
Abstract
With the increasing availability of biosimilars, the practice of switching therapies for non-medical reasons between an originator biologic and an analogous biosimilar has become more common. The evidence to support this practice mostly comes from single-switch randomized controlled trials (RCTs) and real-world (RW) evidence studies. However, as more biosimilars of the same originator enter the market, multiple switching events between originators and biosimilars is becoming a reality, despite limited evidence to support the efficacy and safety of such practice. Some countries have established guidelines, policies, or laws related to interchangeability and/or automatic substitution, whereas others have left these practices unregulated or controlled by other components of the healthcare system. Collectively, guidelines on single non-medical switching are often vague, with even less focus given to multiple non-medical switching, leaving this practice mostly unregulated. This narrative review will first discuss the current regulatory perspectives on non-medical switching and challenges associated with switching therapies, particularly with the availability of multiple biosimilars. We will then review the current evidence from RCTs and RW studies in the light of three different multiple-switch scenarios currently taking place in clinical practice: switching between an originator and a single biosimilar, switching between biosimilars of the same originator, and the clinical practice of switching back to the originator (i.e., switchbacks) after a failure of the initial non-medical switch to the analogous biosimilar.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The current evidence on the safety, efficacy, and immunogenicity of switching multiple times from an originator to the analogous biosimilar or from a biosimilar to another biosimilar is sparse, and comes from a limited number of randomized-controlled trials and real-world evidence studies. |
More robust, well-designed, long-term studies are needed to investigate the consequences of multiple or biosimilar-to-biosimilar switching. |
Any decision to switch therapies (single or multiple times) should be based on clinical judgement and made jointly between the patient and the treating physician. |
Introduction
The availability of biosimilars has increased the practice of switching between originator biologics and analogous biosimilars for non-medical reasons in the treatment of immune-mediated inflammatory diseases [1,2,3]. In clinical practice, non-medical switching, defined as switching therapy for economic or other reasons not related to patient care, may include both switching from an originator to the analogous biosimilar and vice versa. Furthermore, as more biosimilars become available, switching between biosimilars of the same originator is a possibility. After the initial non-medical switch, changes in pharmaceutical pricing or administrative/reimbursement policies may trigger subsequent switches, leading to a complex multiple-switching environment [1]. A multiple switch can also occur when the initial non-medical switch from an originator to its biosimilar results in worsening of disease or tolerability issues, at which point the patient may be switched back to the originator for medical reasons [4]. In many countries, biologics are included as part of tenders and, with physicians unable to opt out, mandated treatment switches are likely to increase; in some cases, switching currently occurs as often as every 4 months [5]. Multiple switching for economic reasons has already occurred in Hungary, Sweden, and Norway [6,7,8], while in countries such as Australia where automatic substitution of certain biologics is allowed, patients can switch products as often as every month [9, 10]. Similar policies may also be introduced in other countries, e.g., in Germany, a new law—the Act for Greater Security in the Pharmaceutical Supply System—went into effect in 2019 and could provide a list of products approved for substitution by 2022, allowing pharmacists to substitute biologics with biosimilars (the framework and conditions for the new law are still being resolved). Overall, these policies are likely to increase the incidence of non-medical and multiple switching [11].
From a global perspective, although multiple switching between originator products and biosimilars already occurs, it is notable that both the quality and quantity of the evidence supporting this practice are limited. The majority of the non-medical switching studies conducted to date [randomized controlled trials (RCTs) and real-world (RW) studies] have assessed the safety and efficacy of single switches, not multiple, between the originator and one or more of its biosimilars. In addition, these studies do not meet the criteria for a robust, non-medical switching trial [1], and the results from these studies and systematic reviews of the data [2, 12] vary in their conclusions regarding the effect of multiple switching on efficacy and safety. Also, it is important when assessing the evidence to recognize that data from these trials should not be generalized to other originator–biosimilar combinations or to switching between biosimilars of the same originator [13]. Thus, the effect of multiple switching on efficacy, safety, and immunogenicity, as well as other issues (e.g., pharmacovigilance), remains largely unknown [13].
The first objective of this narrative review is to discuss the current regulatory policies and non-medical switching practices, as well as discuss the evolving treatment landscape with multiple biosimilars. The second objective is to review the challenges and potential risks involved with multiple switching based on three different switching scenarios that already occur in clinical practice: multiple switching between an originator and a single biosimilar, switching between two or more biosimilars, and the clinical practice of switching back to the originator (i.e., switchbacks) after a failed non-medical switch to its biosimilar.
Literature Search
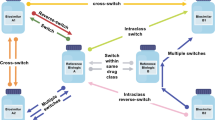
A literature search of databases, including Embase® and MEDLINE, was performed to identify multiple-switching studies. The search was limited to English language studies in human participants with publication dates from January 1, 2012, to February 17, 2020. Included studies involved one of the following three switching scenarios: multiple switching between originator and a single biosimilar, biosimilar-to-biosimilar switching, and switchbacks. Multiple switching was defined as > 1 switch between an originator and a single biosimilar for non-medical reasons (Fig. 1a). Biosimilar-to-biosimilar switching was defined as switching treatment between ≥ 2 different biosimilars of the same originator (Fig. 1b). Switchback was defined as switching treatment back to the originator for medical reasons after a failure of the initial non-medical switch from the originator to the analogous biosimilar (Fig. 1c).

Multiple non-medical switching scenarios: a multiple switching between originator and a single biosimilar, b biosimilar-to-biosimilar switching, and c switchbacks. Dashed line indicates the potential for ongoing switching between originator and single biosimilar. Originator product = blue (dot pattern; a, c); biosimilar products = orange (with vertical line pattern; a, b, c), and green (with horizontal line pattern; b). aMedical switch
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Regulatory Perspectives on Non-medical Switching
Specific guidelines related to biosimilars and/or interchangeability have been developed by regulatory authorities, such as the European Medicines Agency (EMA), the US Food and Drug Administration (FDA), and Health Canada [1, 14,15,16,17].
The EMA does not include recommendations on interchangeability, and the decision-making authority related to substitution policies (including automatic substitution) rests within the EU member states [15]. According to an information guide prepared jointly by the EMA and the European Commission, interchangeability refers to the practice of changing one medicine for another that is expected to have the same clinical effect. This includes switching an originator product with the analogous biosimilar (or vice versa) or switching a biosimilar with another biosimilar, as well as automatic substitution [18]. Switching is carried out by the physician; automatic substitution may be carried out by a pharmacist (e.g., substitution for a generic) without the need for physician consultation [18].
Unlike the EMA, the FDA grants a designation of interchangeability separately from biosimilarity [19]. To support a demonstration of interchangeability, the FDA guidelines note that the product must first show biosimilarity to the originator product, as defined in the Biologics Price Competition and Innovation Act of 2009 [19]. The biosimilar product must also show that it can be expected to produce the same clinical result as the originator product in any given patient, and, for those biologics administered more than once, that the risk in terms of safety or diminished efficacy of alternating or switching between the originator and the biosimilar is not greater than the risk of using the originator product without such alternation or switch [19]. Furthermore, to demonstrate interchangeability, a switching study should employ a study design that alternates 1 treatment arm ≥ 2 times (i.e., ≥ 3 switches) between the originator and biosimilar product (i.e., to show the effect of multiple switching) while the other arm includes only the originator product (non-switching arm) [19]. Importantly, guidance from the FDA notes that multiple exposures in a switching study may potentially prime the immune system to recognize subtle structural differences between the 2 products, leading to an overall increased immune response [19]. Once a product is deemed interchangeable by the FDA, it can be substituted for the originator without intervention by the physician [19, 20]. Currently, no biosimilar has been granted interchangeability by the FDA [21]. However, even without a formal interchangeability designation, patients can be switched for non-medical reasons, and it is up to each state to determine policies on automatic substitution [19, 20].
Like the FDA, Health Canada’s authorization of interchangeability is independent of biosimilarity [17]. In Canada, the term interchangeability usually refers to the pharmacist’s ability to switch a patient from one drug to another equivalent drug without intervention from the prescribing physician. Health Canada’s authorization of a biosimilar is not a declaration of interchangeability, and the authority to declare two products interchangeable ultimately rests with each province and territory [17].
Globally, some countries have established guidelines, policies, or laws related to interchangeability and/or automatic substitution; in other countries, these practices remain unregulated or have been established by other components of the healthcare system [5, 10, 22,23,24]. Collectively, guidelines on non-medical switching are often vague, and even less focus has been given to multiple switching for non-medical reasons, leaving this practice mostly unregulated.
Challenges with Multiple Switching
Although biosimilar products are deemed similar, regulatory agencies acknowledge that they are not structurally identical to the originator [14, 15]. Structural variations may arise from differences in post-translational modifications (such as glycosylation) or higher-order structure (such as protein folding) [14, 25]. When patients are switched from one product to another once or multiple times, these differences in the drug substance (i.e., structure) or the drug product (including process- or product-related impurities) raise the potential for negative outcomes, such as loss of efficacy (including by increasing the risk of immunogenicity) [26] and the emergence or worsening of adverse events (AEs). To better ensure the quality of biosimilars, the International Psoriasis Council suggests guidelines to standardize preclinical analytical assessments determining similarity between a biosimilar and its originator [27]. Of note, manufacturing changes can also lead to relevant quality differences within different batches of the originator product [28]; however, discussing these are beyond the scope for this paper. Also, it is important to note that, although scientific principles for biosimilarity comparability assessments are based on those that assess manufacturing changes for already approved biologics, both regulatory pathways are distinct and should not be confused or used in lieu of the other [14, 15].
Other potential challenges with multiple switching between products include complexity of pharmacovigilance (tracking and tracing) and patient-related challenges owing to differences in delivery devices, presentation, drug formulation, or dosage, and the availability and quality of patient-support programs. Because of these challenges, most global, regional, and national (including US) medical societies urge caution when considering non-medical switching, recommending that any decision to switch be made with the knowledge and consent of the prescribing physician and the patient [29,30,31,32,33,34]. Those same medical and scientific societies may not be in favor of [32,33,34,35] or specifically express opposition to [29,30,31, 36,37,38] multiple switching. We will next look at these challenges in more detail.
Treatment Failures and Discontinuations
Treatment failure, particularly at the individual patient level, is a concern following a non-medical switch, and discontinuation rates ranging from 0 to 87% have been reported in single-switch RW studies [1, 39,40,41]. One reason for treatment failures after switching is tolerability issues and the emergence of new or worsening AEs, potentially caused by unanticipated differences in safety between the 2 products [39, 41, 42]. Treatment failures can also occur due to loss of efficacy when the biosimilar is not clinically functioning the same way as the originator product, potentially because of subtle drug- or product-related differences. Most importantly, these effects may be amplified in the context of multiple switches. Of note, it is important to clarify that, when a patient’s disease is controlled, there is generally no expectation for improvement when switching from an originator to a biosimilar; however, based upon the current evidence, switching for non-medical reasons may carry risks such as treatment failure/discontinuation of therapy [1, 39, 40, 42]. Overall, considering the large variation in discontinuation rates across the current single-switch studies, and because none of the studies meet the minimum requirements for a robust switching study, the evidence on the occurrence of treatment failures following single or multiple non-medical switches remains inconclusive [1].
Immunogenicity Concerns
Immunogenicity, mostly defined by the development of anti-drug antibodies (ADAs), is another concern when switching therapies because of a possible immune response against the different antigens (not detected during or after biosimilar development) that may be present across products, both at the drug substance and at the drug product level [43,44,45,46]. Although ADAs can be transient and without consequences, the persistence of ADAs can have negative effects, including loss of response, type I and type III hypersensitivity, worsening of disease, increased drug toxicity, and tolerability issues, such as injection-site or infusion reactions [42, 43, 47]. Several challenges and gaps remain in the assessment of immunogenicity [48]. In vitro testing and animal studies cannot fully predict immune response in humans, and empiric data from clinical trials are needed to demonstrate whether ADAs are altering the pharmacokinetic and pharmacodynamic properties of a product, thereby affecting clinical outcome, including possible treatment failure [48,49,50,51,52]. An equally important consideration is the variability and heterogeneity present in ADA detection, which should be taken into account when developing ADA assays [50]. Although current RCTs show no difference in immunogenicity after a single-switch from originator to its biosimilar [53,54,55,56], the patient populations in these studies were limited and post-switch follow-up durations were short, suggesting that the studies may not be robust enough to reveal conclusive immunogenicity profiles. In RW studies, 2–14% of patients have discontinued therapy due to “ADAs” or “ADAs and inefficacy” after a single non-medical switch [41, 57,58,59,60,61]. However, because most RW studies conducted to date either do not report immunogenicity data or give no specific reasons for discontinuation (including whether loss of efficacy or AEs that led to discontinuation were ADA driven), the data are currently inconclusive. Thus, the concern of immunogenicity exists and is amplified when a switch is carried out multiple times between products [57]. Longer-term data from studies with ≥ 1 year follow-up are needed to assess immunogenicity that may not be detected for some time following drug administration or treatment switches [1, 14, 62, 63].
The Nocebo Effect
Treatment failures following a non-medical switch can also occur because of the nocebo effect, defined as a negative outcome or failure of treatment resulting from a patient’s negative expectations [41, 64,65,66]. Nocebo response is a problem in both clinical trials and real-life practice. In clinical switching trials, nocebo response may affect both trial outcomes and interpretation, while in clinical practice, nocebo response could lead to higher relapse rates, which have the potential to increase healthcare costs and the number of additional therapies needed to manage individual patients. Although the nocebo response is a well-characterized phenomenon [64,65,66], the current evidence regarding a role in treatment failure following a non-medical switch between biologics and their biosimilars is limited and based on studies that lack adequate design or do not collect all the necessary data to assess it properly [41]. Despite these limitations, a few studies have suggested that some treatment failures following a non-medical switch can be attributed to the nocebo response [7, 67,68,69]. In contrast, a retrospective study that included a blinded control group did not find any evidence of a nocebo response following a non-medical switch [70]. However, some limitations must be noted with this study; namely that no direct comparison of data for patients who knew about the switch (n = 24) versus those who did not (n = 60) was reported [70]. Thus, the frequency and impact of nocebo response following a non-medical switch remains inconclusive.
Pharmacovigilance Concerns
Subsequent to drug approval, pharmacovigilance systems continuously assess the risk–benefit profile of an agent to detect new safety signals and to minimize risks [71, 72]. Switching between an originator and biosimilars, especially multiple times and sometimes at intervals of only a few weeks, has the potential to complicate the accurate assessment and traceability of AEs. One reason for this is the delay that may occur between starting a biologic drug and the occurrence and identification of AEs, due to the relatively long latency in the development of some of these adverse reactions, including those related to immunogenicity [72,73,74,75]. Traceability is likely to get more complicated when automatic substitution and non-medical switching become more widely used. Pharmacy-level substitution of originators/biosimilars is already possible in some EU countries (Czech Republic, Latvia, Turkey, Poland, Serbia, and Estonia) and in Australia, although physicians can opt out from the practice in each country. In addition, non-medical switching is allowed in 12 EU countries without the treating physician’s consent [5, 9, 10]. To add complexity, biosimilars are mostly given the same non-proprietary name as their originator product, and the lack of a unique identifier may lead to AEs being inappropriately attributed to the wrong product [71, 72]. One potential solution is to generate unique identifiers, or qualifiers, for the originator and corresponding biosimilars to distinguish them from one another [72, 76]. To monitor AEs and to attribute them to the correct product, improved pharmacovigilance efforts are still largely needed in the era of biosimilars, although some advances have been made [77]. Accordingly, a number of medical societies have proposed requirements for postmarketing surveillance, registries, and traceability [30, 32, 33, 78, 79].
Other Patient-Related Concerns
Several additional concerns are relevant when patients are switched for non-medical reasons, including the availability and quality of patient-support programs, which may differ between originator and biosimilars or not be available at all with some products. Differences in patient-support programs can affect how patients are educated on the use and acceptance of the biologic; it is suggested that patient-support programs from the biosimilar should be just as good (i.e., in terms of patient offerings, drug-use training, disease education, etc.) as those programs offered by the originator product [80]. Switching patients once or multiple times between therapies can also lead to concerns related to product delivery devices and injection-site reactions because the biosimilar may not use the same delivery device, formulation, dosage, or dosing interval as the originator [81,82,83,84]. Differences in formulation may also influence patient satisfaction and adherence [81]. Of note, fewer injection-site reactions were reported with biosimilar etanercept SB4 (3.7%) compared with the originator etanercept (17.2%) in a phase 3 RCT in biologic-naive patients with rheumatoid arthritis [85]. The authors concluded that the only difference in drug formulation between the products was the absence of l-arginine in SB4 [85]. Although they noted that l-arginine has not been associated with an increased risk of injection-site reactions, the authors could not exclude this difference in formulation as the cause of injection-site reactions [85]. Issues related to product delivery devices can also lead to increased healthcare utilization if patients need to be reeducated after each switch or critical handling errors when patients are not trained in the use of or confuse the different devices [81]. Such issues can also reduce treatment adherence and thereby compromise disease control and increase the toll on healthcare services [81, 83].
Multiple Switching Between Originator and a Single Biosimilar (Scenario 1)
As previously described, the act of non-medical switching from an originator product to a biosimilar or vice versa is mandated in multiple jurisdictions in select countries driven by economic reasons [6, 7, 67]. This provides the framework whereby constant fluctuations in pharmaceutical pricing and changes in reimbursement policies can lead to patients being switched multiple times between an originator and the analogous biosimilar (Fig. 1a). This has already happened in the EU, and data from two RW multiple-switch observational studies in clinical settings are available [6, 7]. In Hungary, biosimilar infliximab was mandated in 2014 to patients naive to tumor necrosis factor antagonists or those with a ≥ 1-year drug holiday. However, all patients underwent a mandated reverse switch from the biosimilar to the originator in 2017 because of changes in the National Health Insurance Fund policy [6]. In Sweden, patients who were switched from the etanercept originator to its biosimilar in 2016 were later switched back to the originator based upon pricing considerations [7]. Although in both of these studies efficacy was maintained after the switches, these studies evaluated only short-term outcomes and included small numbers of patients or patients who had been on drug holiday for an extended time before the first switch (Table 1) [6, 7].
Often, non-medical switching policies have been implemented in clinical practice based on a few single-switch studies concluding that switching from a tumor necrosis factor inhibitor originator to a biosimilar was safe and efficacious [54,55,56]. However, to date, only a few RCTs have assessed the safety and efficacy of multiple non-medical switches (Table 1) [86,87,88,89]. Importantly, none of these studies meet the criteria for a robust switching trial as defined by the FDA [1, 19].
One such study (ADACCESS) compared the efficacy and safety of adalimumab originator with its biosimilar, GP2017, in patients with moderate-to-severe plaque psoriasis [86]. Patients were initially randomized to receive either GP2017 or the originator adalimumab for 17 weeks, after which patients were sequentially switched 4 times between the 2 products (switch groups) or continued the originator or GP2017 (non-switch groups) for an additional 34 weeks. The study showed similar efficacy (defined by a Psoriasis Area and Severity Index 75 response), immunogenicity, and safety in the switch groups compared with the non-switch groups over time. However, although the study was powered to assess biosimilarity, it was not powered to assess the effect of treatment switching; therefore, no meaningful insights on the effect of multiple switches can be extrapolated from this study [86]. Another limitation of the study was the relatively short follow-up duration (16 weeks) after the last switch.
Another study performed in patients with moderate-to-severe rheumatoid arthritis compared the efficacy and safety of originator adalimumab with the biosimilar FKB327 [87]. Patients were initially randomized to receive either the originator or FKB327, rerandomized either to switch therapies or continue treatment during the first part of an open-label extension phase, and randomized again to receive FKB327 during the second part of the open-label extension [87]. As a result, one group experienced a multiple switch, from FKB327 to the originator adalimumab and back to FKB327; 2 groups experienced a single switch; and 1 group received FKB327 throughout the study. An interim analysis at week 30 showed that safety and efficacy profiles were similar for all treatment groups; however, this study was not adequately powered to show statistical differences between treatments [87]. Limitations included that this was an open-label extension study and no originator–biosimilar–originator switch or continuous–originator treatment groups were included for comparison. Definitive interpretation of the findings from this study is pending publication of the final results.
A third multiple-switching study is the EGALITY trial that evaluated multiple switching between originator etanercept and the biosimilar GP2015 in patients with moderate-to-severe chronic plaque-type psoriasis [88, 89]. Patients were initially randomized either to receive the etanercept originator or GP2015 for 12 weeks, then rerandomized to continue the same treatment or undergo 3 treatment switches during the 18-week treatment period 2, followed by a 22-week extension period [88]. A prespecified analysis was performed to evaluate the effect of multiple switches compared with continued treatment in pooled treatment arms, and the study showed no difference in efficacy in the pooled switch and non-switch groups [89]. However, limitations of the study included a short follow-up duration after the last switch and pooling of the study data for the switch and non-switch groups, complicating the interpretation of the efficacy and safety results [89].
As mentioned above, immunogenicity is another concern that should be evaluated, because single or multiple switching could trigger an immune response to different antigenic determinants that might be present between an originator product and analogous biosimilars [43,44,45,46]. Only one of the two RW studies evaluated immunogenicity, and it was limited to 16 weeks and assessment of 14 patients (Table 1) [6]. In contrast, all three RCTs assessed the development of ADAs (Table 1). In the ADACCESS study, 39% and 47% of patients in the switch groups developed ADAs versus 36% and 45% in the non-switch groups by week 51, of which 100%, 75%, 86%, and 85%, respectively, were neutralizing ADAs [86]. In the FKB327 biosimilar study, 54% of patients in the multiple-switch group (from biosimilar to originator adalimumab back to biosimilar) had ADAs at week 78 compared with 51% in the treatment continuation group, and 43% and 48% in the single-switch groups; the authors concluded that long-term immunogenicity was comparable between the products but provided no statistical comparison [90]. In the 52-week EGALITY study, 1.9% (5/267) of patients in the group that continued treatment with originator etanercept had confirmed positive non-neutralizing ADAs within the first 4 weeks of treatment, and an additional patient (1/90) in the group that switched from originator etanercept was positive for non-neutralizing ADAs at week 36; no other confirmed ADAs were observed, and all respective patients with previous positive ADAs were ADA-negative at all subsequent visits [88]. It is worth noting, however, that etanercept has historically presented with a low rate of immunogenicity [91, 92], which raises questions regarding the relevance of the reported ADA rates to clinical practice. Overall, the follow-up duration in these studies varied and might not have been long enough to detect immune reactions or all AEs, which can have implications on pharmacovigilance as well [73].
Overall, the evidence regarding the efficacy, safety, and immunogenicity of multiple switching from an originator to a single biosimilar is currently based on two RW studies [6, 7] and three multiple-switch RCT studies, none of which were powered to show a difference between switchers and non-switchers and were limited in their follow-up durations after the last switch [86,87,88,89]. Furthermore, to our knowledge, no study to date has investigated the potential implications of the differences in patient-support programs, devices, or drug formulations in a single- or multiple-switch scenario. Thus, until more robust, longer-term RCTs, supported by well-designed RW studies, are conducted, any decision to switch, especially multiple times, should be made by the treating physician based on clinical judgment and with informed patient consent.
Switching from Biosimilar to Biosimilar (Scenario 2)
With an array of biosimilars entering the market, the prospect of single or multiple switches between biosimilars of the same originator is already a reality that is likely to increase in the future (Fig. 1b) [1, 93,94,95,96,97,98]. However, comparability studies for product registration are performed between a biosimilar and its originator product and not between biosimilars, which raises the potential for broader differences between biosimilars of the same originator product [1, 99, 100]. As such, for any given comparison, each biosimilar could be on different portions of the similarity or equivalence margin as defined for the specific originator product and, at least theoretically, there is a chance that the products would not meet a comparability standard if both biosimilars were compared head-to-head (Fig. 2) [101]. For this reason, data from switching studies with an originator and the analogous biosimilar are unique to those particular products and should not be generalized to other switching scenarios between the originator and its biosimilars or between different biosimilars of the same originator. Importantly, the FDA guidelines clearly stipulate that interchangeability is defined only against the originator product and not against another biosimilar of the same reference product [19]. Therefore, the similarity between two biosimilars of the same originator product is currently unknown.

Equivalence margin between a reference product (i.e., originator) and two different biosimilar products. aCastaneda-Hernandez et al. [101]
To our knowledge, only five RW studies have evaluated switching between two biosimilars of the same originator product on efficacy or safety, and one additional study has evaluated the immunogenic effect of such a switch (Table 1) [93,94,95,96,97,98]. The first study conducted in the United Kingdom assessed clinical outcomes in 133 patients with inflammatory bowel disease who agreed to transition from infliximab biosimilar, CT-P13 (mean treatment duration of 3 years), to infliximab biosimilar, SB2 [96]. The results demonstrated that disease activity did not significantly change after 16 or 18 weeks with SB2 nor were there any significant difference in drug persistence between a historical CT-P13 cohort and the SB2 switch group. Eighteen patients (14%) stopped treatment after the switch (therapeutic failure, n = 7; adverse drug reactions, n = 6; withdrew consent, n = 2; lost to follow-up, n = 2; withdrew for other reasons, n = 1). The limitations of this study include short follow-up duration, lack of a matched or adjusted control group of non-switchers for comparison of disease activity, and lack of immunogenicity or safety data.
The second study (PERFUSE) investigated the efficacy and safety of switching from CT-P13 to SB2 in patients with rheumatoid arthritis, psoriatic arthritis, or ankylosing spondylitis across 6 clinical practices in France [94]. The interim results demonstrated no clinically meaningful differences in disease activity from baseline to month 6 (n = 96) or 12 (n = 67) in patients previously treated with infliximab originator or CT-P13 (only pooled data were reported). SB2 persistence at 12 months ranged from 92 to 100%. Four serious AEs were reported (no details on treatment history were reported for these patients). Limitations of the study include a small patient population and pooling of the data.
The third study investigated the safety and efficacy of cross-switching from CT-P13 to SB2 in 24 patients with psoriasis in Italy [95]. The mean (SD) previous exposure to CT-P13 was 23.2 (7.5) months. No difference in efficacy was observed 6 months post-switch and only 4 AEs were reported. However, the lack of immunogenicity data and a control group, as well as the small patient population and short follow-up duration, are limitations of this study.
The fourth study, conducted in France, March–May 2018, reported retention rate for 18 patients switching from CT-P13 to SB2 [98]. Data were compared with 45 patients switching from originator infliximab to SB2 during the same time period at the same rheumatology department. The retention rate after switching from CT-P13 to SB2 was 12/18 (66.7%), suggesting that 6 (33.3%) patients discontinued after the switch. In comparison, the retention rate following a switch from originator infliximab to SB2 was 41/45 (91.1%) with a median follow-up of 34 weeks. No efficacy, safety or immunogenicity data were reported and the study was limited by a small patient population and short follow-up duration.
The fifth study was a descriptive analysis by the US Department of Veterans Affairs of outpatients receiving infliximab and switched to/from the originator or one of two biosimilars between January 2016 and January 2019 [93]. Of the 607 patients who switched from biosimilar to biosimilar, 138 (23%) patients had at least one AE, and 22 (4%) had at least one hospitalization. Among all single switchers, 335/1024 (33%) and 69/1024 (7%) patients had at least one AE or hospitalization compared with 172/666 (26%) and 38/666 (6%) multiple switchers, respectively, during the same time period. No details on disease, disease activity, discontinuation, or immunogenicity were reported.
The only study reporting immunogenicity data included 265 patients with chronic inflammatory diseases receiving maintenance treatment with infliximab in France [97]. The patients were switched to an initial infliximab biosimilar in October/December 2015; of these, 140 patients were switched to a second infliximab biosimilar starting in December 2017, 26 remained on the first biosimilar, and 55 switched back to the originator. At the end of the 3-year observation period, the biosimilar retention rate was 58% (154/265; 131 receiving the second biosimilar and 23 receiving the first biosimilar). During the study, 29 patients had ADAs at baseline; of the 236 patients without ADAs, 20 developed ADAs during the observation period, corresponding to an immunogenicity rate of 3/100 patient-years. Of these 20 patients, 4 were switched back to the originator at the time of ADA detection, 10 were exposed only to the first biosimilar, and 6 to the second. Although the risk of treatment discontinuation was significantly higher among patients who were ADA positive at baseline or during follow-up, a Kaplan–Meier analysis showed that immunogenicity was not influenced by the number of infliximab biosimilars received. However, as above, these results are based on a single study with no control group of matched or adjusted non-switchers, and data were pooled from multiple indications.
Because only six studies conducted to date have assessed the efficacy, safety, or immunogenicity of biosimilar-to-biosimilar switches and because none were powered to detect differences or investigated pharmacokinetic parameters, no definite conclusions can be drawn about the potential risks of such switches. Furthermore, concerns regarding pharmacovigilance, patient-support programs, and other patient-related concerns remain unknown. In cases in which ≥ 2 biosimilars are added into the scenario, these concerns are amplified because patients can be switched multiple times between several different products, and each switch may be associated with different delivery systems, drug dosages, dosing intervals, and formulations. Until more evidence is available, patients should not be forced to undergo a biosimilar-to-biosimilar switch, and patients and their physicians should be fully informed and allowed to opt out of such switches.
Switchback (Scenario 3)
As stated above, numerous studies have investigated the safety and efficacy of a single non-medical switch from an originator to a biosimilar [1, 39, 40]. Although many of these studies showed that such a switch is generally feasible and well-tolerated, 0–87% of patients discontinued therapy after the switch. Those patients for whom the switch fails and who discontinue treatment are sometimes allowed to switch back to the originator product, leading to a multiple-switch scenario. A meta-analysis of 62 RW studies (with almost half of the studies reporting switchback data) showed an annualized (95% confidence interval) switchback rate of 14% (10–17%) among all non-medical switchers and 62% (44–80%) among those who discontinue therapy, suggesting that switchback is occurring in a considerable number of patients who are initially switched from originator to a biosimilar for non-medical reasons [4]. However, no details were reported on the success of switchback in this analysis.
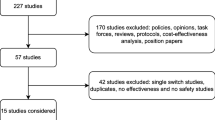
To assess this further, we identified 57 RW studies that reported switchback to the originator treatment after a non-medical switching failure (Table 2) [7, 58, 68,69,70, 93, 102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152]. The switchback rate varied widely between studies, with 1–72% of patients switching back after biosimilar failure either due to loss of efficacy, AEs, assumed nocebo effect, or subjective reasons. Only 24 studies reported outcomes following the switchback with the majority of patients (50–100%) regaining disease control following reinstitution of the originator treatment [58, 68, 70, 104, 107, 109, 115, 118,119,120, 123, 124, 128, 129, 133, 134, 138,139,140, 143,144,145,146, 148]. However, most of these studies do not provide an objective, blinded assessment of treatment failure after the switch or regaining response after switchback, and therefore it is important to note that there is a potential for bias in these studies.
In a few of these studies, the authors speculated that a nocebo effect was one explanation for treatment failure and the subsequent switchback success [7, 58, 67,68,69, 124]. However, this assumption may be unwarranted because none of the studies were adequately designed to test this hypothesis [41]. Furthermore, efforts to reduce the nocebo effect, such as patient education, have produced inconsistent results, and treatment failures have been observed even after patients were informed and educated before switching [41].
As mentioned previously, because patients may regain disease control after switching back to the originator, switchbacks can be considered a viable alternative compared with switching to another biologic within the same therapeutic class or a different drug class altogether (Table 2). However, it is important to note that all treatment failures and discontinuations carry a risk to the patient (including those resulting from the nocebo effect), and not all patients were successfully switched back to the originator treatment in these studies. Thus, treatment failure remains a concern.
Importantly, unlike the two other multiple-switching scenarios, switchbacks should be considered medical switches. Because the switchback is usually medically driven and sometimes requested by the patient after treatment failure, it is not necessarily associated with the same challenges as the other two scenarios (e.g., issues with patient support programs and other patient-related concerns). However, immunogenicity remains a key concern, as with any multiple-switch practice. Only a few of the switchback studies [58, 69, 109, 136, 141, 148] reported immunogenicity data before and after the initial switch, and the long-term consequences after switchback remain unknown (Table 2).
Pharmacovigilance concerns also exist, particularly in those cases in which switchbacks happen relatively quickly after the initial switch, making it difficult to distinguish to which product an AE should be attributed. Patient-reported problems, such as more pain or injection/infusion reactions, and issues with delivery devices, were reported in 12 studies following a non-medical switch, often leading to a switchback to the originator [102, 110, 111, 113,114,115, 121, 122, 128, 134, 140, 142]. However, this evidence is based on a limited number of RW studies not powered to investigate differences in injection/infusion reactions or issues with the delivery device after a switch, and it was not disclosed whether patients were adequately educated on the use of the new device.
Overall, the current evidence regarding switchbacks mainly comes from RW studies that were not robust enough or properly designed to investigate the risks of switchbacks. Considering the limitations of the current evidence, the practice of non-medical switching from originator to biosimilar (especially in patients who are doing well with the originator treatment) should be conducted with reservations, if at all, and should be jointly decided by the patient and the physician, with the patient having the option to refuse such a switch.
Conclusions
With the increasing availability of biosimilars, multiple switching between originator and its biosimilar, biosimilar-to-biosimilar switching, and switchbacks are becoming more widely used practices; however, the current evidence regarding the potential risks of these practices remains limited and is not robust enough to dispel all the concerns related to loss of efficacy, immunogenicity, and safety. Although no substantial safety/efficacy concerns have been reported to date at the population level, important concerns have been identified at the individual patient level (including loss of efficacy and emergence/worsening of AEs). Multiple switching and biosimilar-to-biosimilar switching carry the most concerns, and more studies are needed to fully evaluate the risks involved with such switching. More data are available for switching back to the originator after failure of a non-medical switch, and this practice is generally successful; however, not all patients regain disease control after switchback, and immunogenicity concerns remain. Because of this, it is important that regulators, policy-makers, and healthcare providers consider potential safety and efficacy concerns before making crucial treatment, regulatory, or policy decisions that involve or could lead to switching patients multiple times for non-medical reasons. Any decision to switch, whether single or multiple times, should be made by the treating physician based on clinical judgment and with informed patient consent. Until further scientific understanding is gained through robust RCTs, supported by well-designed RW studies, the potential short- and long-term risks of multiple switching, biosimilar-to-biosimilar switching, and switchbacks remain largely unknown.
References
Numan S, Faccin F. Non-medical switching from originator tumor necrosis factor inhibitors to their biosimilars: systematic review of randomized controlled trials and real-world studies. Adv Ther. 2018;35:1295–332.
Moots R, Azevedo V, Coindreau JL, et al. Switching between reference biologics and biosimilars for the treatment of rheumatology, gastroenterology, and dermatology inflammatory conditions: considerations for the clinician. Curr Rheumatol Rep. 2017;19:37.
McKinnon RA, Cook M, Liauw W, et al. Biosimilarity and interchangeability: principles and evidence: a systematic review. Biodrugs. 2018;32:27–52.
Yifei L, Skup M, Yang M, Qi C, Doctor T. Prevalence rates of biosimilar discontinuation and switchback to originator biologics following non-medical switching: a meta-analysis of real-world studies [abstract P445]. J Crohns Colitis. 2019;13:S334.
Reiland J-B, Freischem B, Roediger A. What pricing and reimbursement policies to use for off-patent biologicals in Europe? Results from the second EBE biological medicines policy survey. GaBI J. 2017;6:1–18.
Ilias A, Szanto K, Gonczi L, et al. Outcomes of patients with inflammatory bowel diseases switched from maintenance therapy with a biosimilar to remicade. Clin Gastroenterol Hepatol. 2019;17:2506–13.
Sigurdardottir VR, Svärd A. Multiswitching—from reference product etanercept to biosimilar and back again—real-world data from a clinic-wide multiswitch experience [abstract THU0222]. Ann Rheum Dis. 2018;77:A331.
Småstuen MC, Madla R, Solli O, Hjelvin E. OP0311 Retrospective analysis of prescription dynamics of etanercept after introduction of biosimilars based on Norwegian prescription database. An interim analysis. Ann Rheum Dis. 2019;78:237–8.
Australia’s PBAC recommends substitution of biosimilars. GaBI Online. http://www.gabionline.net/Biosimilars/General/Australia-s-PBAC-recommends-substitution-of-biosimilars. Accessed Dec 2, 2019.
Who chooses whether the biosimilar medicine or the reference biological medicine is used? Canberra, Australia: Australian Government Department of Health; Feb 22, 2019.
Automatic pharmacist substitution of biosimilars in Germany. GaBI Online. http://www.gabionline.net/Policies-Legislation/Automatic-pharmacist-substitution-of-biosimilars-in-Germany. Accessed Aug 22, 2019.
Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78:463–78.
Faccin F, Tebbey P, Alexander E, Wang X, Cui L, Albuquerque T. The design of clinical trials to support the switching and alternation of biosimilars. Expert Opin Biol Ther. 2016;16:1445–53.
Scientific considerations in demonstrating biosimilarity to a reference product: Guidance for Industry. Silver Springs: US Food and Drug Administration; 2015.
Committee for Medicinal Products for Human Use. Guideline on similar biological medicinal products. London: European Medicines Agency; 2014. CHMP/437/04 Rev 1.
Kurki P, van Aerts L, Wolff-Holz E, Giezen T, Skibeli V, Weise M. Interchangeability of biosimilars: a European perspective. Biodrugs. 2017;31:83–91.
Biosimilar biologic drugs in Canada: fact sheet. Ottawa: Health Canada; Aug 23, 2019.
Biosimilars in the EU: information guide for healthcare professionals. London: European Medicines Agency/European Commission; Apr 27, 2017.
Considerations in demonstrating interchangeability with a reference product. Guidance for Industry. Silver Springs: US Food and Drug Administration; May, 2019.
State laws and legislation related to biologic medications and substitution of biosimilars. July 2015—covering 2013-2015 legislative sessions. Washington, DC: National Conference of State Legislature; Rev. July 27, 2015.
Center for Drug Evaluation and Research. List of licensed biological products with (1) reference product exclusivity and (2) biosimilarity or interchangeability evaluations to date. https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/HowDrugsareDevelopedandApproved/ApprovalApplications/TherapeuticBiologicApplications/Biosimilars/UCM560162.pdf. Accessed Mar 10, 2020.
Remuzat C, Dorey J, Cristeau O, Ionescu D, Radiere G, Toumi M. Key drivers for market penetration of biosimilars in Europe. J Mark Access Health Policy. 2017;5:1272308.
Fact sheet: biosimilars. Ottawa: Health Canada; Aug 3, 2017.
Nagai S, Yanagihara R, Kishioka Y. Japanese regulatory authority’s perspective on biosimilars. Lancet Oncol. 2015;16:e101.
Kunkel JP, Jan DC, Butler M, Jamieson JC. Comparisons of the glycosylation of a monoclonal antibody produced under nominally identical cell culture conditions in two different bioreactors. Biotechnol Prog. 2000;16:462–70.
Scallon BJ, Tam SH, McCarthy SG, Cai AN, Raju TS. Higher levels of sialylated Fc glycans in immunoglobulin G molecules can adversely impact functionality. Mol Immunol. 2007;44:1524–34.
Blauvelt A, Cohen AD, Puig L, Vender R, van der Walt J, Wu JJ. Biosimilars for psoriasis: preclinical analytical assessment to determine similarity. Br J Dermatol. 2016;174:282–6.
Kim S, Song J, Park S, et al. Drifts in ADCC-related quality attributes of Herceptin(R): impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–14.
de Ridder L, Assa A, Bronsky J, et al. Use of biosimilars in pediatric inflammatory bowel disease: an updated position statement of the Pediatric IBD Porto Group of ESPGHAN. J Pediatr Gastroenterol Nutr. 2019;68:144–53.
Danese S, Fiorino G, Raine T, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease–an update. J Crohns Colitis. 2017;11:26–34.
Crohn’s and Colitis Foundation. Biosimilars: position statement. https://www.crohnscolitisfoundation.org/sites/default/files/2019-06/biosimilars-statement-needs_0.pdf. Accessed July 26, 2019.
American College of Rheumatology. American College of Rheumatology position statement. Biosimilars. https://www.rheumatology.org/Portals/0/Files/Biosimilars-Position-Statement.pdf. Accessed June 18, 2019.
El Zorkany B, Al Ani N, Al Emadi S, et al. Biosimilars in rheumatology: recommendations for regulation and use in Middle Eastern countries. Clin Rheumatol. 2018;37:1143–52.
European Biopharmaceutical Enterprises, European Federation of Pharmaceutical Industries and Associations, International Federation of Pharmaceutical Manufacturers and Associations. Considerations for physicians on switching decisions regarding biosimilars. https://www.ebe-biopharma.eu/wp-content/uploads/2017/04/considerations-for-switching-decisions_biosimilars-and-rbps-final-branded-1.pdf. Accessed June 18, 2019.
Tabernero J, Vyas M, Giuliani R, et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open. 2016;1:e000142.
Australian Rheumatology Association. ARA position statement on the introduction of biosimilars for the treatment of rheumatic diseases. http://rheumatology.org.au//gps/documents/ARAbiosimilarsPANov16-updateJun17.pdf. Accessed Oct 4, 2019.
Brazilian Society of Rheumatology. Brazilian Society of Rheumatology position statement. http://www.reumatologia.org.br/download/posicionamento-da-sociedade-brasileira-de-reumatologia-sobre-aprovacao-e-o-uso-de-medicamentos-biossimilares-no-brasil. Accessed Oct 4, 2019.
Blauvelt A, Puig L, Chimenti S, et al. Biosimilars for psoriasis: clinical studies to determine similarity. Br J Dermatol. 2017;177:23–33.
Bakalos G, Zintzaras E. Drug discontinuation in studies including a switch from an originator to a biosimilar monoclonal antibody: a systematic literature review. Clin Ther. 2019;41(155–73):e13.
Chaparro M, Garre A, Guerra Veloz MF, et al. Effectiveness and safety of the switch from Remicade® to CT-P13 in patients with inflammatory bowel disease [abstract #P0978]. United Eur Gastroenterol J. 2018;6:A453.
Fleischmann R, Jairath V, Mysler E, Nicholls D, Declerck P. Nonmedical switching from originators to biosimilars: does the nocebo effect explain treatment failures and adverse events in rheumatology and gastroenterology? Rheumatol Ther. 2020.
Azevedo V, Dorner T, Strohal R, et al. Biosimilars: considerations for clinical practice. Consider Med. 2017;1:13–8.
Pradeu T, Jaeger S, Vivier E. The speed of change: towards a discontinuity theory of immunity? Nat Rev Immunol. 2013;13:764–9.
Schellekens H. Immunogenicity of therapeutic proteins: clinical implications and future prospects. Clin Ther. 2002;24:1720–40 (discussion 19).
Scott BJ, Klein AV, Wang J. Biosimilar monoclonal antibodies: a Canadian regulatory perspective on the assessment of clinically relevant differences and indication extrapolation. J Clin Pharmacol. 2015;55:S123–32.
Toussirot E, Marotte H. Switching from originator biological agents to biosimilars: what is the evidence and what are the issues? RMD Open. 2017;3:e000492.
Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16:658–73.
Wadhwa M, Knezevic I, Kang HN, Thorpe R. Immunogenicity assessment of biotherapeutic products: an overview of assays and their utility. Biologicals. 2015;43:298–306.
World Health Organization. Guidelines on evaluation of similar biotherapeutic products (SBPs). https://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=1. Accessed Dec 9, 2019.
Tatarewicz SM, Mytych DT, Manning MS, Swanson SJ, Moxness MS, Chirmule N. Strategic characterization of anti-drug antibody responses for the assessment of clinical relevance and impact. Bioanalysis. 2014;6:1509–23.
Wadhwa M, Skog AL, Bird C, et al. Immunogenicity of granulocyte-macrophage colony-stimulating factor (GM-CSF) products in patients undergoing combination therapy with GM-CSF. Clin Cancer Res. 1999;5:1353–61.
Hjelm Skog AL, Wadhwa M, Hassan M, et al. Alteration of interleukin 2 (IL-2) pharmacokinetics and function by IL-2 antibodies induced after treatment of colorectal carcinoma patients with a combination of monoclonal antibody 17-1A, granulocyte macrophage colony-stimulating factor, and IL-2. Clin Cancer Res. 2001;7:1163–70.
Smolen JS, Choe JY, Prodanovic N, et al. Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: results of a randomised, double-blind, phase III transition study. Ann Rheum Dis. 2018;77:234–40.
Yoo DH, Prodanovic N, Jaworski J, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76:355–63.
Park W, Yoo DH, Miranda P, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76:346–54.
Jorgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389:2304–16.
Bhandare AP, Crooks B, Nigam GB, Limdi JK. Switching from originator infliximab to CT-P13: single centre experience from the UK. J Crohns Colitis. 2019;13:S270–1.
Boone NW, Liu L, Romberg-Camps MJ, et al. The nocebo effect challenges the non-medical infliximab switch in practice. Eur J Clin Pharmacol. 2018;74:655–61.
Plevris N, Jones GR, Jenkinson PW, et al. Implementation of CT-P13 via a managed switch programme in Crohn’s disease: 12-month real-world outcomes. Dig Dis Sci. 2019;64:1660–7.
Ala K, Avery P, Wilson R, et al. PTU-059 early experience with biosimilar infliximab at a district general hospital for an entire Crohns disease patient cohort switch from Remicade to Inflectra. Gut. 2016;65.
Nugent S, Nugent M, Mullane D, Kelly C. P430 EirSwitch echoes of NorSwitch: switching biosimilar therapy in an IBD cohort: an Irish experience. J Crohn’s Colitis. 2017;11:S295.
Guidance for Industry. Immunogenicity assessment for therapeutic protein products. Silver Springs: US Food and Drug Administration; 2014.
Committee for Medicinal Products for Human Use. Guideline on immunogenicity assessment of therapeutic proteins. London: European Medicines Agency; May 18, 2017. EMEA/CHMP/BMWP/14327/2006 Rev 1.
Colloca L, Miller FG. The nocebo effect and its relevance for clinical practice. Psychosom Med. 2011;73:598–603.
Kristensen LE, Alten R, Puig L, et al. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. Biodrugs. 2018;32:397–404.
Pouillon L, Socha M, Demore B, et al. The nocebo effect: a clinical challenge in the era of biosimilars. Expert Rev Clin Immunol. 2018;14:739–49.
Glintborg B, Sorensen IJ, Loft AG, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76:1426–31.
Glintborg B, Loft AG, Omerovic E, et al. To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis. 2019;78:192–200.
Nikiphorou E, Kautiainen H, Hannonen P, et al. Clinical effectiveness of CT-P13 (infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther. 2015;15:1677–83.
Kiltz U, Tsiami S, Baraliakos X, Braun J. Non-medical switching from originator to biosimilar etanercept—no evidence for a relevant nocebo effect—a retrospective analysis of real-life data. Ann Rheum Dis. 2019;78:716–7.
Zuniga L, Calvo B. Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19:661–9.
Casadevall N, Edwards IR, Felix T, et al. Pharmacovigilance and biosimilars: considerations, needs and challenges. Expert Opin Biol Ther. 2013;13:1039–47.
Grampp G, Felix T. Pharmacovigilance considerations for biosimilars in the USA. Biodrugs. 2015;29:309–21.
Castaneda-Hernandez G, Sandoval H, Coindreau J, Rodriguez-Davison LF, Pineda C. Barriers towards effective pharmacovigilance systems of biosimilars in rheumatology: a Latin American survey. Pharmacoepidemiol Drug Saf. 2019;28:1035–44.
Edwards CJ, Hercogova J, Albrand H, Amiot A. Switching to biosimilars: current perspectives in immune-mediated inflammatory diseases. Expert Opin Biol Ther. 2019;19:1001–14.
Biological qualifier: an INN proposal. Programme on International Nonproprietary Names (INN). Geneva: World Health Organization; October, 2015. INN Working Doc. 14.342.
Directive 2010/84/EU of the European Parliament and of the Council. European Union; Dec 15, 2010.
BSG guidance on the use of biosimilar infliximab CT-P13 in IBD. London: British Society of Gastroenterology; Feb 1, 2016.
British Society of Rheumatology. British Society for Rheumatology position statement on biosimilar medicines (revised January 2017). https://www.nras.org.uk/data/files/revised_bsr_biosimilars_position_statement_jan_2017.pdf. Accessed June 18, 2019.
Manolis CH, Rajasenan K, Harwin W, McClelland S, Lopes M, Farnum C. Biosimilars: opportunities to promote optimization through payer and provider collaboration. J Manag Care Spec Pharm. 2016;22:S3–9.
Bjornsdottir US, Gizurarson S, Sabale U. Potential negative consequences of non-consented switch of inhaled medications and devices in asthma patients. Int J Clin Pract. 2013;67:904–10.
Fenwick S, Thakur K, Munro D. Nurse and patient perceptions and preferences for subcutaneous autoinjectors for inflammatory joint or bowel disease: findings from a European survey. Rheumatol Ther. 2019;6:195–206.
Thakur K, Biberger A, Handrich A, Rezk MF. Patient perceptions and preferences of two etanercept autoinjectors for rheumatoid arthritis: findings from a patient survey in Europe. Rheumatol Ther. 2016;3:245–56.
Zharkov A, Barton B, Heinzmann D, Bakalos G, Schreitmüller T. Development pathways for subcutaneous formulations of biologics versus biosimilar development. Expert Rev Precis Med Drug Dev. 2019. https://doi.org/10.1080/23808993.2019.1585806.
Emery P, Vencovsky J, Sylwestrzak A, et al. A phase III randomised, double-blind, parallel-group study comparing SB4 with etanercept reference product in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis. 2017;76:51–7.
Blauvelt A, Lacour JP, Fowler JF Jr, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179:623–31.
Genovese MC, Glover J, Matsunaga N, Chisholm D, Alten R. Effcacy, safety and immunogenicity in randomized, double-blind (DB) and open-label extension (OLE) studies comparing FKB327, an adalimumab biosimilar, with the adalimumab reference product (Humira®; RP) in patients (pts) with active rheumatoid arthritis (RA). Arthritis Rheumatol. 2017;69:abstract 2799.
Griffiths CEM, Thaci D, Gerdes S, et al. The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2017;176:928–38.
Gerdes S, Thaci D, Griffiths CEM, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32:420–7.
Alten R, Markland C, Kawakami K, et al. Immunogenicity of a proposed adalimumab biosimilar, FKB327, and the reference product in patients with rheumatoid arthritis [abstract P422]. J Crohns Colitis. 2019;13:S320.
Strand V, Balsa A, Al-Saleh J, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. Biodrugs. 2017;31:299–316.
ENBREL (etanercept) Prescribing Information. Thousand Oaks: Amgen; 2019.
Cunningham F, Shurmunov A, Dong D, Salone C, Zacher J, Glassman P. Biosimilar safety dashboard to assess switching in veterans. Pharmacoepidemiol Drug Saf. 2019;28:474–5.
Fautrel B, Bouhnik Y, Desjeux G, Freudensprung U, Addison J, Brigui A. PERFUSE: a French prospective/retrospective non-interventional cohort study of infliximab-naive and transitioned patients receiving infliximab biosimilar SB2; an interim analysis. Arthritis Rheumatol. 2019;71:abstract P1809.
Gisondi P, Virga C, Girolomoni G. Cross-switch from CT-P13 to SB2 infliximab biosimilars in patients with chronic plaque psoriasis (abstract P049). Presented at: 6th Congress of Skin Inflammation and Psoriasis International Network; Apr 25–27, 2019, Paris.
Harris C, Harris R, Young D, et al. IBD biosimilar to biosimilar infliximab switching study: preliminary results (abstract P0419). Presented at: 27th United European Gastroenterology Week; Oct 19–23, 2019, Barcelona.
Lauret A, Molto A, Abitbol V, et al. Effects of successive switches to different biosimilars infliximab on immunogenicity in chronic inflammatory diseases in daily clinical practice [abstract #OP0227]. Am Rheum Dis. 2019;78:190.
Petit J, Antignac M, Poilverd RM, et al. How to reduce the nocebo effect when switching from originator infliximab to a biosimilar: positive results of a multidisciplinary team intervention. Ann Rheum Dis. 2019;78:1447–9.
World Health Organization. 56th Consultation on International Nonproprietary Names for Pharmaceutical Substances. Geneva, 15–17 April 2013: executive summary. https://www.who.int/medicines/services/inn/56th_Executive_Summary.pdf?ua=1. Accessed July 24, 2019.
Grampp G, Ramanan S. The diversity of biosimilar design and development: implications for policies and stakeholders. Biodrugs. 2015;29:365–72.
Castaneda-Hernandez G, Gonzalez-Ramirez R, Kay J, Scheinberg MA. Biosimilars in rheumatology: what the clinician should know. RMD Open. 2015;1:e000010.
Abdalla A, Byrne N, Conway R, et al. Long-term safety and efficacy of biosimilar infliximab among patients with inflammatory arthritis switched from reference product. Open Access Rheumatol. 2017;9:29–35.
Ali SS, Hill D, Sofat N. Audit examining the difference in clinical outcomes amongst originator biologic treated patients with RA, PsA and axSpA who were switched to biosimilar versions and monitored routinely at St George’s university hospital NHS trust. Rheumatology. 2019;58.
Alkoky H, Pakozdi A, Tahir H. Benepali switches in clinical practice—a positive single centre experience. Arthritis Rheumatol. 2018;70:2788–9.
Alten R, Tarallo M, Gray C, Miglio C. Real world switching patterns of etanercept original and biosilmilar in Germany (abstract 1443). Arthritis Rheumatol. 2019;71.
Avouac J, Molto A, Abitbol V, et al. Systematic switch from innovator infliximab to biosimilar infliximab in inflammatory chronic diseases in daily clinical practice: the experience of Cochin University Hospital, Paris, France. Semin Arthritis Rheumatol. 2018;47:741–8.
Babai S, Akrout W, Le-Louet H. Reintroduction of reference infliximab product in patients showing inefficacy to its biosimilar [abstract]. Drug Saf. 2017;40:1027–8.
Baganz L, Strangfeld A, Herzer P, Krause A, Tony H-P, Zink A. SAT0134 Comparing real-world retention rates in a matched cohort of rheumatoid arthritis patients who either remained on the etanercept originator or switched to a biosimilar. Ann Rheum Dis. 2019;78:1136.
Binkhorst L, Sobels A, Stuyt R, Westerman EM, West RL. Switching to a infliximab biosimilar: short-term results of clinical monitoring in patients with inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2018;30:699–703.
Dahanayake C, Shah K, Al-Abdulla A, Carulli M. Assessing reasons for patients switching back from biosimilar etanercept therapy to originator product. Rheumatology. 2019;58:abstract 102.
Davies R, Kearsley-Fleet L, Lunt M, Watson KD, Hyrich KL, BSRBR-RA Contributors Group. Frequency and reasons for switching back to biologic originator following initial switch to biologic biosimilar [abstract #SAT0141]. Ann Rheum Dis. 2019;78:1141.
De Cock D, Kearsley-Fleet L, Watson K, Hyrich KL. Switching from RA originator to biosimilar in routine clinical care: early data from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis [abstract #2439]. Arthritis Rheumatol. 2017;69:3489–91.
De Cock D, Davies R, Kearsley-Fleet L, et al. Biosimilar use in children and young people with juvenile idiopathic arthritis in a real-world setting in the United Kingdom. Rheumatology. 2017;57:abstract P34.
Dyball S, Hoskins V, Christy-Kilner S, Haque S. Effectiveness and tolerability of Benepali in rheumatoid arthritis patients switched from Enbrel [abstract]. Arthritis Rheumatol. 2017;69:3505–6.
Felis-Giemza A, Chmurzyńska K, Nałęcz-Janik J, et al. Observational study of inflammatory arthritis treatment by etanercept originator switched to an etanercept biosimilar. Reumatologia. 2019;57:257–63.
Fernández S, Gonzalez S, Ordas-Calvo C, García-Fernández E, Rodríguez-De-Castro B, Babío J. AB0376 Results of a mandatory switching from originator to biosimilar etanercept in 117 patients with inflammatory arthritis from a single center. Ann Rheum Dis. 2019;78:1648.
Forejtová S, Zavada J, Szczukova L, Jarosova K, Philipp T, Pavelka K. A non-medical switch from originator infliximab to biosimilar CT-P13 in 36 patients with ankylosing spondylitis: 6-months clinical outcomes from the Czech biologic registry ATTRA [abstract]. Arthritis Rheumatol. 2017;69:2198–201.
Gentileschi S, Barreca C, Bellisai F, et al. Switch from infliximab to infliximab biosimilar: efficacy and safety in a cohort of patients with different rheumatic diseases. Response to: Nikiphorou E, Kautiainen H, Hannonen P, et al. Clinical effectiveness of CT-P13 (infliximab biosimilar) used as a switch from Remicade (infliximab) in patients with established rheumatic disease. Report of clinical experience based on prospective observational data. Expert Opin Biol Ther. 2015;15:1677–1683. Expert Opin Biol Ther. 2016;16:1311–2.
Germain V, Scherlinger M, Barnetche T, Schaeverbeke T, Federation Hospitalouniversitaire Acronim. Long-term follow-up after switching from originator infliximab to its biosimilar CT-P13: the weight of nocebo effect. Ann Rheum Dis. 2020;79:e11.
Hendricks O, Hørslev-Petersen K. When etanercept switch fails—clinical considerations [abstract]. Arthritis Rheumatol. 2017;69:3570–1.
Holroyd C, Parker L, Bennett S, et al. Switching to biosimilar infliximab: real-world data from the Southampton Biologic Therapies Review Service [abstract #O52]. Rheumatology. 2016;55:i60–1.
Hoque T, Suddle A, Herdman L, Kitchen J. Patient perceptions of switching to biosimilars [abstract #O72]. Rheumatology. 2018;57:iii67.
Jung YS, Park DI, Kim YH, et al. Efficacy and safety of CT-P13, a biosimilar of infliximab, in patients with inflammatory bowel disease: a retrospective multicenter study. J Gastroenterol Hepatol. 2015;30:1705–12.
Kaltsonoudis E, Pelechas E, Voulgari PV, Drosos AA. Maintained clinical remission in ankylosing spondylitis patients switched from reference infliximab to its biosimilar: an 18-month comparative open-label study. J Clin Med. 2019;8:956.
Klink A, Sadik K, Lee C, Pires A, Ellis L, Feinberg B. Real-world treatment patterns of rheumatoid arthritis patients who switched from infliximab to iinfliximab-dyyb. J Manag Care Spec Pharm. 2019;25:S81–2.
Layegh Z, Ruwaard J, Hebing RC, et al. Efficacious transition from reference product infliximab to the biosimilar in daily practice [abstract #AB1279]. Ann Rheum Dis. 2018;77:A1733.
Lee S, Szeto M, Galloway J. Bio-similar to bio-originator switchback: not a reliable quality indicator [abstract #AB1267]. Ann Rheum Dis. 2018;77:1727–8.
Madenidou AV, Jeffries A, Varughese S, et al. Switching patients with arthritis from etanercept (Enbrel) to the biosimilar drug, Benepali: a single center retrospective observational study [abstract #2533]. Arthritis Rheumatol. 2018;70:2817–9.
Mahmmod S, Schultheiss JPD, Mahmmod N, Tan AC, Dijksitra G, Fidder HH. Reverse switching to originator infliximab may be considered in patients with inflammatory bowel diseases experiencing new side effects or loss of response after switching to a CT-P13 biosimilar [abstract P1809] Presented at: 27th United European Gastroenterology Week; 2019, Barcelona.
Malaiya R, McKee Z, Kiely P. Infliximab biosimilars—switching Remicade to Remsima in routine care: patient acceptability and early outcome data [abstract #158]. Rheumatology. 2016;55:i125.
Moorthy A, Hall K, Walton S. Biosimilars switch in a multi-ethnic rheumatology patient group. Rheumatology. 2019;58:abstract 100.
Müskens WD, Rongen-van Dartel SAA, Adang E, van Riel PL. The influence of switching from etanercept originator to its biosimilar on effectiveness and the impact of shared decision making on retention and withdrawal rates [abstract #AB0475]. Ann Rheum Dis. 2018;77:A1399.
Nisar MK. SAT0576 Etanercept biosimilar switch: can it be successful without clinic review? Ann Rheum Dis. 2019;78:1380–1.
Patel D, Ahmed TJ, Levy S, Rajak R, Sathananthan R, Horwood N. Analysis of rheumatoid arthritis patients who failed the switch from originator etanercept to biosimilar etanercept in Croydon [abstract #E55]. Rheumatology. 2018;57:iii199.
Rajamani K, Choy E. Change in disease activity after switching etanercept (originator) to biosimilar (benepali) is associated with active disease at baseline. Arthritis Rheumatol. 2018;70:2791.
Razanskaite V, Bettey M, Downey L, et al. Biosimilar infliximab in inflammatory bowel disease: outcomes of a managed switching programme. J Crohns Colitis. 2017;11:690–6.
Reuber K, Kostev K. Prevalence of switching from two anti-TNF biosimilars back to biologic reference products in Germany. Int J Clin Pharmacol Ther. 2019;57:323–8.
Saxby K, Sanghvi S, Bodalia PN, et al. A novel approach to support implementation of biosimilars within a UK tertiary hospital. Br J Clin Pharmacol. 2020;86:23–8.
Scherlinger M, Langlois E, Germain V, Schaeverbeke T. Acceptance rate and sociological factors involved in the switch from originator to biosimilar etanercept (SB4). Semin Arthritis Rheum. 2019;48:927–32.
Scherlinger M, Germain V, Labadie C, et al. Switching from originator infliximab to biosimilar CT-P13 in real-life: the weight of patient acceptance. Joint Bone Spine. 2018;85:561–7.
Schmitz EMH, Boekema PJ, Straathof JWA, et al. Switching from infliximab innovator to biosimilar in patients with inflammatory bowel disease: a 12-month multicentre observational prospective cohort study. Aliment Pharmacol Ther. 2018;47:356–63.
Shah K, Flora K, Penn H. Clinical outcomes of a multi-disciplinary switching programme to biosimilar etanercept for patients with rheumatoid arthritis [abstract #232]. Rheumatology. 2018;57:iii143.
Sheppard M, Hadavi S, Hayes F, Kent J, Dasgupta B. Preliminary data on the introduction of the infliximab biosimilar (CT-P13) to a real world cohort of rheumatology patients [abstract #AB0322]. Ann Rheum Dis. 2016;75:1011.
Smith R, Fawthrop F. Similar experience of biosimilars: a review of Rotherham Hospital’s experience of switching from Enbrel to Benepali [abstract #O63]. Rheumatology. 2018;57:iii64.
Steel L, Marshall T, Loke M. Real world experience of biosimilar switching at the Norfolk and Norwich University Hospital, United Kingdom [abstract THU0204]. Ann Rheum Dis. 2018;77:321.
Tansley S, Smith S, Bond D, Korendowych E. Apparent high frequency of disease flare following switch from reference biologic rituximab (Mabthera) to a biosimilar (Truxima) in patients with vasculitis [abstract #195]. Rheumatology (United Kingdom). 2019;58:iii121.
Tweehuysen L, Huiskes VJB, van den Bemt BJF, et al. Open-label non-mandatory transitioning from originator etanercept to biosimilar SB4: 6-month results from a controlled cohort study. Arthritis Rheumatol. 2018;70:1408–18.
Tweehuysen L, van den Bemt BJF, van Ingen IL, et al. Subjective complaints as the main reason for biosimilar discontinuation after open-label transition from reference infliximab to biosimilar infliximab. Arthritis Rheumatol. 2018;70:60–8.
Uke PC, Morris C, Chitale S, James J, Miller H, Gladston-Chelliah E. Etanercept (ETN) biosimilar (Benapali—SB4) switches in inflammatory arthritis patients at Wrightington Hospital. Rheumatology. 2019;58:abstract E007.
Valido A, Silva-Dinis J, Saavedra MJ, Bernardo N, Fonseca JE. Efficacy and cost analysis of a systematic switch from originator infliximab to biosimilar CT-P13 of all patients with inflammatory arthritis from a single centre [abstract #AB1231]. Ann Rheum Dis. 2018;77:A1712.
Yazici Y, Xie L, Ogbomo A, et al. A descriptive analysis of real-world treatment patterns in a Turkish rheumatology population that continued innovator infliximab (Remicade) therapy or switched to biosimilar infliximab [abstract #SAT0175]. Arthritis Rheumatol. 2016;68:2903–6.
Yazici Y, Xie L, Ogbomo A, et al. Analysis of real-world treatment patterns in a matched rheumatology population that continued innovator infliximab therapy or switched to biosimilar infliximab. Biologics. 2018;12:127–34.
Acknowledgements
Funding
AbbVie Inc. (North Chicago, IL, USA) funded this review and the journal’s rapid service and open access fees, was involved in the collection, analysis, and interpretation of the data, and in the writing, review, and approval of the publication.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors had the idea for the article and critically revised the work. Literature search was performed by Complete Publication Solutions, LLC (North Wales, PA, USA), and was funded by AbbVie.
Medical Writing and Editorial Assistance
Medical writing support was provided by Madhura Mehta, PhD, and Maria Hovenden, PhD, of Complete Publication Solutions, LLC (North Wales, PA, USA), and was funded by AbbVie.
Disclosures
Brian G. Feagan has received consulting fees from AbbVie, Allergan, ActoGeniX, Albireo, Amgen, AstraZeneca, Atlantic Pharma, Avaxia Biologics, Axcan, Baxter Healthcare Corp., Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Centocor, Elan/Biogen, EnGene, Ferring, Roche/Genentech, GICare, Gilead, Given Imaging, GSK, Ironwood, Johnson & Johnson/Janssen, Kyowa Hakko Kirin Co., Lycera, Lexicon, Lilly, Merck, Mesoblast, Millennium, Nektar, Novartis Novo Nordisk, Prometheus Therapeutics & Diagnostics, Pfizer, Protagonist, Receptos, Salix, Serono, Shire, Sigmoid, Synergy, Takeda, Teva, Tillotts, TiGenix, UCB, Warner-Chilcott, Wyeth, Zealand, and Zyngenia; speaker fees from AbbVie, Johnson & Johnson/Janssen, Takeda, and UCB; financial support for research from AbbVie, Amgen, AstraZeneca/MedImmune, Atlantic Pharmaceuticals, Boehringer Ingelheim, Celgene Corporation, Celltech, Genentech Inc/Hoffmann-La Roche, Gilead Sciences, GlaxoSmithKline (GSK), Janssen Research & Development, Pfizer, Receptos/Celgene International, Sanofi, Santarus, Takeda Development Center Americas, Tillotts Pharma AG, and UCB; and member of the board of directors of Robarts Clinical Trials. Mona Marabani has been an investigator in clinical trials for AbbVie and Roche and has received consultancy fees from Janssen, AbbVie, Pfizer, Roche, BMS, Novartis, Amgen, NPS Australia, Lilly, and Sanofi. Jashin J. Wu is or has been an investigator, consultant, or speaker for AbbVie, Almirall, Amgen, Arcutis, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Dermavant, Dermira, Dr. Reddy’s Laboratories, Eli Lilly, Galderma, Janssen, LEO Pharma, Novartis, Regeneron, Sanofi Genzyme, Sun Pharmaceutical, UCB, and Valeant Pharmaceuticals North America. Freddy Faccin and Claire Spronk are full-time AbbVie employees and may own AbbVie stock/options. Gilberto Castañeda-Hernández has received consultancy and speaker fees from AbbVie, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Eli Lilly, Janssen-Cilag, Laboratorios Sophia, Libbs, Novartis, Pfizer, Roche, Sanofi, Teva, and UCB.
Compliance With Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12770936.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Feagan, B.G., Marabani, M., Wu, J.J. et al. The Challenges of Switching Therapies in an Evolving Multiple Biosimilars Landscape: A Narrative Review of Current Evidence. Adv Ther 37, 4491–4518 (2020). https://doi.org/10.1007/s12325-020-01472-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01472-1