
Abstract
Introduction
A clinical trial evaluated ocular hypotensive efficacy and safety of netarsudil 0.02% once daily (QD) relative to ripasudil 0.4% twice daily (BID).
Methods
This was a single-masked, randomized, phase 3, superiority study. Japanese patients were randomized to either the netarsudil 0.02% group or the ripasudil 0.4% group in a 1:1 ratio and treated for 4 weeks. The primary efficacy variable was mean diurnal intraocular pressure (IOP) (average of diurnal time points at 09:00, 11:00, and 16:00) at Week 4.
Results
A total of 245 patients were included in the primary analysis. At Week 4, least squares (LS) mean of diurnal IOP adjusted for baseline was 15.96 and 17.71 mmHg in the netarsudil 0.02% and ripasudil 0.4% groups, respectively, demonstrating the superiority of netarsudil 0.02% QD over ripasudil 0.4% BID by a margin of − 1.74 mmHg (p < 0.0001). Mean reduction from baseline in mean diurnal IOP at Week 4 was 4.65 and 2.98 mmHg, respectively. Adverse events (AEs) occurred less frequently in netarsudil 0.02% than in ripasudil 0.4%, with the incidence of ocular AEs being 59.8% and 66.7%, respectively. The most frequently reported AE was conjunctival hyperemia in both groups, with an incidence of 54.9% and 62.6%, respectively. No serious eye-related AEs were reported.
Conclusion
Netarsudil ophthalmic solution 0.02% dosed QD (p.m.) was well tolerated and more effective in reducing IOP than ripasudil ophthalmic solution 0.4% dosed BID. Netarsudil 0.02% QD may become an important option for the treatment of Japanese patients with primary open-angle glaucoma (POAG) or ocular hypertension (OHT).
Trial registration
ClinicalTrials.gov identifier, NCT04620135.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Glaucoma, one of the leading causes of blindness in the world, is largely caused by elevated intraocular pressure (IOP). | |
Rho-associated protein kinase (ROCK) inhibitors, such as netarsudil, have a unique mechanism of action relative to other pharmacotherapies aimed at reducing IOP and have been introduced into widespread clinical use in recent years. | |
This Phase 3 study was designed to compare the ocular hypotensive efficacy and safety of netarsudil ophthalmic solution 0.02% once daily (QD) to the active comparator, ripasudil hydrochloride hydrate ophthalmic solution 0.4% twice daily (BID), over a 4-week period in Japanese patients. | |
Netarsudil ophthalmic solution 0.02% dosed QD demonstrated clinically significant efficacy and was superior to ripasudil 0.4% BID in reducing IOP at Week 4 in Japanese patients with primary open-angle glaucoma (POAG) or ocular hypertension (OHT). |
Introduction
Glaucoma is a leading cause of blindness affecting 64 million people in the world and is estimated to rise to 112 million by 2040 [1, 2]. It affects about 4.65 million people in Japan alone [1]. A major risk factor for disease progression is elevated intraocular pressure (IOP) [3], and every millimeter of IOP reduction is reported to significantly delay the disease progression, not only in patients with primary open-angle glaucoma (POAG) with elevated IOP but also in those with normal-tension glaucoma (NTG) [3,4,5,6,7].
Current pharmacotherapies available to lower IOP typically increase uveoscleral outflow (e.g., prostaglandins and alpha agonists) or decrease aqueous humor production (e.g., beta blockers, carbonic anhydrase inhibitors, alpha agonists) [8, 9]. Such pharmacotherapies have demonstrated treatment efficacy, but various classes may not be suitable for use in every patient. Prostaglandin analogs, for example, are often recommended as first-line monotherapy due to the strong IOP lowering effect and once-daily regimen, but are associated with cosmetic adverse reactions, and beta blockers are contraindicated in certain patients with cardiovascular and respiratory comorbidities [9]. Rho-associated protein kinase (ROCK) inhibitors have a unique mechanism of action and have been introduced into widespread clinical use in recent years. These agents increase aqueous outflow through the trabecular meshwork (TM) outflow pathway by multiple mechanisms: decreasing actomyosin-driven cellular contractions, reducing production of fibrogenic extracellular matrix proteins, and decreasing cell stiffness to relax TM outflow tissues and lower IOP [10,11,12,13,14].
In Japan, Glanatec® (ripasudil hydrochloride hydrate 0.4%) ophthalmic solution is the first ROCK inhibitor approved for the treatment of patients with glaucoma or ocular hypertension (OHT), who did not sufficiently respond to or were unable to use other glaucoma drugs. It has a twice-daily (BID) dosing regimen, and adverse reactions such as blepharitis are known to be leading causes of treatment discontinuation [15,16,17,18,19].
Netarsudil mesylate is a ROCK inhibitor being evaluated for potential use in Japan. It inhibits both ROCK1 and ROCK2 and has been shown to increase TM outflow facility [20,21,22,23] and to reduce episcleral venous pressure [23, 24]. Axonal protection by netarsudil has also been recently reported in rats with tumor necrosis factor-induced optic nerve damage [25]. Netarsudil ophthalmic solution 0.02% was approved by the United States (US) Food and Drug Administration in December 2017 and by the European Commission in November 2019 for reduction of elevated IOP in patients with POAG or OHT. The recommended dosing regimen is once daily (QD) [26], with data demonstrating consistent IOP reduction throughout both the nocturnal and diurnal periods [27]. Netarsudil 0.02% was safe and well-tolerated in multiple studies, with significant reduction in IOP levels in patients with POAG or OHT, including those with lower baseline IOP [23, 28,29,30,31,32], an effect of particular relevance in Japan, where there is a high prevalence of POAG with relatively low IOP, including NTG [33, 34].
A recently completed Phase 2 study in Japan showed clinically relevant efficacy and safety, and superiority of netarsudil ophthalmic solution 0.01%, 0.02%, and 0.04% QD to placebo in mean diurnal IOP following 4 weeks of treatment in Japanese subjects with POAG or OHT. Based upon the cumulative efficacy and safety results, 0.02% was determined as the concentration with the optimum efficacy and safety profile for the Japanese population [1]. Indirect, cross-study comparison between this netarsudil Phase 2 study [1] and a ripasudil Phase 2 study [35] suggests superior IOP lowering efficacy of netarsudil ophthalmic solution 0.02% QD to ripasudil hydrochloride hydrate ophthalmic solution 0.4% BID. The current Phase 3 study was therefore designed to directly compare the ocular hypotensive efficacy and safety of netarsudil ophthalmic solution 0.02% QD to the active comparator, ripasudil hydrochloride hydrate ophthalmic solution 0.4% BID, over a 4-week period.
Methods
Study Design
This study was a prospective, single-masked, randomized, multi-center, parallel-group, 4-week, Phase 3 study that evaluated the efficacy and safety of netarsudil ophthalmic solution 0.02% QD compared to ripasudil hydrochloride hydrate ophthalmic solution 0.4% BID in Japanese subjects aged ≥ 20 years with POAG or OHT. The study was conducted at 27 sites in Japan from November 2020 to July 2021 (registered with clinicaltrials.gov as NCT04620135). The elements of study design described below were based on and similar to those of the previous Phase 2 study comparing netarsudil to placebo in Japanese patients [1].
Participants
Key inclusion criteria were: diagnosis of POAG or OHT in both eyes or POAG in one eye and OHT in the other (fellow) eye; age ≥ 20 years; medicated IOP ≥ 14 mmHg in at least one eye and < 30 mmHg in both eyes at the Screening Visit; best corrected visual acuity (BCVA) + 0.7 log MAR or better (20/100 Snellen or better or 0.20 or better in decimal unit) in each eye. Unmedicated (post washout) IOP for eyes with POAG had to be ≥ 15 mmHg and < 35 mmHg in the study eye, and for eyes with OHT it had to be ≥ 22 mmHg and < 35 mmHg in the study eye, for all measurements taken at Qualification Visit 1 (09:00) and Qualification Visit 2 (09:00, 11:00, and 16:00).
Main exclusion criteria, similar to those used in the Phase 2 study, were clinically significant ocular disease; retinal diseases that may progress during the study period, pseudoexfoliation or pigment dispersion glaucoma, history of angle-closure or narrow angles; ocular hyperemia score of moderate (+ 2) or severe (+ 3) at Day 1; previous intraocular glaucoma or refractive surgery in either eye; ocular trauma within 6 months or ocular surgery or non-refractive laser treatment within 3 months prior to screening; evidence of ocular infection or inflammation in either eye, clinically significant blepharitis, conjunctivitis, keratitis or a history of herpes simplex or zoster keratitis in either eye at screening; any corneal disease or condition in either eye that, in the investigator’s opinion, may have confounded assessment of the cornea; current evidence of corneal deposits or cornea verticillata in either eye; planned use of any prohibited concomitant medications in either eye during the study; mean central corneal thickness > 620 μm in either eye at screening; any abnormality preventing reliable applanation tonometry of either eye; known hypersensitivity to any component of netarsudil ophthalmic solution 0.02%, ripasudil hydrochloride hydrate ophthalmic solution 0.4%, or to topical anesthetic; or could not demonstrate proper delivery of the eye drop or, in the investigator’s opinion, would be unable to deliver the eye drop consistently. Patients were also excluded from the study if they had clinically significant systemic disease which might have interfered with the study; had participated in any interventional study within 30 days before screening; or had used systemic medication(s) that could have had a substantial effect on IOP within 30 days before screening, or were anticipated to use such medication during the study, including any corticosteroid-containing drug regardless of route of administration. Women of childbearing potential who were pregnant, nursing, planning a pregnancy, or not using a medically acceptable form of birth control were also excluded from the study. All females of childbearing potential had to have a negative urine pregnancy test result at the screening examination and not intend to become pregnant during the study [1].
Treatments
On study Day 1 (baseline), eligible patients were randomized (1:1) by a computer-generated randomization list using an interactive web response system to receive netarsudil ophthalmic solution 0.02% or ripasudil hydrochloride hydrate ophthalmic solution 0.4%. Netarsudil ophthalmic solution 0.02% was dosed QD in the evening (21:00 ± 1 h), while ripasudil hydrochloride hydrate ophthalmic solution 0.4% was dosed BID in the morning and evening (09:00 ± 1 h /21:00 ± 1 h). In order to be adequately masked, subjects assigned to netarsudil ophthalmic solution were dosed with vehicle solution QD in the morning (09:00 ± 1 h). The study medication was dosed (1 drop) into each eye, beginning on Day 1 and up to and including the evening before the Week 4 visit. At Weeks 1, 2 and 4, the study medication in the morning was dosed at the sites immediately following the morning of IOP measurement.
Treatment assignments were masked to the investigators, the clinical study team (sponsor, monitors, data managers, and statisticians), and the subjects for the duration of the study. A single-masked (evaluator-masked) study design was adopted in the study because the containers of netarsudil and ripasudil ophthalmic solutions differed in the material, and the stability could not be assured if the solutions were refilled in the alternative containers. To ensure masking of the investigators and the clinical study team, study staff responsible for dispensing study medications to the subject did so in a sealed container and instructed subjects that the study medication must be in the container when it was returned.
As in the Phase 2 study, patients were permitted to have intermittent use of artificial tear lubricant products (a gap of at least 10 min between the use of artificial tear lubricant products and the study medication) and wear contact lenses (a gap of 30 min between the contact lens wear and instillation of study medication). Patients were prohibited to use any form of ocular hypotensive medications; miotics; epinephrine-related compounds; carbonic anhydrase inhibitors; alpha agonists; β-adrenoceptor antagonists; muscarinic agonists; ocular prostaglandins analogs; ROCK inhibitors; any ocular or systemic corticosteroids; or systemic medications known to cause corneal deposits or cornea verticillata [1].
Study Endpoints
Efficacy
The primary efficacy endpoint was the comparison of netarsudil ophthalmic solution 0.02% QD to ripasudil ophthalmic solution 0.4% BID for mean diurnal IOP at Week 4, with mean diurnal IOP calculated by averaging IOP measurements at 09:00, 11:00, and 16:00 h for each patient first and then calculating the mean across patients within each treatment group. The secondary efficacy endpoints were mean diurnal IOP at Weeks 1 and 2; mean change and mean percent change from baseline in mean diurnal IOP at each post-treatment visit; mean, mean change, and mean percent change in IOP at each post-treatment time point (09:00, 11:00, and 16:00) at each post-treatment visit; and percentage of patients achieving pre-specified mean diurnal IOP, and mean change and mean percent change in mean diurnal IOP levels.
Safety
The ocular safety endpoints were defined similarly to those in the Phase 2 study, including ocular symptoms/adverse events (AEs); BCVA; objective findings of biomicroscopic examinations; and dilated ophthalmoscopy, including vertical cup–disc ratio measurements. The other safety endpoints were non-ocular AEs and vital signs [1].
Assessments
As described previously, the patients had a total of 6 study visits, including the Screening Visit (Visit 1) and Qualification Visit 1 (Visit 2, after a washout period of pre-study ocular hypotensive medication of 5 days to 6 weeks; 5 days for muscarinic agonists or carbonic anhydrase inhibitors; 2 weeks for adrenergic agonists; 4 weeks for prostaglandins or β adrenoceptor antagonists; 6 weeks for Rho kinase inhibitor). The subsequent visits were Qualification Visit 2/Day 1/Baseline (Visit 3; 2–7 days after Qualification Visit 1), and visits at Week 1 (Visit 4; Day 8 ± 2 days), Week 2 (Visit 5; Day 15 ± 3 days), and Week 4 (Visit 6; Day 29 ± 3 days) [1].
Patients who met the study eligibility criteria at the Screening Visit and Qualification Visits 1 and 2 were considered eligible to participate in the study. The study eye was selected based upon IOP, ocular history, and exam qualifications. For subjects with only one qualified eye, this eye was designated as the study eye. If both eyes of a subject qualified, the eye with the higher IOP at 09:00 h during Visit 3 was designated as the study eye. Both eyes of all subjects were treated.
Efficacy (IOP) was measured at the Screening Visit (any time of the day), Qualification Visit 1 [09:00 (+ 30 min)], and Day 1 [09:00 (+ 30 min), 11:00 (+ 30 min), and 16:00 (± 30 min)], and Weeks 1, 2, and 4 [09:00 (+ 30 min) before the dose in the morning, 11:00: 2 h (± 30 min) after the dose in the morning, and 16:00: 7 h (± 30 min) after the dose in the morning] using a calibrated Goldmann applanation tonometer. Two consecutive IOP measurements of each eye were obtained at each time point. If the two measurements differed by > 2 mmHg, a third measurement was obtained. The IOP was recorded as the mean of two measurements or the median of three measurements.
Safety assessments included ocular symptoms/AEs; BCVA; biomicroscopic examinations of the eyelids, conjunctiva, cornea, anterior chamber, iris, pupil, and lens of the eye; dilated ophthalmoscopy examination of the retina, vitreous, macula, choroid, optic nerve, and vertical cup/disc ratio; vital signs (heart rate and blood pressure); and urine pregnancy test. AEs were collected from the time the subject received the first dose of study medication until Week 4 (Day 29) or study discontinuation. BCVA, biomicroscopy, and vital signs were taken at the Screening Visit, Qualification Visit 1, Day 1, and Weeks 1, 2, and 4. Dilated ophthalmoscopy was examined at the Screening Visit and Week 4. As in the Phase 2 study, BCVA was measured using a Landolt-C chart or its equivalent, at a distance as per the site’s standard practice, with the patient’s best correction from the manifest refraction in place. A decrease in BCVA ≥ 3 lines from baseline was considered clinically significant. The BCVA assessment preceded IOP measurements and the administration of topical anesthetic agents, or any examination requiring contact with the anterior segment. Biomicroscopic abnormal findings were graded on scales of 0 (none) to 3 (severe), or 0 to 4 (cells and cornea verticillata), and lens status was reported as phakic, pseudophakic, or aphakic. Dilated ophthalmoscopy findings were assessed as 0 (normal) or 1 (abnormal). The cup–disc ratio was scored on a scale of 0.1 to 1.0 in 0.1 increments. A change of 0.2 units from baseline in either eye was considered as clinically significant [1].
Statistical Analysis
The statistical analyses were conducted using SAS® (v.9.4; SAS Institute, Cary, NC, USA), . The intent-to-treat (ITT) population was used for efficacy analyses where the ITT population included as randomized patients all who received at least 1 dose of the study medication.
The analysis of the primary efficacy variable employed an analysis of covariance (ANCOVA) model with mean diurnal IOP at Week 4 as the response, baseline mean diurnal IOP as a covariate, and treatment as the main effect, using the ITT population with Monte Carlo Markov Chain multiple imputation techniques to impute the missing data. The least squares (LS) mean difference (netarsudil 0.02% – ripasudil 0.4%) was calculated, as well as two-sided p values and 95% confidence intervals (CIs). Superiority for netarsudil 0.02% was concluded if the 2-sided p value for testing the LS mean difference (netarsudil 0.02% – ripasudil 0.4%) to 0 was ≤ 0.05 and the point estimate of the LS mean difference was < 0 at Week 4. A similar ANCOVA model was used for analysis of the secondary efficacy endpoints mean diurnal IOP at Weeks 1 and 2 and IOP at each post-treatment time point. The change in IOP was tested using a two-sample t test and 95% t distribution CIs on the difference. Fisher’s exact test (two-sided p values) was used to test differences between netarsudil 0.02% versus ripasudil 0.4% in the percentages of patients achieving categorical IOP endpoints.
The safety analyses were performed on the safety population, which included all randomized (as treated) patients who received at least one dose of study medication. The AEs were coded using the Medical Dictionary for Regulatory Activities/Japanese translation, v.23.1. The other safety variables were summarized by descriptive statistics.
The sample size was based on assumptions of a two-sided test with alpha = 0.05 and a difference of the change from baseline in mean diurnal IOP (netarsudil 0.02% – ripasudil 0.4%) of -1.1 mmHg with a common standard deviation of 2.3. It was estimated that 93 subjects (study eyes) per treatment group in the ITT population would yield at least 90% power to demonstrate superiority of netarsudil 0.02% to ripasudil 0.4% in the mean diurnal study eye IOP at Week 4. The target sample size was set to 120 per treatment group (240 in total) to allow for withdrawals and dropouts.
Ethical Conduct
The study was conducted in accordance with ethical principles based on the Declaration of Helsinki and the guidance stipulated in Article 14, Paragraph 3, and Article 80–2 of the Pharmaceuticals, Medical Devices and Other Therapeutic Products Act of Japan, Ministry of Health, Labour and Welfare (MHLW) Ordinance on Good Clinical Practice (MHLW Ordinance No 28 [March 27, 1997]), International Council for Harmonization Guideline E6 (R2), the study protocol, and the standard operating procedures. All patients provided their written informed consent before participating in the study. All study-related documents were reviewed and approved by appropriate ethics committees.
Results
Demographics and Disposition
A total of 245 patients were randomized to treatment (122 and 123 patients in the netarsudil 0.02% and ripasudil 0.4% groups, respectively). The mean ± SD age of the overall population was 61.7 ± 13.06 years, the majority of subjects were female, and most had POAG in the two treatment groups. There was no statistically significant difference in demographics and other baseline characteristics between the treatment groups (Table 1).
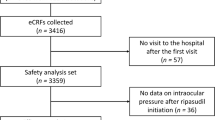
Of 245 patients, 238 (97.1%) completed 4 weeks of treatment and 7 (2.9%) discontinued the study. Of the 7 discontinued patients, 5 discontinued due to AEs [netarsudil 0.02%: 2 patients (1 had conjunctival hyperemia, conjunctival oedema, and eyelid oedema; 1 had blepharitis), ripasudil 0.4%: 3 patients (1 had conjunctival hyperemia; 1 had pneumonia; 1 had conjunctivitis)], and 2 patients discontinued due to withdrawal of consent (Fig. 1).

Study disposition. N total number of patients in the given treatment, n number of patients in a given category. ITT intent-to-treat, PP per protocol
Efficacy
All 245 patients were included in the primary efficacy analysis (ITT population). The mean diurnal IOP of study eyes at baseline was 20.48 and 20.83 mmHg in the netarsudil 0.02% and ripasudil 0.4% arm, respectively, and showed no statistically significant difference between the treatment groups. At Week 4, the LS mean diurnal IOP adjusted for baseline of study eyes treated with netarsudil 0.02% and ripasudil 0.4% was 15.96 and 17.71 mmHg, respectively. The adjusted LS mean difference between netarsudil 0.02% and ripasudil 0.4% [LS mean difference (95% CI)] was − 1.74 (− 2.17, − 1.31) mmHg, with the superiority of netarsudil 0.02% to ripasudil 0.4% in mean diurnal IOP at Week 4, the primary endpoint of the study, achieving statistical significance (p < 0.0001 and the point estimate of the LS mean difference < 0). Similar results were reported at Weeks 1 and 2 (Table 2).
There was a statistically significantly greater reduction in mean diurnal IOP from baseline in the netarsudil 0.02% group (ranging from 4.34 to 4.65 mmHg) than the ripasudil group (ranging from 2.81 to 2.98 mmHg) at Weeks 1, 2, and 4 (all p < 0.0001) (Table 3), which corresponded to statistically significantly higher percent reductions in mean diurnal IOP from baseline in the netarsudil group (ranging from 21.0 to 22.6%) in comparison with the ripasudil group (13.5 to 14.3%) at Weeks 1, 2, and 4 (all p < 0.0001) (Table 4).
The mean IOP was also statistically significantly lower in the netarsudil group (ranging from 15.36 to 16.69 mmHg) when compared with the ripasudil 0.4% group (ranging from 16.93 to 19.11 mmHg) across all post-treatment time points (09:00, 11:00, and 16:00) at each study visit (all p < 0.001 except for p = 0.0028 at 11:00 Week 1) (Fig. 2).

Mean IOP ± SE of study eye at each timepoint (09:00, 11:00, and 16:00) at baseline, Week 1, Week 2, and Week 4 (ITT population, observed data). IOP intraocular pressure, ITT intent-to-treat, SE standard error, **p value < 0.01, ***p value < 0.001
In responder analyses, the patients in the netarsudil 0.02% group generally achieved lower mean diurnal IOP compared to patients in the ripasudil 0.4% group at Week 4. The data also showed a greater magnitude of IOP reduction and percent reduction in mean diurnal IOP from baseline at Week 4 for patients treated with netarsudil 0.02% compared to those treated with ripasudil 0.4% (Fig. 3).

Percentage of patients with (A) mean diurnal IOP ≤ 17 mmHg at Week 4, B mean diurnal IOP reduction ≥ 4 mmHg from baseline at Week 4, C) mean diurnal IOP ≥ 20% reduction from baseline at Week 4 in the study eye. IOP intraocular pressure
In order to assess the ocular hypotensive efficacy of netarsudil 0.02% by baseline mean diurnal IOP, mean change and mean percent change from baseline in mean diurnal IOP were calculated for subgroups of baseline mean diurnal IOP of ≥ 20 mmHg or < 20 mmHg as a post hoc analysis. In the subgroup with baseline mean diurnal IOP of ≥ 20 mmHg (baseline mean diurnal IOP of 22.57 mmHg), netarsudil 0.02% produced a 24% reduction in mean diurnal IOP (5.37 mmHg) at Week 4. In the subgroup with baseline mean diurnal IOP of < 20 mmHg (baseline mean diurnal IOP of 17.75 mmHg), all of whom were confirmed to be POAG patients, netarsudil 0.02% produced a 21% reduction in mean diurnal IOP (3.80 mmHg) (Fig. 4).

Efficacy of netarsudil 0.02% in patient subgroups stratified by baseline mean diurnal IOP of ≥ 20 mmHg or < 20 mmHg. Mean and mean change/mean percent change from baseline in mean diurnal IOP (mean ± SE) at Week 4 (ITT population, observed data). BL baseline, IOP intraocular pressure, ITT intent-to-treat, SE standard error
Safety
AEs
The incidence of AEs was 59.8% and 69.1% in the netarsudil 0.02% and ripasudil 0.4% groups, respectively. All the cases were assessed as mild in severity other than 2 moderate cases in the netarsudil 0.02% (1 patient with conjunctival hyperemia and 1 patient with conjunctival edema, eyelid edema, and conjunctival hyperemia) and 4 moderate cases in ripasudil 0.4% groups [pneumonia, erythema multiforme exudativum, conjunctivitis, and conjunctival hyperemia (1 patient each)]. Treatment-related AEs were reported in 59.0% (72/122) and 65.9% (81/123) in the netarsudil 0.02% and ripasudil 0.4% groups, respectively. Two serious AEs (pneumonia and erythema multiforme exudativum) were reported in the study and no deaths.
The ocular AEs present in > 1.0% of patients are summarized in Table 5. The most common ocular AE was conjunctival hyperemia (n = 144), with a slightly lower incidence in the netarsudil 0.02% group compared to the ripasudil 0.4% group [netarsudil 0.02%: 54.9% (67/122), ripasudil 0.4%: 62.6%[(7/123), not statistically significant]. All events of conjunctival hyperemia were assessed as mild except 2 moderate events in the netarsudil 0.02% group and one moderate event in the ripasudil 0.4% group. The majority of conjunctival hyperemia cases [99.3% (143/144)] were considered as related to the treatment and 2 of 143 treatment-related conjunctival hyperemia cases led to patient discontinuation from the study (netarsudil 0.02%: 1 patient, ripasudil 0.4%: 1 patient).
The next most common ocular AEs were eye irritation and conjunctival hemorrhage, with conjunctival hemorrhage only being reported in the netarsudil 0.02% group. All cases of eye irritation [netarsudil 0.02%: 5.7% (7/122), ripasudil 0.4%: 5.7% (7/123)] and conjunctival hemorrhage [netarsudil 0.02%: 4.9% (6/122)] were assessed as mild. All 14 cases of eye irritation and 2 of 6 cases of conjunctival hemorrhage were assessed as related to the study drugs. During the study, two cases of cornea verticillata were reported in the netarsudil 0.02% group. Cornea verticillata was diagnosed at Week 4 in both cases.
Eight non-ocular AEs were reported in the study [netarsudil 0.02%: 2.5% (3/122), ripasudil 0.4%: 4.1% (5/123)]. All cases of non-ocular AEs were assessed as mild except for 2 SAE cases, which were both assessed as moderate (pneumonia and erythema multiforme exudativum in the ripasudil 0.4% group). Among non-ocular AEs, only one case of oropharyngeal discomfort in the ripasudil 0.4% group and one case of stomach discomfort in the netarsudil 0.02% were reported as related to the study drug.
Other Safety Measures
There were no clinically relevant changes reported in visual acuity for the study eyes or the fellow eyes at any post-treatment visits. There were no statistically significant differences between the netarsudil 0.02% and ripasudil 0.4% groups for biomicroscopy findings other than conjunctival hyperemia. The mean of the conjunctival hyperemia score evaluated on a 4-point scale from 0 to 3 (none, mild, moderate, and severe) was below 0.4 at every post-treatment time point (09:00, 11:00, and 16:00 at Weeks 1, 2, and 4) in both the netarsudil 0.02% and ripasudil 0.4% groups; however, the hyperemia score was statistically significantly higher in the netarsudil 0.02% group than in the ripasudil 0.4% group except for 11:00 at Weeks 2 and 4 (Fig. 5). No clinically significant changes in vital signs were reported between the treatment groups from baseline to any post-treatment visit.

Mean conjunctival hyperemia score in the study eye at each timepoint (09:00, 11:00, and 16:00) at baseline, Week 1, Week 2, and Week 4, N.S. not significant, SE standard error, *p value < 0.05, **p value < 0.01, ***p value < 0.001
Discussion
The current Phase 3 clinical trial is the first to compare the ocular hypotensive effects and clinical safety profile between two commercially available ROCK inhibitor eye drops. In the trial, netarsudil ophthalmic solution 0.02% dosed QD (p.m.) demonstrated clinically significant efficacy and met the primary endpoint of superiority to ripasudil 0.4% BID in mean diurnal IOP at Week 4 in Japanese patients with POAG or OHT by a margin of -1.74 mmHg (p < 0.0001). Netarsudil 0.02% achieved a mean reduction in mean diurnal IOP at Week 4 of 4.65 mmHg that was a 22.62% reduction from baseline in this study population with relatively low baseline mean diurnal IOP (≈ 20.5 mmHg). In responder analyses, 66.1% of the patients in the netarsudil 0.02% group achieved ≥ 20%reduction from baseline in mean diurnal IOP at Week 4 compared to 22.5% in the ripasudil 0.4% group. The overall ocular hypotensive efficacy of netarsudil 0.02% was significantly greater than that of ripasudil 0.4%.
The ocular hypotensive efficacy of netarsudil 0.02% shown in the present study was consistent with the results of the Phase 3 studies in the US [28,29,30,31] as well as the dose finding Phase 2 study in Japan [1]. In the US Phase 3 studies, when baseline IOP was < 25 mmHg, netarsudil 0.02% QD demonstrated noninferiority to timolol 0.5% BID, a first-line drug for POAG or OHT worldwide, including Japan, in lowering IOP. Ripasudil 0.4%, in the same ROCK inhibitor class as netarsudil, is broadly used in Japan for glaucoma or OHT as one of the second-line drugs when other therapeutic agents are not effective or cannot be administered. Results of the noninferiority studies suggest that netarsudil 0.02% QD could have the potential of comparable ocular hypotensive efficacy to timolol 0.5% BID and of becoming a second-generation ROCK inhibitor.
Previous clinical studies have shown that netarsudil 0.02% can produce significant reductions in IOP in patients with lower baseline IOP [23, 28,29,30,31,32]. Consistent with previous studies, a post hoc analysis in this study showed that netarsudil 0.02% has significant IOP-lowering efficacy in Japanese patients with low baseline IOPs (< 20 mmHg), reducing mean diurnal IOP from a baseline of 17.75 mmHg to 13.96 mmHg (− 3.8 mmHg). Although there are limitations to interpreting post hoc analyses, this result suggests that netarsudil 0.02% may be well-suited for treating POAG patients in Japan given the relatively high prevalence of POAG patients with low IOP, including patients with NTG [33, 34].
In terms of safety, netarsudil 0.02% was safe and generally well-tolerated in Japanese subjects with POAG or OHT. There was a lower overall incidence of AEs in the netarsudil 0.02% group compared with the ripasudil 0.4% group. As expected, given the known vasodilatory effects of ROCK inhibition [11, 14, 36], conjunctival hyperemia was the predominant reported AE and the most common ocular examination finding in both groups, and was observed at a slightly lower rate with netarsudil 0.02%. The biomicroscopy assessments suggested that hyperemia may last during the day with netarsudil 0.02% administered at night, though its severity was typically categorized as mild. Conjunctival hemorrhage was limited to the netarsudil 0.02% group, and all cases were assessed as mild; none led to study drug interruption or discontinuation.
Two patients developed cornea verticillata at Week 4 in the study. Cornea verticillata, benign lipid microdeposits typically localized to the basal corneal epithelium [37, 38], was a commonly reported ocular AE in the netarsudil US Phase 3 studies (netarsudil 0.02% QD; 5.4–25.5%) [28,29,30,31] and was reported to rarely impact vision and typically resolved upon discontinuation of medication [38]. Cornea verticillata in both cases in the study was confirmed to be resolved within 5 weeks of stopping the study treatment with no impacts on visual function including BCVA. Further clinical data would be needed to evaluate cornea verticillata in Japanese patients, since cornea verticillata findings have generally been reported only after 6 weeks or more of treatment in the clinical studies in the US. Other ocular AEs were generally observed at a similar rate between the two groups without statistically significant differences. There were no other findings in the study which constituted a safety concern for netarsudil.
Overall, the safety profile of netarsudil in our study for the Japanese population was consistent with observations from the Phase 2 study [1] in Japan and the Phase 3 studies [28,29,30,31] in the US, with no new safety issues reported. It needs to be noted that the 28-day study duration did not provide information on the long-term efficacy and safety of netarsudil. Another limitation was the single-masked design necessitated by differences in the containers for netarsudil and ripasudil ophthalmic solutions; however, measures were taken as described to minimize bias.
Conclusion
Netarsudil 0.02% was superior to ripasudil 0.4% in the reduction of IOP following 4 weeks of treatment, and was generally well-tolerated and showed a favorable safety profile in Japanese subjects. Netarsudil ophthalmic solution 0.02% QD could provide an important new treatment for patients with POAG or OHT with its clinically significant IOP reduction as a second-generation ROCK inhibitor.
References
Araie M, Sugiyama K, Aso K, et al. Phase 2 randomized clinical study of netarsudil ophthalmic solution in Japanese patients with primary open-angle glaucoma or ocular hypertension. Adv Ther. 2021;38(4):1757–75.
Tham YC, Li X, Wong TY, Quigley HA, Aung T, Cheng CY. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–90.
The AGIS Investigators. The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol. 2000;130:429–40.
Heijl A, Leske MC, Bengtsson B, et al. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch Ophthalmol. 2002;120:1268–79.
Kass MA, Heuer DK, Higginbotham EJ, et al. The ocular hypertension treatment study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:701–13 (Discussion 829–30).
Kass MA, Gordon MO, Gao F, et al. Delaying treatment of ocular hypertension: the ocular hypertension treatment study. Arch Ophthalmol. 2010;128:276–87.
Collaborative Normal-Tension Glaucoma Study Group. The effectiveness of intraocular pressure reduction in the treatment of normal-tension glaucoma. Am J Ophthalmol. 1998;126:498–505.
Bucolo C, Salomone S, Drago F, Reibaldi M, Longo A, Uva MG. Pharmacological management of ocular hypertension: current approaches and future prospective. Curr Opin Pharmacol. 2013;13:50–5.
Sambhara D, Aref AA. Glaucoma management: relative value and place in therapy of available drug treatments. Ther Adv Chronic Dis. 2014;5:30–43.
Chen J, Runyan SA, Robinson MR. Novel ocular antihypertensive compounds in clinical trials. Clin Ophthalmol. 2011;5:667–77.
Kopczynski CC, Epstein DL. Emerging trabecular outflow drugs. J Ocul Pharmacol Ther. 2014;30:85–7.
Tokushige H, Inatani M, Nemoto S, et al. Effects of topical administration of Y-39983, a selective rho-associated protein kinase inhibitor, on ocular tissues in rabbits and monkeys. Invest Ophthalmol Vis Sci. 2007;48:3216–22.
Wang RF, Williamson JE, Kopczynski C, Serle JB. Effect of 0.04% AR-13324, a ROCK and norepinephrine transporter inhibitor, on aqueous humor dynamics in normotensive monkey eyes. J Glaucoma. 2015;24:51–4.
Rao PV, Pattabiraman PP, Kopczynski C. Role of the Rho GTPase/Rho kinase signaling pathway in pathogenesis and treatment of glaucoma: bench to bedside research. Exp Eye Res. 2017;158:23–32.
Tanihara H, Inoue T, Yamamoto T, et al. One-year clinical evaluation of 0.4% ripasudil (K-115) in patients with open-angle glaucoma and ocular hypertension. Acta Ophthalmol. 2016;94:e26–34.
Saito H, Kagami S, Mishima K, Mataki N, Fukushima A, Araie M. Long-term side effects including blepharitis leading to discontinuation of ripasudil. J Glaucoma. 2019;28:289–93.
Tanihara H, Kakuda T, Sano T, Kanno T, Gunji R. Safety and efficacy of ripasudil in Japanese patients with glaucoma or ocular hypertension: 12-month interim analysis of ROCK-J, a post-marketing surveillance study. BMC Ophthalmol. 2020;20(1):275.
Maruyama Y, Ikeda Y, Mori K, et al. Safety and efficacy of long-term ripasudil 0.4% instillation for the reduction of intraocular pressure in Japanese open-angle glaucoma patients. J Ocul Pharmacol Ther. 2020;36(4):229–33.
Tanihara H, Kakuda T, Sano T, Kanno T, Kurihara Y. Long-term intraocular pressure-lowering effects and adverse events of ripasudil in patients with glaucoma or ocular hypertension over 24 months. Adv Ther. 2022;39(4):1659–77.
Li G, Mukherjee D, Navarro I, et al. Visualization of conventional outflow tissue responses to netarsudil in living mouse eyes. Eur J Pharmacol. 2016;787:20–31.
Lin CW, Sherman B, Moore LA, et al. Discovery and preclinical development of netarsudil, a novel ocular hypotensive agent for the treatment of glaucoma. J Ocul Pharmacol Ther. 2018;34:40–51.
Ren R, Li G, Le TD, Kopczynski C, Stamer WD, Gong H. Netarsudil increases outflow facility in human eyes through multiple mechanisms. Invest Ophthalmol Vis Sci. 2016;57:6197–209.
Kazemi A, McLaren JW, Kopczynski CC, Heah TG, Novack GD, Sit AJ. The effects of netarsudil ophthalmic solution on aqueous humor dynamics in a randomized study in humans. J Ocul Pharmacol Ther. 2018;34:380–6.
Kiel JW, Kopczynski C. Effect of AR-13324 on episcleral venous pressure in Dutch Belted rabbits. J Ocul Pharmacol Ther. 2015;31:146–51.
Kitaoka Y, Sase K, Tsukahara C, et al. Axonal protection by netarsudil, a ROCK inhibitor, is linked to an AMPK-autophagy pathway in TNF-induced optic nerve degeneration. Invest Ophthalmol Vis Sci. 2022;63(1):4.
Hoy SM. Netarsudil ophthalmic solution 0.02%: first global approval. Drugs. 2018;78:389–96.
Peace JH, McKee HJ, Kopczynski CC. A randomized, phase 2 study of 24-h efficacy and tolerability of netarsudil in ocular hypertension and open-angle glaucoma. Ophthalmol Ther. 2021;10(1):89–100.
Singh IP, Fechtner RD, Myers JS, et al. Pooled efficacy and safety profile of netarsudil ophthalmic solution 0.02% in patients with open-angle glaucoma or ocular hypertension. J Glaucoma. 2020;29(10):878–84.
Kahook MY, Serle JB, Mah FS, et al. Long-term safety and ocular hypotensive efficacy evaluation of netarsudil ophthalmic solution: rho kinase elevated IOP treatment trial (ROCKET-2). Am J Ophthalmol. 2019;200:130–7.
Khouri AS, Serle JB, Bacharach J, et al. Once-daily netarsudil versus twice-daily timolol in patients with elevated intraocular pressure: the randomized phase 3 ROCKET-4 study. Am J Ophthalmol. 2019;204:97–104.
Serle JB, Katz LJ, McLaurin E, et al. Two phase 3 clinical trials comparing the safety and efficacy of netarsudil to timolol in patients with elevated intraocular pressure: rho kinase elevated IOP treatment trial 1 and 2 (ROCKET-1 and ROCKET-2). Am J Ophthalmol. 2018;186:116–27.
Bacharach J, Dubiner HB, Levy B, Kopczynski CC, Novack GD, AR-13324-CS202 Study Group. Double-masked, randomized, dose-response study of AR-13324 versus latanoprost in patients with elevated intraocular pressure. Ophthalmology. 2015;122:302–7.
Iwase A, Suzuki Y, Araie M, et al. The prevalence of primary open-angle glaucoma in Japanese: the Tajimi Study. Ophthalmology. 2004;111:1641–8.
Yamamoto T, Sawaguchi S, Iwase A, et al. Primary open-angle glaucoma in a population associated with high prevalence of primary angle-closure glaucoma: the Kumejima Study. Ophthalmology. 2014;121:1558–65.
Tanihara H, Inoue T, Yamamoto T, Kuwayama Y, Abe H, Araie M, K-115 Clinical Study Group. Phase 2 randomized clinical study of a Rho kinase inhibitor, K-115, in primary open-angle glaucoma and ocular hypertension. Am J Ophthalmol. 2013;156(4):731–6.
Watabe H, Abe S, Yoshitomi T. Effects of Rho-associated protein kinase inhibitors Y-27632 and Y-39983 on isolated rabbit ciliary arteries. Jpn J Ophthalmol. 2011;55:411–7.
Mantyjarvi M, Tuppurainen K, Ikäheimo K. Ocular side effects of amiodarone. Surv Ophthalmol. 1998;42:360–6.
Hollander DA, Aldave AJ. Drug-induced corneal complications. Curr Opin Ophthalmol. 2004;15:541–8.
Acknowledgements
The authors thank the participants of the study.
Funding
This study, including the Rapid Service Fee and the Open Access Fee, was sponsored by Aerie Pharmaceuticals Ireland Limited.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Kenji Aso, Koji Kanemoto, and Ryo Iwata contributed to concept and design of the clinical study and analysis and interpretation of the data; drafted and revised the manuscript; approved the final version to be published; and agreed to be accountable for all aspects of the manuscript. Makoto Araie, Kazuhisa Sugiyama, David A. Hollander, Michelle Senchyna, and Casey C. Kopczynski contributed to concept and design of the clinical study and interpretation of the data; revised the manuscript; approved the final version to be published; and agreed to be accountable for all aspects of the manuscript.
List of Investigators
Sakae Matsuzaki (Tokyo), Misato Adachi (Tokyo), Motohiro Kiyosawa (Tokyo), Setsuko Hashida (Tokyo), Yoshiyuki Ichihashi (Tokyo), Takuji Kato (Tokyo), Isao Sato (Tokyo), Kayo Kure (Chiba), Takuya Kikuchi (Kanagawa), Norihiko Honda (Kanagawa), Naoki Hamada (Saitama), Yuko Shibuya (Saitama), Toru Nakajima (Shizuoka), Masahito Imai (Yamanashi), Junji Ono (Shizuoka), Tomoyuki Muramatsu (Shizuoka), Yuichi Inamoto (Osaka), Torao Sugiura (Osaka), Mitsuko Sugao (Osaka), Hidetaka Maeda (Osaka), Yasutaka Kubota (Osaka), Kazuko Hijikuro (Hyogo), Mikki Arai (Fukuoka), Hiroko Ueda (Tokyo), Kiyoshi Shimizu (Saitama), Kazukuni Kakinoki (Tokyo), Fumihide Abe (Oita).
Prior Presentation
The results of this study were presented at the 126th Annual Meeting of Japanese Ophthalmological Society held from Apr 14 to Apr 17, 2022 in Osaka, Japan; presentation O1-044.
Disclosures
Makoto Araie is a consultant for Aerie and has received fees from Heidelberg Engineering, Alcon, Pfizer, Santen, Topcon Medical Systems, Otsuka, Senju, and Kowa, and holds patents/royalties with Topcon Medical Systems. Kazuhisa Sugiyama is a consultant for Aerie and has received lecture/manuscript fees from Otsuka, Santen, Senju, Viatris, Kowa, Novartis, Bayer, and Inami. Ryo Iwata, Casey Kopczynski, Michelle Senchyna are the employees of and stockholders in Aerie Pharmaceuticals (now Alcon). Kenji Aso (Sun Pharma Japan Ltd.), Koji Kanemoto (Alexion Pharma GK), and David A Hollander (Revance Therapeutics, Inc) were employees of Aerie Pharmaceuticals during the conduct of the study.
Compliance with Ethics Guidelines
The study was approved by appropriate ethics committees. See Supplementary Materials for Institutional Review Board (IRB) information. The study was conducted in accordance with ethical principles based on the Declaration of Helsinki and the guidance stipulated in Article 14, Paragraph 3, and Article 80–2 of the Pharmaceuticals, Medical Devices and Other Therapeutic Products Act of Japan, MHLW Ordinance on Good Clinical Practice (MHLW Ordinance No 28 [March 27, 1997]), International Council for Harmonization Guideline E6 (R2), the study protocol, and the standard operating procedures. All patients provided the written informed consent prior to their participation into the study.
Data Availability
The analysis results of the current study are available in the ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT04620135.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Araie, M., Sugiyama, K., Aso, K. et al. Phase 3 Clinical Trial Comparing the Safety and Efficacy of Netarsudil to Ripasudil in Patients with Primary Open-Angle Glaucoma or Ocular Hypertension: Japan Rho Kinase Elevated Intraocular Pressure Treatment Trial (J-ROCKET). Adv Ther 40, 4639–4656 (2023). https://doi.org/10.1007/s12325-023-02550-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02550-w