-
SBP decrease of >35 mm Hg
-
SBP ≥200 mm Hg
-
SBP increase of ≥50 mm Hg
-
SBP ≥180 mm Hg and increase of ≥20 mm Hg from baseline
-
DBP <50 mm Hg
-
DBP decrease of >25 mm Hg
-
DBP ≥115 mm Hg
-
DBP increase of ≥30 mm Hg.
Continuous 12-lead ECG monitoring and pulse oximetry began 2 h before study drug administration and continued until 2-h post-dose. Pulse oximetry was also conducted at 3-7 and 8 h after drug administration. Additional evaluations conducted at screening, baseline, and follow-up included serum chemistry (sodium, potassium, blood urea nitrogen, bicarbonate, calcium, phosphate, chloride, glucose, and creatinine); kidney function, using the MDRD equation to calculate eGFR from serum creatinine;19 hemoglobin level; and physical examination.
Statistical Analyses
A total of 450 subjects enrolled with 300 receiving regadenoson was considered appropriate clinically to assess the safety and tolerability of the drug. This sample size was deemed sufficient to detect an adverse event rate of 54 per 10,000 subjects with approximately 80% probability and an adverse event rate of 76 per 10,000 subjects with approximately 90% probability. Safety analyses were conducted on all randomized subjects who received any amount of study drug.
Protocol Amendments
Protocol amendments of note that were made after trial commencement included the addition of current or history of cigarette smoking (minimum 10 pack-years exposure) as a qualifying CAD risk factor and an update of the race component of the MDRD equation used to calculate eGFR. Both of these amendments were introduced after the enrollment of 17 regadenoson and 11 placebo subjects.
Results
Subject Characteristics
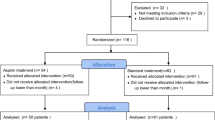
Of 511 subjects randomized, 504 received study drug and were included in the safety analysis (3 subjects randomized to regadenoson and 4 randomized to placebo did not receive study drug; Figure 1). This safety analysis set comprised 432 subjects with stage 3 CKD (regadenoson n = 287; placebo n = 145) and 72 with stage 4 CKD (regadenoson n = 47; placebo n = 25). No significant differences were observed between the regadenoson and placebo groups in demographic or baseline clinical characteristics (Table 1). The 2 treatment groups were also comparable with regard to therapies used during the 21 days prior to study drug administration and those used from the time of study drug administration to the follow-up visit.

Subject disposition
Serious Adverse Events
No serious adverse events, including deaths, were reported with regadenoson or placebo from the time of study drug administration to the follow-up visit at 24 h (primary outcome measure). One subject died during a cardiopulmonary arrest 20 days after receiving placebo; this event was considered unrelated to the study drug by the investigator.
All Adverse Events
The overall incidence of adverse events was significantly higher with regadenoson than placebo: adverse events were reported by 62.6% (209/334) subjects receiving regadenoson and 21.2% (36/170) receiving placebo (P < .0001). None of the adverse events resulted in study discontinuation. Table 2 lists the adverse events that occurred in ≥5% of subjects in the regadenoson group. Of these, headache, dyspnea, chest discomfort, nausea, flushing, and dizziness were significantly more common with regadenoson than placebo (P < .0001). Adverse events considered possibly or probably related to study medication by the investigator were also more common with regadenoson (202/334 subjects; 60.5%) than placebo (29/170 subjects; 17.1%) (P < .0001).
Most adverse events were mild. However, 7 severe adverse events (i.e., resulted in an inability to perform daily activities) were reported in 5 (1.5%) regadenoson subjects (headache, n = 2; dyspnea, n = 2; nausea, n = 1; sensation of heaviness, n = 1; neck pain, n = 1). None of the placebo subjects experienced severe adverse events. The majority of adverse events occurred within 2 h of study drug administration: 204/334 (61.1%) subjects receiving regadenoson and 25/170 (14.7%) receiving placebo had adverse events during this period.
The stage of CKD did not affect adverse event rates for either study drug. Adverse events occurred in 182/287 (63.4%) of regadenoson-treated subjects with stage 3 and 27/47 (57.4%) with stage 4 CKD, and in 30/145 (20.7%) of placebo-treated subjects with stage 3 and 6/25 (24.0%) with stage 4 CKD. These rates were similar to the overall incidence for each treatment group.
Five adverse events were considered clinically important. Hemiparesis occurred in a 67-year-old white male with a history of CKD, prior myocardial infarction with stent placement, hypercholesterolemia, hypertension, hypothyroidism, right facial numbness, and sleep apnea. His concomitant medications included aspirin, levothyroxine, atorvastatin, lisinopril, omega-3 fatty acids, fenofibric acid, and ezetimibe. The subject experienced transient left-sided weakness starting 2 minutes after the administration of regadenoson and lasting for 28 minutes. Blood pressure was 92/58 mm Hg at baseline (prior to regadenoson administration), 100/58 mm Hg at 5 minutes post-dose, and 98/70 mm Hg at 30 minutes post-dose, when the hemiparesis resolved. Pulse oximetry showed that his oxygen saturation, which was 97% at 1 minute pre-dose, decreased to 86% at 5 minutes post-dose, but recovered to 94% at 10 minutes post-dose. The subject recovered fully, required no treatment, and had no sequelae. The event was deemed possibly related to the study drug by the investigator.
Another subject (a 73-year-old white male with a history of CKD, cigarette smoking, hypercholesterolemia, hypertension, obesity, and CAD) experienced non-sustained ventricular tachycardia 2 h after regadenoson administration. This asymptomatic event was reported as part of the continuous Holter monitor data and was recorded as an adverse event of <1 minute in duration. Concomitant medications included desloratadine, metoprolol, rabeprazole, atorvastatin, doxazosin, potassium, furosemide, warfarin, fenofibrate, and levothyroxine. His blood pressure remained stable throughout the procedure, being 128/70 mm Hg at baseline and 122/80 mm Hg 2 h after regadenoson administration, when the episode of ventricular tachycardia occurred. This event was considered to be mild in severity, not related to study drug, and required no treatment.
Non-sustained ventricular tachycardia also was reported in a placebo subject after discharge (22-h post-dose). This was a single episode, which was considered mild and possibly related to study drug (placebo). No adverse events were reported in this subject following documentation of the arrhythmia. Second-degree atrioventricular node block occurred in a placebo subject 60 minutes post-dose, and sinus arrest of approximately 4 seconds occurred in a placebo subject 13 minutes post-dose.
One subject in the regadenoson group received 25 mg of aminophylline orally to attenuate dyspnea. None of the placebo-treated subjects experienced any adverse reactions that were sufficiently severe and/or persistent to require either IV or oral aminophylline.
Serum Chemistry
Mean changes from baseline were numerically small and similar with regadenoson and placebo for all serum chemistry parameters, including serum creatinine, and for hemoglobin.
Estimated Glomerular Filtration Rate
Mean eGFR at baseline was similar in the regadenoson and placebo groups for all subjects and for those with either stage of CKD (Table 3). Mean changes from baseline in eGFR, irrespective of CKD stage, were numerically small and comparable in the placebo and regadenoson groups (Table 3). Ten subjects (all with stage 3 CKD) had a reduction in eGFR of >10 mL/minute/1.73 m2 from baseline: eGFR decreased by 11-12 mL/minute/1.73 m2 in 2 placebo subjects with no associated adverse events; eGFR decreased by 11-17 mL/minute/1.73 m2 in 7 regadenoson subjects, 4 of whom experienced mild adverse events typical of regadenoson; and eGFR decreased by 42 mL/minute/1.73 m2 (from 73 to 31 mL/minute/1.73 m2) in 1 regadenoson subject, without any associated adverse events. A Fisher’s exact test showed that the number of regadenoson and placebo subjects with a >10 mL/minute/1.73 m2 decrease in eGFR was not statistically significant, whether considering only subjects with stage 3 CKD (P = .5072) or all subjects (P = .5068).
Vital Signs and 12-Lead ECGs
Mean changes from baseline in SBP and DBP were generally similar between the regadenoson and placebo groups (Figure 2). However, HR increased to >100 bpm in the 1-h post-dose period in more subjects receiving regadenoson (21/332 [6.3%]) than placebo (3/168 [1.8%]) (P = .03). The percentages of subjects meeting other prespecified vital sign criteria were comparable between the regadenoson and the placebo groups. Changes in respiratory rate were also similar between the 2 groups.

Mean (±SD) SBP, DBP, and HR following the administration of regadenoson or placebo
The percentage of subjects with an ECG abnormality was similar in the regadenoson and placebo groups at all the post-dose intervals assessed. No clinically meaningful differences were observed between treatments in the mean change from baseline in HR, QT interval (including measurement using Bazett’s and Fridericia’s corrections), PR interval, or QRS interval. Some differences were noted between the treatment groups at 5 minutes post-dose: mean HR increased from baseline with regadenoson (13.9 bpm) but not placebo (−0.4 bpm) and mean RR interval decreased from baseline with regadenoson (−163.6 ms) but not placebo (3.2 ms); these differences diminished at each successive post-dose interval. Continuous 12-lead ECG monitoring showed no apparent differences in the distribution of ECG abnormalities from baseline to 2-h post-dose with either regadenoson or placebo.
Pulse Oximetry and Physical Examination Findings
No statistically significant differences were observed between treatment groups in the mean change in oxygen saturation from baseline to 2-h post-dose for subjects with stage 3 (regadenoson −0.2% ± 1.6%; placebo −0.6% ± 2.7%) or stage 4 (regadenoson −0.4% ± 1.4%; placebo −0.3% ± 1.0%) CKD. The proportions of subjects in the stages 3 and 4 CKD subgroups with oxygen saturation <92% (considered clinically meaningful) at any time during the 2-h continuous post-dose pulse oximetry collection period were also similar with regadenoson and placebo: 188/276 (68.1%) of stage 3 CKD subjects receiving regadenoson and 84/142 (59.2%) receiving placebo (difference −9.0%; 95% confidence interval −18.7%, 0.8%), and 32/45 (71.1%) of stage 4 CKD subjects receiving regadenoson and 15/23 (65.2%) receiving placebo (difference −5.9%; 95% confidence interval −29.4%, 17.6%). Interestingly, the proportions of subjects with oxygen saturation <92% at any time during the 2-h continuous pre-dose monitoring period were also similar between the treatment groups: 199/276 (72.1%) of stage 3 CKD subjects receiving regadenoson and 98/142 (69.0%) receiving placebo (difference −3.1%; 95% confidence interval −12.4%, 6.2%), and 31/45 (68.9%) of stage 4 CKD subjects receiving regadenoson and 14/23 (60.9%) receiving placebo (difference −8.0%; 95% confidence interval −32.1%, 16.1%). None of the changes in oxygen saturation resulted in any clinically meaningful adverse events requiring intervention. Physical examination findings were unremarkable.
Discussion
A previous Phase 1 study in healthy male volunteers showed that renal excretion was a major elimination pathway for regadenoson, with an average of 57% (range 19%-77%) of a given IV dose being excreted unchanged in the urine.22 It was also shown that the average plasma renal clearance of regadenoson (approximately 450 mL/minute) exceeded the normal GFR, indicating that active renal tubular secretion plays a role in the elimination of the drug.22 A subsequent Phase 1 study investigated the pharmacokinetics and tolerability of regadenoson (single IV 0.4 mg bolus) in subjects (n = 24) with varying degrees of kidney function (creatinine clearance <30 mL/minute, n = 5; 30-49 mL/minute, n = 6; 50-79 mL/minute, n = 7; 80-140 mL/minute, n = 6).12 An inverse correlation was observed between the severity of CKD and regadenoson urinary clearance, total clearance, and terminal elimination half-life. However, plasma concentration-time curves directly post-dose (when the pharmacodynamic effect is most important for radiotracer uptake) were unaffected by creatinine clearance, as evidenced by similar maximum plasma concentrations and similar volumes of distribution estimates across the categories. The number and severity of adverse events were also comparable among this small sample of subjects. This study was conducted to further explore the safety and tolerability of regadenoson among individuals with decreased renal function.
This randomized controlled trial of regadenoson versus placebo demonstrated that the safety profile of regadenoson in individuals with stage 3 or 4 CKD was similar to the safety profile observed in patients participating in previous Phase 3 clinical trials with the drug.17,18 For example, a total of 80% of those who received regadenoson in the pivotal, Phase 3 clinical trials reported an adverse event6 compared with 62.6% in this study. No serious adverse events were reported from the time of administration of a single IV 0.4 mg dose of regadenoson or placebo to the follow-up visit, 24-h post-dose. Of the 2 adverse events of clinical importance that occurred in the regadenoson group, only 1 was considered possibly related to study drug. This was a 28-minute episode of left hemiparesis beginning 2 minutes after the administration of regadenoson, which coincided with a period of low oxygen saturation (documented as 86% at 5 minutes post-dose, the first time it was measured after dosing). This individual had had sleep apnea and right facial numbness since 2009, but he was not receiving treatment for either condition at the time of the study. People with sleep apnea, especially those not on continuous positive airway pressure therapy, are susceptible to strokes and transient ischemic attacks.23 The adverse event profile of regadenoson in subjects with stage 3 or 4 CKD was consistent with previous trials with the drug,17,18,24 with no unexpected adverse events being reported. Headache, dyspnea, chest discomfort, nausea, flushing, and dizziness were among the notable side effects that were significantly more common with regadenoson than placebo, as expected. The frequency of these side effects was comparable to previously published data from pivotal regadenoson Phase 3 trials.17,18 Adverse event profiles were also similar between subjects with stage 3 and 4 CKD for regadenoson and for placebo (i.e., the types and frequencies of adverse events with regadenoson [or placebo] were similar regardless of CKD stage). Although a slightly higher proportion of subjects with stage 3 than stage 4 CKD experienced an adverse event, this difference likely reflects the small number of subjects with stage 4 CKD. With greater numbers of subjects with stage 4 CKD, the differences would most likely be less. Using the overall adverse event rates, comparing the number of subjects in the stage 3 versus the stage 4 CKD group with adverse events and the number of subjects without adverse events, the results are not statistically significant (Fisher’s exact test). It is also not statistically significant for placebo-treated subjects in the stage 3 versus stage 4 CKD groups. There is also no difference in the safety profile from a clinically meaningful standpoint. Most adverse events occurred in the 2 h following regadenoson administration, consistent with the pharmacodynamic effect and terminal half-life of the drug.6,22,25
The proportion of subjects whose HR exceeded 100 bpm was greater with regadenoson than placebo; all other vital sign criteria were comparable between the 2 treatment groups. The tachycardic effect was noted despite 55.1% of regadenoson subjects being on beta-blockers at the time of the study. Two mechanisms appear to account for the tachycardia induced by regadenoson. First, peripheral vasodilation following drug administration leads to a fall in blood pressure, triggering a baroreflex-mediated activation of the sympathetic nervous system and a subsequent increase in HR.26 Second, evidence from animal models suggests that regadenoson also causes direct sympathoexcitation through stimulation of arterial chemoreceptors via A2A receptors, such that the regadenoson-induced tachycardia is dissociated from the decrease in mean arterial pressure.26
No clinically important differences were apparent between the regadenoson and the placebo groups with regard to changes from baseline in clinical laboratory findings or ECG abnormalities following drug administration. The changes in these parameters, when observed, were not clinically meaningful and none required intervention. One subject experienced left hemiparesis, which was considered possibly related to regadenoson administration, concurrently with a decrease in oxygen saturation.
Limitations
Several limitations should be considered when evaluating the results of this study. First, of the 504 subjects that were randomized, only 72 (14%) had stage 4 CKD. Thus, the results of this study in this subgroup of subjects should be interpreted with caution. Second, the trial did not include subjects with stage 5 CKD (on dialysis). Third, no pharmacokinetic data were collected to support the previously published findings of Gordi et al,12 which included only a small number of patients with impaired renal function. For drugs that are eliminated by the kidney (i.e., ≥30% of the administered dose eliminated unchanged in the urine), renal impairment often results in a significant increase in drug exposure, manifested as an increase in the area under the concentration-time curve.27 In some cases, this necessitates a recommendation for dose adjustment according to the degree of renal dysfunction.27 Regadenoson plasma concentrations display a tri-exponential decline after administration of an IV bolus dose, with rapid distribution phases followed by a relatively longer elimination phase.22 Any increase in exposure to regadenoson in patients with renal impairment would be expected to occur during the final phase of clearance. However, most adverse events observed with regadenoson occur shortly after dosing and resolve within approximately 15 minutes (except for headache, which may last for 30 minutes), reflecting the timing of the drug’s pharmacodynamic effect.6,28,29 Thus, it is unlikely that an increase in regadenoson exposure among patients with renal impairment would affect the adverse event profile, a speculation supported by the results of this study.
Conclusion
This study demonstrated that regadenoson, an A2A receptor agonist, has a safety profile in individuals with stage 3 or 4 CKD that is comparable to the safety profile in the general patient population, although it should be noted that only 14% of subjects included had stage 4 CKD. These results indicate that regadenoson is suitable for use as a coronary vasodilator for pharmacologic stress MPI in patients with stage 3 or 4 CKD.
References
Shryock JC, Belardinelli L. Adenosine and adenosine receptors in the cardiovascular system: Biochemistry, physiology, and pharmacology. Am J Cardiol 1997;79:2-10.
Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 2001;53:527-52.
Cerqueira MD. The future of pharmacologic stress: Selective A2A adenosine receptor agonists. Am J Cardiol 2004;94:33D-40D.
Zablocki J, Palle V, Blackburn B, Elzein E, Nudelman G, Gothe S, et al. 2-substituted pi system derivatives of adenosine that are coronary vasodilators acting via the A2A adenosine receptor. Nucleosides Nucleotides Nucleic Acids 2001;20:343-60.
Gao Z, Li Z, Baker SP, Lasley RD, Meyer S, Elzein E, et al. Novel short-acting A2A adenosine receptor agonists for coronary vasodilation: Inverse relationship between affinity and duration of action of A2A agonists. J Pharmacol Exp Ther 2001;298:209-18.
Lexiscan® (regadenoson injection) [prescribing information]. Deerfield, IL: Astellas Pharma US Inc. April 2011.
Buhr C, Gossl M, Erbel R, Eggebrecht H. Regadenoson in the detection of coronary artery disease. J Vasc Health Risk Manag 2008;4:337-40.
Hendel RC. Update on myocardial perfusion imaging: Role of regadenoson. Rep Med Imaging 2009;2:13-23.
Al Jaroudi W, Iskandrian AE. Regadenoson: A new myocardial stress agent. J Am Coll Cardiol 2009;54:1123-30.
Garnock-Jones KP, Curran MP. Regadenoson. Am J Cardiovasc Drugs 2010;10:65-71.
Thompson CA. FDA approves pharmacologic stress agent. Am J Health Syst Pharm 2008;65:890.
Gordi T, Blackburn B, Lieu H. Regadenoson pharmacokinetics and tolerability in subjects with impaired renal function. J Clin Pharmacol 2007;47:825-33.
De Winter O, Van de Veire N, Gemmel F, Goethals I, De Sutter J. Myocardial perfusion imaging in the elderly: A review. Nucl Med Commun 2006;27:529-34.
Wetzels JF, Kiemeney LA, Swinkels DW, Willems HL, den Heijer M. Age- and gender-specific reference values of estimated GFR in Caucasians: The Nijmegen Biomedical Study. Kidney Int 2007;72:632-7.
Stevens LA, Li S, Wang C, Huang C, Becker BN, Bomback AS, et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: Results from the Kidney Early Evaluation Program (KEEP). Am J Kidney Dis 2010;55:S23-33.
Leoncini G, Viazzi F, Rosei EA, Ambrosioni E, Costa FV, Leonetti G, et al. Chronic kidney disease in hypertension under specialist care: The I-DEMAND study. J Hypertens 2010;28:156-62.
Iskandrian AE, Bateman TM, Belardinelli L, Blackburn B, Cerqueira MD, Hendel RC, et al. Adenosine versus regadenoson comparative evaluation in myocardial perfusion imaging: Results of the ADVANCE phase 3 multicenter international trial. J Nucl Cardiol 2007;14:645-58.
Cerqueira MD, Nguyen P, Staehr P, Underwood SR, Iskandrian AE, on behalf of the ADVANCE-MPI Trial Investigators. Effects of age, gender, obesity, and diabetes on the efficacy and safety of the selective A2A agonist regadenoson versus adenosine in myocardial perfusion imaging. J Am Coll Cardiol Imaging 2008;1:307-16.
Levey AS, Coresh J, Greene T, Stevens LA, Zhang YL, Hendriksen S, et al. Using standardized serum creatinine values in the modification of diet in renal disease study equation for estimating glomerular filtration rate. Ann Intern Med 2006;145:247-54.
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D. A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 1999;130:461-70.
Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, et al. National Kidney Foundation practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Ann Intern Med 2003;139:137-47.
Gordi T, Frohna P, Sun HL, Wolff A, Belardinelli L, Lieu H. A population pharmacokinetic/pharmacodynamic analysis of regadenoson, an adenosine A2A-receptor agonist, in healthy male volunteers. Clin Pharmacokinet 2006;45:1201-12.
Yaggi HK, Concato J, Kernan WN, Lichtman JH, Brass LM, Mohsenin V. Obstructive sleep apnea as a risk factor for stroke and death. N Engl J Med 2005;353:2034-41.
Hendel RC, Bateman TM, Cerqueira MD, Iskandrian AE, Leppo JA, Blackburn B, et al. Initial clinical experience with regadenoson, a novel selective A2A agonist for pharmacologic stress single-photon emission computed tomography myocardial perfusion imaging. J Am Coll Cardiol 2005;46:2069-75.
Lieu HD, Shryock JC, von Mering GO, Gordi T, Blackburn B, Olmsted AW, et al. Regadenoson, a selective A2A adenosine receptor agonist, causes dose-dependent increases in coronary blood flow velocity in humans. J Nucl Cardiol 2007;14:514-20.
Dhalla AK, Wong MY, Wang WQ, Biaggioni I, Belardinelli L. Tachycardia caused by A2A adenosine receptor agonists is mediated by direct sympathoexcitation in awake rats. J Pharmacol Exp Ther 2006;316:695-702.
Zhang Y, Zhang L, Abraham S, Apparaju S, Wu TC, Strong JM, et al. Assessment of the impact of renal impairment on systemic exposure of new molecular entities: Evaluation of recent new drug applications. Clin Pharmacol Ther 2009;85:305-11.
Cerqueira MD, Mahmarian JJ, Kinser C, Papatheofanis FJ, Segreti A. Frequency of aminophylline use to attenuate adverse reactions to regadenoson and adenosine. J Nucl Cardiol 2009;16:658. (abstract 10.01).
Johnson SG, Peters S. Advances in pharmacologic stress agents: Focus on regadenoson. J Nucl Med Technol 2010;38:163-71.
Acknowledgments
The study and publication process was sponsored by Astellas Pharma Global Development, Inc. Dr Bukofzer, Ms Klauke, Ms Feaheny, and Dr McNutt are employees of Astellas Pharma Global Development, Inc. Dr Ananthasubramaniam has received research grants from GE Healthcare, Molecular Insight Pharmaceuticals, the American Society of Echocardiography, and Astellas Global Pharma Development, Inc.; he is a member of the Speakers’ Bureau of Astellas Pharma US and Lantheus Medical Imaging; and is a consultant for Lantheus Medical Imaging and the US Department of Justice. Dr Weiss has received research grants from Merck, Novartis, Johnson & Johnson, Boehringer Ingelheim, Regeneron, Amgen, Pfizer, Gilead, PGx, and Astellas Global Pharma Development, Inc. Writing assistance was provided by Elaine F. Griffin, MA, DPhil (a medical writer at Envision Scientific Solutions), funded by Astellas Pharma Global Development, Inc. The authors would like to thank the 32 investigators who participated in this study, of whom 26 have given permission to be acknowledged, as follows: Azazuddin A. Ahmed (Apex Medical Research, Inc., Chicago, IL); Sivakumar Amar (Advanced Research Institute, Trinity, FL); Karthik Ananthasubramaniam (Henry Ford Hospital, Detroit, MI); Ahmed Arif (Apex Medical Research, Flint, MI); Brad Bart (Hennepen County Medical Center, Minneapolis Medical Research Foundation, Minneapolis, MN); Andrew Cash (Ilumina Clinical Associates, Indiana, PA); Clinton N. Corder (COR Clinical Research, LLC, Oklahoma City, OK); Oscar DeValle (West Houston Clinical Research Service, Houston, TX); Steven Edell (Delaware Clinical Trials, LP, Wilmington, DE); Mohamed El-Shahawy (Academic Medical Research Institute, E Los Angeles, CA); Faisal A. Fakih (Florida Pulmonary Research Institute, LLC, Winter Park, FL); James V. Felt (Alta Bates Summit Medical Center, Oakland, CA); Charles Fogarty (Spartanburg Medical Research, Spartanburg, SC); Almena Free (Pinnacle Research Group, Anniston, AL); Michael Halter (Ilumina Clinical Associates, Tyrone Hospital, Tyrone, PA); Joe L. Hargrove (Cardiology and Medicine Clinic, Little Rock, AR); Alan Kivitz (Altoona Center for Clinical Research, Duncansville, PA); Thomas C. Marbury (Orlando Clinical Research Center, Orlando, FL); James L. Pearle (California Research Medical Group, Inc., San Diego, CA); Ronald Polinsky Jr (Berks Cardiologists Ltd., Wyomissing, PA); Galal N. Salem (SunRise Clinical Research, Bell Gardens, CA); Anil V. Shah (Coastal Multi-Specialty Research, Santa Ana, CA); William Smith (New Orleans Center for Clinical Research, Knoxville, TN); Gregory S. Thomas (Mission Internal Medical Group, Mission Viejo, CA); Frederick Weiland (Sutter Roseville Medical Center, Roseville, CA); and Robert Weiss (Maine Research Associates, Auburn, ME).
Author information
Authors and Affiliations
Corresponding author
Additional information
ClinicalTrials.gov number, NCT00863707.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ananthasubramaniam, K., Weiss, R., McNutt, B. et al. A randomized, double-blind, placebo-controlled study of the safety and tolerance of regadenoson in subjects with stage 3 or 4 chronic kidney disease. J. Nucl. Cardiol. 19, 319–329 (2012). https://doi.org/10.1007/s12350-011-9508-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12350-011-9508-3