
Abstract
Introduction
Low-dose methoxyflurane, administered via a hand-held inhaler, has been used for short-term pain relief in emergency medicine in Australia and New Zealand for over 40 years, and was recently approved in Europe for the rapid relief of moderate-to-severe trauma-related pain in adults. There is currently a lack of data for methoxyflurane versus active comparators, therefore this trial will investigate the efficacy and safety of inhaled methoxyflurane compared with standard of care (SoC) in the treatment of acute trauma-related pain in pre-hospital and ED settings in Italy.
Methods
MEDITA (Methoxyflurane in Emergency Department in ITAly) is a Phase IIIb, prospective, randomised, active-controlled, parallel-group, open-label, multicentre trial. A total of 272 adult patients with moderate-to-severe pain [score ≥ 4 on the Numerical Rating Scale (NRS)] due to limb trauma will be randomised 1:1 to receive 3 mL methoxyflurane (self-administered by the patient via inhalation under supervision of a trained person) or medications that currently comprise the SoC in Italy [intravenous (IV) morphine for severe pain (NRS ≥ 7); IV paracetamol or ketoprofen for moderate pain (NRS 4–6)], administered as soon as possible after randomisation.
Planned Outcomes
Pain intensity will be measured using a 100-mm visual analogue scale (VAS) at baseline (time of randomisation) and at intervals up to 30 min. Time of onset of pain relief as reported by the patient and use of rescue medication will be recorded. The patient will rate the efficacy and the healthcare professional will rate the practicality of study treatment at 30 min after randomisation using a 5-point Likert scale. Adverse events will be recorded until safety follow-up at 14 ± 2 days. Vital signs will be measured at baseline, 10 and 30 min. The primary aim is to demonstrate non-inferiority of methoxyflurane versus SoC for the change in VAS pain intensity from baseline (randomisation) to 3, 5 and 10 min.
Trial Registration
EudraCT number: 2017-001565-25. Clinicaltrials.gov identifier: NCT03585374.
Funding
Mundipharma Pharmaceuticals srl.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Plain language summary
An estimated 38 million people attend European Emergency Departments (EDs) each year due to injuries, and many experience pain that is inadequately treated. Currently available painkillers (analgesics) have limitations such as being slow to work (oral medications), requiring needles (intravenous medications) or side effects (e.g., opioids). A new analgesic (methoxyflurane) is available in Europe for emergency relief of moderate-to-severe acute pain due to injury. Methoxyflurane is administered using a disposable hand-held inhaler, through which the patient can inhale and control their own level of analgesia. Methoxyflurane has been used in Australia and New Zealand for over 40 years, with nearly 6 million uses to date, but no large-scale trials have assessed its effectiveness compared with other analgesics in the emergency setting. A new trial (MEDITA—Methoxyflurane in Emergency Department in ITAly) will investigate the effectiveness and safety of methoxyflurane versus standard analgesic treatment in Italian emergency medical centres.
The MEDITA trial will enroll approximately 272 adult patients with limb injuries and/or suspected fractures who have moderate-to-severe pain when they are assessed in the ED or are assisted through the 118 Emergency system (pre-hospital and ambulance service). Patients will be randomly allocated to treatment with inhaled methoxyflurane or standard analgesic treatment; this will be intravenous morphine for patients with severe pain and intravenous paracetamol or ketoprofen for patients with moderate pain. The trial will compare reductions in patients’ pain intensity, how quickly pain relief is achieved, whether additional analgesics are required and the number and type of side effects between the two treatment groups.
Introduction
The health burden of injury and trauma pain is significant; each year there are an estimated 38 million injury-related emergency department (ED) attendances across the European Union [1]. The prevalence of trauma-related pain has been reported as 70% of patients in the pre-hospital setting [2] and up to 91% in the ED [3, 4], with many patients still experiencing moderate-to-severe pain at discharge [3, 5]. Undertreatment of acute pain (oligoanalgesia) in trauma patients remains a widespread problem in the pre-hospital and ED settings; a study in the pre-hospital setting found the prevalence of oligoanalgesia to be 43% [6], while a study in the ED found that only 35.7% of patients received analgesia and 12.5% received adequate pain management [7]. Reasons for the undertreatment of pain include inadequate or under-assessment of pain, lack of training or guidance on pain management, insufficient time and resources, and reluctance to administer opioids [8,9,10].
In Europe, commonly used analgesics for trauma pain include paracetamol, non-steroidal anti-inflammatory drugs (NSAIDs), nitrous oxide (N2O) and opioids [2, 11], although there are regional and institutional variations. Current analgesics present several limitations in the treatment of trauma pain with respect to their routes of administration, strength of analgesia, side effect profiles and pharmacokinetic properties [8]. Paracetamol and NSAIDs are weak analgesics often used as first-line treatment of mild-to-moderate pain, but onset of action is slow when administered orally, while parenteral administration requires additional resources for cannulation and may be difficult in the pre-hospital emergency setting. There is also risk of overdose if the patient has already self-medicated before receiving medical assistance. N2O is fast acting, but its canisters are bulky and heavy, making them unwieldy to transport and manoeuvre in emergency situations, [12] and must also be used with caution in patients with chest injury due to the risk or accumulation of gas and rapid worsening of pneumothorax if present [13]. Opioids are a highly effective treatment option for severe pain, but require additional resources for prescribing, administration, monitoring and observation due to their controlled status and challenging safety profile. A recent review has estimated the cost per patient of intravenous (IV) morphine administration in Europe at €18–28 (of which €14–22 is accounted for by workforce costs), rising to €121–132 when morphine-related adverse events (AEs) and IV-related complications are also considered [14]. Furthermore, there is often a general reluctance on the part of healthcare providers to prescribe opioids due to concerns over safety, dependence and abuse. According to the Italian Inter-societary Recommendations (SIAARTI, SIMEU, SIS 118, AISD, SIARED, SICUT, IRC) on emergency pain management, “The ideal pre-hospital analgesic should be simple to use, safe, effective, unaffected by transport times, have a rapid onset, a short duration of action, and be able to be titrated to achieve the desired effect in all patients” [15].
Low-dose methoxyflurane, a non-opioid, volatile fluorinated hydrocarbon, administered via a lightweight, disposable, hand-held inhaler (Penthrox®; Medical Developments International Limited, Scoresby, Australia) has recently been approved in Europe for the emergency relief of moderate-to-severe pain in conscious adults with trauma and associated pain [16]. While use of methoxyflurane at higher doses for general anaesthesia was discontinued in the 1970s due to concerns about nephrotoxicity [17], administration of lower, analgesic doses of methoxyflurane is not typically associated with renal side effects [18]. Low-dose methoxyflurane has been widely used in Australia and New Zealand for over 40 years for short-term pain relief in both adults and children in emergency medicine, minor surgical and dental procedures [19, 20], with a total of over 5 million administrations worldwide to date [21].
The European approval for low-dose methoxyflurane was primarily based on a UK-based randomised, controlled trial (STOP!), which demonstrated significantly greater reductions in pain intensity, a rapid onset of action (median 4 min), along with significantly less use of rescue medication and high patient satisfaction with methoxyflurane compared with placebo in 300 patients aged ≥ 12 years with acute trauma pain [22], as well as in the adult-only subgroup [23]. With the exception of two smaller studies versus intramuscular tramadol [24] and placebo [25], other clinical data supporting low-dose methoxyflurane in the treatment of trauma pain are mainly from real-world observational or retrospective studies outside Europe [26,27,28,29,30]. There is a need for better evidence from randomised trials using clinically relevant active controls. Therefore, a randomised controlled trial aiming to investigate the efficacy and safety of low-dose methoxyflurane analgesia compared with standard of care (SoC) in the treatment of acute trauma-related pain has been initiated in emergency units in Italy and is described in this paper.
Methods
Study Design
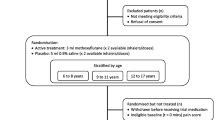
MEDITA (Methoxyflurane in Emergency Department in ITAly) is a Phase IIIb, prospective, randomised, active-controlled, parallel-group, open-label, multicentre trial of the efficacy and safety of methoxyflurane versus SoC in adult patients with acute trauma-related pain (study code MR311-3504; EudraCT 2017-001565-25; NCT03585374). The study is being conducted at 16 emergency medical centres in Italy (in the pre-hospital or ED settings).
Adult, stable, alert and collaborative patients with limb trauma in a single area and a pain score ≥ 4 on the 11-point Numerical Rating Scale (NRS) presenting at the hospital for triage, or who are rescued in the pre-hospital environment through the 118 system of the participating sites, will be assessed for eligibility in the trial. Full inclusion and exclusion criteria are presented in Table 1. A total of 272 patients are expected to be enrolled and randomised 1:1 to receive methoxyflurane or medications that currently comprise the SoC in Italy [intravenous (IV) morphine for the treatment of severe pain (NRS ≥ 7) and IV paracetamol or ketoprofen for the treatment of moderate pain (NRS 4–6)]. Patients will receive the assigned study treatment and undergo study assessments in a single day (the day of enrolment and randomisation), with a follow-up telephone call 14 ± 2 days later to assess safety and record any concomitant therapies.
Treatments
Patients will be randomised in a 1:1 ratio to receive either inhaled methoxyflurane or SoC. Treatment randomisation will be through a block randomisation scheme of four prepared by before the start of the trial. No stratification for any characteristic or balancing between the two treatment arms will be performed. After patient details are entered into the electronic case report form (eCRF) and eligibility is confirmed, patients will be randomised automatically through an Interactive Web Response System set up within the eCRF. Treatment will be administered in an open-label fashion commencing as soon as possible after randomisation.
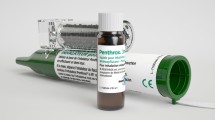
Patients randomised to methoxyflurane will receive one hand-held inhaler with a 3-mL vial of methoxyflurane (Fig. 1). Prior to use, the methoxyflurane liquid is added to the inhaler via a one-way valve and is absorbed by a polypropylene wick; once absorbed, the liquid vaporizes and the patient inhales the vapour through the mouthpiece. The patient also exhales back into the mouthpiece, so that any exhaled methoxyflurane is captured by the activated charcoal (AC) chamber, which adsorbs methoxyflurane and prevents fugitive emissions resulting in occupational exposure. The patient will be instructed to inhale intermittently from the device to obtain adequate analgesia. If a greater analgesic effect is required, the patient will be instructed to cover the diluter hole at the mouthpiece end, which, when covered with the patient’s index finger, allows the patient to inhale a higher concentration of study medication. One inhaler (3 mL methoxyflurane) will provide approximately 1 h of analgesia under suggested intermittent inhalation conditions; continuous inhalation reduces the duration of use to approximately 25 min of analgesia.

Methoxyflurane inhaler with activated carbon chamber
For patients randomised to SoC, treatment will be determined based on the intensity of the patient’s pain. Patients with severe pain (NRS ≥ 7) will receive IV morphine (0.10 mg/kg). Patients with moderate pain (NRS 4–6) will receive IV paracetamol (1 g) or ketoprofen (100 mg), chosen by the investigator on the basis of availability and local practice, and considering any prior history of allergy in the patient.
If a patient in either treatment group experiences insufficient analgesia, rescue medication will be permitted according to local practice from 25 min after randomisation (after measurement of pain relief for this time point). However, the investigator may administer rescue medication at any time at his/her discretion in the event that pain worsens, or improvement is insufficient.
Study Objectives and Endpoints
The primary objective of the trial is to assess the efficacy of inhaled methoxyflurane in the treatment of moderate-to-severe acute trauma-related pain compared with SoC. The primary, secondary and exploratory objectives of the trial and associated endpoints are presented in Table 2. Baseline will be taken as the time of randomisation. This is intended to take into account not only the intrinsic efficacy of the trial treatments but also the speed of drug administration in the emergency setting, since the aim of treatment is to relieve the patient’s pain as rapidly as possible and allow continuation of the diagnostic–therapeutic process.
Pain intensity (as a trial endpoint) will be assessed using a 100-mm visual analogue scale (VAS; 0 = no pain to 100 = maximum pain) at baseline and at 3, 5, 10, 15, 20, 25 and 30 min, or until rescue medication is administered. The patient will be asked to respond to the question “How much pain do you feel at this moment?” by independently marking a vertical line on the VAS that best represents their state and signing and dating the record. Given the emergency/rescue setting and the short detection times for the variable, a specially trained healthcare professional may assist the patient by administering the VAS and recording the value and time if required, but the patient must authenticate this with a signature and date as soon as they are able. The time of onset of pain relief as reported by the patient will be recorded. For patients randomised to methoxyflurane, it will be noted (yes/no) whether the patient covered the diluter hole during inhalation. Use of rescue medication (yes/no), including the type, dose and time after randomisation (if applicable), will be recorded.
At 30 min after randomisation, the patient will be asked to rate the overall efficacy of the study treatment and the healthcare professional will rate the practicality of using the study treatment. Both will be rated on a 5-point Likert qualitative scale (“Poor”, “Fair”, “Good”, “Very Good’, or “Excellent”).
Adverse events (not related to the trauma presentation) will be recorded from the time of randomisation until the safety follow-up telephone call at 14 ± 2 days after treatment. Vital signs (supine systolic and diastolic blood pressure, heart rate and respiration rate) will be measured at baseline (if not already assessed during the selection phase) and at 10 and 30 min after randomisation. Concomitant therapies taken by the patient within 7 days prior to inclusion in the trial and during the following 14 days, including rescue medication use, will be recorded. Additionally, the final diagnosis of the trauma category (fracture, dislocation, crushing, contusion) will be recorded during the safety follow-up telephone call. All data collected during the trial will be entered into an eCRF system accessible via the internet.
Sample Size Estimate
The primary objective to show non-inferiority of methoxyflurane compared to SoC will be evaluated on the basis of the difference in the change from baseline (randomisation) in VAS pain intensity at 3, 5 and 10 min. Assuming a non-inferiority margin of 1.0, a standard deviation of 2.5 [22] and a significance level of 0.05, a sample size of 108 patients per treatment group will provide a power of 90%. Allowing for 20% of patients being non-evaluable, a total of 136 patients per treatment group are required to be randomised. The co-primary objective was not considered in the sample size calculation because enrolment will not be balanced by baseline pain severity, and the number of patients enrolling with severe pain (NRS ≥ 7) will be unpredictable. The robustness of the obtained estimates for the co-primary endpoint will be appropriately studied and commented on during analysis.
Statistical Analyses
The primary objective is to demonstrate non-inferiority of methoxyflurane versus SoC with regard to the change in VAS pain intensity from baseline (randomisation) to 3, 5 and 10 min (primary endpoint). The primary endpoint will be analysed using a repeated-measures analysis of covariance (ANCOVA) adjusted for baseline VAS score, and the interaction between time point and treatment. The statistical model will be used to calculate the treatment difference (methoxyflurane–SoC) and associated 95% confidence interval (CI) at each time point; the primary analysis is the overall test for treatment effect considering all three time points. Non-inferiority will be concluded if the estimated upper 95% confidence limit is below the non-inferiority margin of 1.0. The co-primary objective to demonstrate superiority of methoxyflurane versus IV paracetamol/ketoprofen in patients with moderate pain (NRS 4–6) at baseline, and the secondary objective to demonstrate superiority of methoxyflurane versus SoC across all patients, will each be evaluated using a similar repeated-measures ANCOVA on the primary endpoint. For secondary and exploratory VAS pain intensity endpoints, the mean change from baseline at each time point will be estimated with 95% CI in each treatment group.
For other secondary endpoints, the percentage of patients who resort to additional analgesia (rescue medication) will be compared between the treatment groups using a Z test. The time from randomisation to the onset of pain relief will be described for each treatment group using Kaplan–Meier curves and, if appropriate, compared between the treatment groups using a Cox proportional hazards model. For patient ratings of efficacy and healthcare professional ratings of practicality of study treatment, the percentage of patients in each response category will be calculated for each treatment group and, if appropriate, the proportions will be compared between the treatment groups using a Chi squared test. The proportion of patients in the methoxyflurane treatment group who resort to closure of the diluter hole (exploratory endpoint) will be evaluated as a relative frequency with 95% CI.
Adverse events will be coded using the Medical Dictionary for Regulatory Activities and summarised descriptively (using absolute frequencies and percentages) for each treatment group. The mean change from baseline (randomisation) to 10 and 30 min for each vital sign parameter will be calculated with relative 95% CIs for each treatment group. All efficacy and safety analyses will be performed using the intention-to-treat population, defined as all randomised and eligible patients who received study treatment. No imputation of missing data is anticipated, therefore only patients with available data will be considered for each analysis.
Strengths and Limitations of the Study
The trial is designed to compare the efficacy and safety of low-dose methoxyflurane versus the SoC for acute trauma-related pain. The medications which constitute the SoC in Italy are IV morphine for severe pain (NRS ≥ 7) and IV paracetamol or ketoprofen for moderate pain (NRS 4–6). Morphine is generally accepted as the “gold standard” for the treatment of severe pain. The choice of two comparators for the treatment of moderate pain reflects the heterogeneity of treatment available in the country and also allows for a therapeutic alternative in the event that the patient is allergic to paracetamol or NSAIDs. These treatments are commonly implemented in pain management guidelines [11, 14] and have been shown to be effective options for the management of moderate-to-severe musculoskeletal pain [31,32,33]. The definitive SoC in Italy will allow for a clear and consistent comparative treatment arm, while the co-primary objective will allow a separate comparison of methoxyflurane versus IV paracetamol/ketoprofen for moderate pain. A similar randomised controlled trial has recently completed in Spain [34], where there is no well-established standard of analgesic treatment or clinical guidelines for managing trauma pain, therefore methoxyflurane was compared with the SoC for the individual study site.
Risks for the patient randomised to treatment with methoxyflurane are considered to be minimal. With over 40 years of clinical use as an analgesic in Australia and New Zealand and over 5 million administrations to date, methoxyflurane has an established safety profile and has no clinically significant effects on systolic blood pressure, pulse rate, respiratory rate or consciousness levels [35, 36]. The most common adverse events are headache and dizziness; adverse events are generally mild and transient, resolving after inhalation is stopped [16]. Although nephrotoxicity was previously a concern with much higher anaesthetic doses of methoxyflurane [17], clinical experience suggests that a low but effective analgesic dose is not associated with the risk of renal adverse events [18]. Laboratory evidence shows a large safety margin for analgesic use of methoxyflurane; the maximum exposure from a single inhaler is 0.3 alveolar concentration (MAC)-hours, which is well below the reported level of risk of nephrotoxicity of 2.0 MAC-hours [18]. Furthermore, a retrospective linkage study of 17,629 patients exposed to at least one dose of methoxyflurane for pre-hospital analgesia showed that methoxyflurane was not associated with an increased risk of renal or hepatic failure [37].
Pain assessment for the purposes of verifying trial eligibility will be performed using the 11-point NRS (ranging from 0 “no pain” to 10 “worst possible pain”). The NRS score is the most frequently used clinical and emergency tool for pain surveys due to the ease with which the measurement can be administered and collected [38]. For this reason, many of the guidelines on the use of pain therapy are based on the NRS score and the related classification (NRS 1–3 = mild pain; NRS 4–6 = moderate pain; NRS ≥ 7 = severe pain). The 100-mm pain VAS (0 = no pain to 100 = maximum pain) will be used for assessing pain intensity as an efficacy endpoint in this trial, as it is a more sensitive tool (allowing the patient to mark a point on the scale that is measured to the nearest mm), and will permit direct comparison of data from this study with data from the STOP! study [22]. The pain VAS is frequently used in pain studies because is easy to use, requires no verbal or reading skills, and is sufficiently versatile to be employed in a variety of settings [39, 40].
A limitation of the trial is its open-label design. A double-blind study, while ideal from a methodological perspective, was not feasible due to the different routes of administration and unique characteristics of the trial treatments (methoxyflurane has a distinctive fruity smell). Although this could potentially be overcome using a double-dummy design, this is not practical nor ethical in the context of the trial setting, where the additional time taken to dispense and administer two treatments would delay treatment and prolong pain for the trauma patient.
Ethics and Dissemination
The MEDITA trial is sponsored by Mundipharma Pharmaceuticals S.r.l. and will be conducted in accordance with International Council on Harmonization Good Clinical Practice adhering to the ethical principles of the Declaration of Helsinki (1964 and subsequent amendments), as well as national and local guidelines. The trial has been approved by the Italian Medicines Agency and is registered with the European Union Clinical Trials Register (EudraCT number 2017-001565-25) and ClinicalTrials.gov (NCT03585374). All trial documents and procedures will be reviewed and approved by the appropriate Ethics Committees at each site. Written informed consent will be obtained from all patients before initiation into the trial. Given the emergency setting and the requirement for rapid analgesia, where the patient is unable to provide written informed consent, witnessed verbal consent will be obtained, with the patient signing the informed consent as soon as they are able.
Conclusion
Inhaled analgesic methoxyflurane has a number of characteristics that make it an attractive option as a pre-hospital and ED analgesic. It is portable, non-invasive, non-narcotic, simple to use and the patient can manage their own level of analgesia [16]. The STOP! study demonstrated a fast onset of pain relief for methoxyflurane (within 4 min) [22], comparable with the onset of both IV morphine [41] and inhaled N2O [42], and high patient satisfaction with the efficacy and tolerability of treatment. Methoxyflurane does not interfere with most other anaesthetic or analgesic agents, and its effects are rapidly reversed once inhalation stops (within 3–20 min), thus it does not limit subsequent assessment and treatment options [16]. Low-dose methoxyflurane analgesia may therefore address an unmet need in the emergency setting and provide simple, fast and effective pain relief able to avoid unnecessary suffering and improve patient flow. With methoxyflurane having recently been licensed in Europe for use in adult patients with trauma-associated pain, it is anticipated that the results of this study, along with findings of other recent studies [34, 43], can provide additional clinical evidence to help inform treatment choices for acute trauma-related pain in the ED.
References
EuroSafe. Injuries in the European Union: summary of injury statistics for the years 2012–2014, 6th Edition. 2016. http://www.eurosafe.eu.com/uploads/inline-files/EuropeSafe_Master_Web_02112016%20%282%29.pdf. Accessed 06 Sept 2018.
Berben SAA, Schoonhoven L, Meijs THJM, van Vugt AB, van Grunsven PM. Prevalence and relief of pain in trauma patients in emergency medical services. Clin J Pain. 2011;27:587–92.
Berben SA, Meijs TH, van Dongen RT, et al. Pain prevalence and pain relief in trauma patients in the Accident and Emergency Department. Injury. 2008;39:578–85.
Cordell WH, Keene KK, Giles BK, Jones JB, Jones JH, Brizendine EJ. The high prevalence of pain in emergency medical care. Am J Emerg Med. 2002;20:165–9.
Todd KH, Ducharme J, Choiniere M, et al. Pain in the emergency department: results of the pain and emergency medicine initiative (PEMI) multicenter study. J Pain. 2007;8:460–6.
Albrecht E, Taffe P, Yersin B, Schoettker P, Decosterd I, Hugli O. Undertreatment of acute pain (oligoanalgesia) and medical practice variation in prehospital analgesia of adult trauma patients: a 10 year retrospective study. Br J Anaesth. 2013;110:96–106.
Pierik JG, IJzerman MJ, Gaakeer MI, et al. Pain management in the emergency chain: the use and effectiveness of pain management in patients with acute musculoskeletal pain. Pain Med. 2015;16:970–84.
Diβmann PD, Maignan M, Cloves PD, Gutierrez Parres B, Dickerson S, Eberhardt A. A review of the burden of trauma pain in emergency settings in Europe. Pain Ther. 2018. https://doi.org/10.1007/s40122-018-0101-1 (Epub ahead of print).
Pierik JGJ, IJzerman MJ, Gaakeer MI, Vollenbroek-Hutten MMR, Doggen CJM. Painful discrimination in the emergency department: risk factors for underassessment of patients’ pain by nurses. J Emerg Nurs. 2017;43:228–38.
Motov SM, Khan AN. Problems and barriers of pain management in the emergency department: are we ever going to get better? J Pain Res. 2008;2:5–11.
The College of Emergency Medicine. Management of pain in adults. 2014. https://www.rcem.ac.uk/docs/College%20Guidelines/5w.%20Management%20of%20Pain%20in%20Adults%20(Revised%20December%202014).pdf. Accessed 06 Sept 2018.
Komessaroff D. Pre-hospital pain relief: penthrane or entonox. Aust J Emerg Care. 1995;2:28–9.
Entonox summary of product characteristics. 2018. http://www.mhra.gov.uk/spc-pil/?prodName=ENTONOX%20MEDICINAL%20GAS&subsName=NITROUS%20OXIDE&pageID=SecondLevel. Accessed 06 Sept 2018.
Casamayor M, DiDonato K, Hennebert M, Brazzi L, Prosen G. Administration of intravenous morphine for acute pain in the emergency department inflicts an economic burden in Europe. Drugs Context. 2018;7:212524. https://doi.org/10.7573/dic.212524 (eCollection 2018).
Raccomandazioni Intersocietarie Italiane (SIAARTI, SIMEU, SIS 118, AISD, SIARED, SICUT, IRC) sulla gestione del dolore in emergenza. 2014. http://www.aisd.it/e107_files/downloads/raccintersocietarie_it_complete31052014.pdf. Accessed 06 Sept 2018.
Penthrox® summary of product characteristics. 2016. https://www.medicines.org.uk/emc/medicine/31391. Accessed 06 Sept 2018.
Cousins MJ, Mazze RI. Methoxyflurane nephrotoxicity. A study of dose response in man. JAMA. 1973;225:1611–6.
Dayan AD. Analgesic use of inhaled methoxyflurane: evaluation of its potential nephrotoxicity. Hum Exp Toxicol. 2016;35:91–100.
Australian Therapeutic Goods Administration approved Product Information for Penthrox. 2016. https://www.ebs.tga.gov.au/ebs/picmi/picmirepository.nsf/PICMI?OpenForm&t=pi&q=methoxyflurane. Accessed 06 Sept 2018.
New Zealand Datasheet for Penthrox. 2017. http://www.medsafe.govt.nz/profs/datasheet/p/penthroxinh.pdf. Accessed 06 Sept 2018.
Medical Developments International. European Penthrox® Update, 12 August 2016. 2016. http://www.medicaldev.com/wp-content/uploads/2016/08/ASX-Announcement_European-Penthrox-Update_120816.pdf. Accessed 06 Sept 2018.
Coffey F, Wright J, Hartshorn S, et al. STOP!: a randomised, double-blind, placebo-controlled study of the efficacy and safety of methoxyflurane for the treatment of acute pain. Emerg Med J. 2014;31:613–8.
Coffey F, Dissmann P, Mirza K, Lomax M. Methoxyflurane analgesia in adult patients in the Emergency Department: a subgroup analysis of a randomized, double-blind, placebo-controlled study (STOP!). Adv Ther. 2016;33:2012–31.
Konkayev AK. Evaluation of clinical effectiveness of inhalatory analgesic “Penthrox” for pain relief in ankle injuries. Arch Balkan Med Union. 2013;48:239–43.
Chin R, Maccaskill M, Browne G. A randomised control trial of inhaled methoxyflurane pain relief, in children with upper limb fracture. J Paediatr Child Health. 2002;38:A13.
Middleton PM, Simpson PM, Sinclair G, Dobbins TA, Math B, Bendall JC. Effectiveness of morphine, fentanyl, and methoxyflurane in the prehospital setting. Prehosp Emerg Care. 2010;14:439–47.
Bendall JC, Simpson PM, Middleton PM. Effectiveness of prehospital morphine, fentanyl, and methoxyflurane in pediatric patients. Prehosp Emerg Care. 2011;15:158–65.
Buntine P, Thom O, Babl F, Bailey M, Bernard S. Prehospital analgesia in adults using inhaled methoxyflurane. Emerg Med Australas. 2007;19:509–14.
Babl F, Barnett P, Palmer G, Oakley E, Davidson A. A pilot study of inhaled methoxyflurane for procedural analgesia in children. Paediatr Anaesth. 2007;17:148–53.
Babl FE, Jamison SR, Spicer M, Bernard S. Inhaled methoxyflurane as a prehospital analgesic in children. Emerg Med Australas. 2006;18:404–10.
Craig M, Jeavons R, Probert J, Benger J. Randomised comparison of intravenous paracetamol and intravenous morphine for acute traumatic limb pain in the emergency department. Emerg Med J. 2012;29:37–9.
Sin B, Wai M, Tatunchak T, Motov SM. The use of intravenous acetaminophen for acute pain in the Emergency Department. Acad Emerg Med. 2016;23:543–53.
Eken C, Serinken M, Elicabuk H, Uyanik E, Erdal M. Intravenous paracetamol versus dexketoprofen versus morphine in acute mechanical low back pain in the emergency department: a randomised double-blind controlled trial. Emerg Med J. 2014;31:177–81.
Borobia Pérez AM, Capilla Pueyo R, Casal Codesido JR, InMEDIATE Group, et al. Phase IIIb, open label randomised clinical trial to compare pain relief between methoxyflurane and standard of care for treating patients with trauma pain in Spanish Emergency Units (InMEDIATE): Study protocol. IBJ Clin Pharmacol. 2017;1:e0008.
Johnston S, Wilkes GJ, Thompson JA, Ziman M, Brightwell R. Inhaled methoxyflurane and intranasal fentanyl for prehospital management of visceral pain in an Australian ambulance service. Emerg Med J. 2011;28:57–63.
Oxer HF. Effects of Penthrox® (methoxyflurane) as an analgesic on cardiovascular and respiratory functions in the pre-hospital setting. J Mil Veterans Health. 2016;24:14–20.
Jacobs IG. Health effects of patients given methoxyflurane in the prehospital setting: a data linkage study. Open Emerg Med J. 2010;3:7–13.
Hjermstad MJ, Fayers PM, Haugen DF, et al. Studies comparing Numerical Rating Scales, Verbal Rating Scales, and Visual Analogue Scales for assessment of pain intensity in adults: a systematic literature review. J Pain Symptom Manag. 2011;41:1073–93.
Jensen MP, Karoly P. The measurement of clinical pain intensity: a comparison of six methods. Pain. 1986;27:117–26.
Ho K, Spence J, Murphy M. Review of pain measurement tools. Ann Emerg Med. 1996;27:427–31.
Tveita T, Thoner J, Klepstad P, Dale O, Jystad A, Borchgrevink PC. A controlled comparison between single doses of intravenous and intramuscular morphine with respect to analgesic effects and patient safety. Acta Anaesthesiol Scand. 2008;52:920–5.
Ducassé JL, Siksik G, Durand-Béchu M. Nitrous oxide for early analgesia in the emergency setting: a randomized, double-blind multicentre prehospital trial. Acad Emerg Med. 2013;20:178–84.
Medical Developments International Limited. A randomised, double-blind, multicentre, placebo controlled study to evaluate the safety and efficacy of methoxyflurane (PENTHROX®) for the treatment of acute pain in children and adolescents from 6 to less than 18 years of age (presenting to an Emergency Department with minor trauma). 2017. https://www.clinicaltrialsregister.eu/ctr-search/trial/2016-004290-41/GB. EudraCT number 2016-004290-41. Accessed 06 Sept 2018.
Acknowledgements
The investigators of the MEDITA Study Group are: Germana Ruggiano, Isabella Bartoli, Giuseppe Carpinteri, Andrea Fabbri, Francesco Bermano, Maurizio Chiesa, Daniela Mura, Mario Oppes, Peppino Masciari, Davide Torti, Vittorio Iorno, Antonio Voza, Piero Paolini, Sossio Serra, Gianfilippo Gangitano, Raffaella Francesconi.
® Penthrox is a registered trade mark of Medical Developments International Limited and used under license.
Funding
The trial is funded by Mundipharma Pharmaceuticals srl. Costs for article processing and Open Access were funded by Mundipharma Pharmaceuticals srl. All authors had access to the protocol and Statistical Analysis Plan.
Editorial Assistance
Editorial assistance in the preparation of this manuscript was provided by Karen Mower of Scientific Editorial. Support for this assistance was funded by Mundipharma Research Limited.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published.
Disclosures
Andrea Fabbri, Giuseppe Carpinteri and Germana Ruggiano have nothing to disclose. Elisabetta Bonafede is an employee of the clinical research organization conducting the study. Antonella Sblendido is an employee of Mundipharma Pharmaceuticals srl. Amedeo Soldi is an employee of Mundipharma Pharmaceuticals srl. Alberto Farina is an employee of Mundipharma Pharmaceuticals srl.
Compliance with Ethics Guidelines
The trial will be conducted in accordance with the principles established in the Declaration of Helsinki and the International Conference on Harmonization guidelines for good clinical practice. All trial documents and procedures will be approved by the appropriate ethics committees at each site, and written informed consent (or verbal witnessed consent) will be obtained from all patients before initiation into the study.
Data Availability
Data sharing is not applicable to this article as no datasets have been generated or analyzed during the current study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The members of the MEDITA Group are listed in acknowledgements.
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7252880.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fabbri, A., Carpinteri, G., Ruggiano, G. et al. Methoxyflurane Versus Standard of Care for Acute Trauma-Related Pain in the Emergency Setting: Protocol for a Randomised, Controlled Study in Italy (MEDITA). Adv Ther 36, 244–256 (2019). https://doi.org/10.1007/s12325-018-0830-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0830-x