
Abstract
In the novel WHO classification 2008, the classification of aggressive B-cell lymphoma has been revised for several categories with the aim to define “clean” entities. Within large B-cell lymphoma, a few distinct clinico-pathological entities have been recognized with more clinically defined entities than pathologically defined ones. The majority of known morphological variations were not considered to merit more than classification as a variant of DLBCL, not otherwise specified. Specifically, a biological subgrouping of DLBCL on the basis of molecular (activated B-cell versus germinal center B-cell) or immunophenotypic (CD5+) features was felt to be too immature to include at this stage. The role of EBV in aggressive B-cell lymphoma has been explored in more depth with the recognition of several novel and re-defined clinico-pathological entities. Also, in these diseases, clinical definitions play a very dominant role in the WHO classification 2008.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Aggressive B-cell lymphomas encompass several categories within diffuse large B-cell lymphoma (DLBCL), Burkitt lymphoma (BL), and various morphologically, biologically, and clinically distinct diseases of large B-cells. Over the past years, research data on the relationships, overlapping features, and essential differences between various categories of aggressive B-cell lymphomas have served as the basis of discussions to separate new categories or to lump together others in the WHO classification 2008 [1]. EBV-associated B-cell lymphoma and overlapping features between DLBCL and BL on one hand and cHL on the other hand have been a focus of attention. In this paper, issues in DLBCL and EBV-associated B-cell lymphoma will be discussed. The issue of lymphomas with overlapping features between DLBCL and BL and DLBCL and cHL will be discussed separately.
Diffuse large B-cell lymphoma
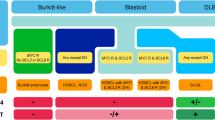
Proliferations of large B-cells can be classified in different ways. Morphology-based classification has been the classical approach for pathologists. However, more modern approaches may focus on biological categories, clinical categories, or even categories that reflect responsiveness to therapies. For the WHO classification 2008, clinico-pathological categories have been separated, and an attempt has been made to define biological categories. As compared to the WHO classification 2001, the prior “waste basket” of neoplastic proliferations of large B-cells has been more extensively subdivided into biologically homogeneous entities with the introduction of five new entities and the promotion of three from variants status of DLBCL to independent distinct entities in recognition of their biological uniqueness (Table 1).
The large group of proliferations of large B-cells is lumped together as diffuse large B-cell lymphoma, not otherwise specified in parallel to the nomenclature in peripheral T-cell lymphoma. In situations where there is insufficient evidence to justify delineation as a separate entity, the term “variants”, “subgroups”, and “subtypes” are used. The variants of DLBCL contain morphologically remarkable features, but do not represent biologically distinct groups. It is noteworthy that some believe that the so-called immunoblastic lymphoma category from previous classifications merits recognition as a distinct entity since it includes the activated B-cell (ABC) type DLBCL and is characterized by a worse prognosis. The only real importance to define such features is to consciously highlight the morphological spectrum of the disease and thereby avoid some pitfalls in daily practice (e.g., T-cell-rich B-cell lymphoma mistaken for T-cell lymphoma or the anaplastic variant of DLBCL mistaken for metastatic carcinoma).
T-cell/histiocyte-rich large B-cell lymphoma (THRBCL) holds a special position by its morphological overlap and possible biological relation to nodular lymphocyte predominant Hodgkin lymphoma (NLPHL). The authors of the DLBCL chapter consider diffuse areas of large B-cells in a background of small T-cells and histiocytes in a lymph node that is otherwise involved in NLPHL as “THRBCL-like areas” not indicative of transformation to aggressive disease NLPHL as a variant of DLBCL, while the authors of the NLPHL chapter leave this issue open and thus it remains controversial. It is agreed upon, however, that the detection of only a single nodule with features of NLPHL suffices to exclude a diagnosis of primary THRBCL, and in this sense, the definition is much sharper than in the WHO classification 2001.
Molecular and immunophenotypic subgrouping in DLBCL, NOS
It has been debated if it would be timely to introduce gene-expression based biological subgrouping of DLBCL in the WHO classification 2008. In 2000, DLBCL with gene-expression profiles of germinal center B-cells and activated B-cell-like were first described. Subsequently, it was found that both groups carry characteristic genetic aberrations and yet may be morphologically indistinguishable to a large extent. Since gene-expression profiling is not generally available for diagnostic purposes in daily practice, “translations” to immunohistochemical algorithms have been defined, including the so-called Hans-algorithm [2]. However, the immunophenotypic subdivision only correlates to the gene-expression subgrouping in around 80% of the cases; thus, the reproducibility of the immunophenotypic classification is suboptimal, and the prognostic impact is conflicting. Similar considerations regarding a lack of reproducibility and biological and prognostic impact can apply for the CD5+ DLBCL, considered a immunohistochemical subgroup in the WHO classification 2008 [3–5]. Therefore, immunohistochemical and molecular subgroups of DLBCL, NOS are only considered as variants with a similar level of relative (un)importance as morphologic variants, but not advocated as separate entities. Indeed, there is no justification to use this information for treatment stratification and for use in routine clinical practice. This situation may be quite different in experimental treatment programs, and it can be expected that introduction of some form of “biomarker”-based treatment stratification will find its way into practice in the coming few years. Up to that time, however, it is debatable whether to include this type of information in routine pathology reports without extensive comments on its interpretation for clinicians.
Clinico-pathological subtypes of large B-cell lymphomas
Both in the listing of “DLBCL, subtypes” and the “other lymphomas of large B-cells”, clinico-pathological subtypes that may be mostly defined by clinical features are found: primary DLBCL of CNS, primary cutaneous DLBCL, leg-type, EBV-positive DLBCL of the elderly, and intravascular large B-cell lymphoma. Of these, intravascular large B-cell lymphoma was also described in the WHO classification 2001. The others are newly introduced entities.
Primary DLBCL of CNS has been introduced to describe primary intracerebral and intraocular DLBCL, excluding lymphomas of the dura, intravascular large B-cell lymphoma, secondary CNS localizations and lymphomas in immunodeficient patients. Apart from the typical clinical situation, these lymphomas are mostly distinctive by biological features related to the immune-privileged character of the primary site. These include microenvironment modulation by IL-4, loss of HLA class I and II protein expression, and immunoglobulin structure (extreme Ig hypermutation in concert with retention of an open reading frame) [6]. Identical features are found in lymphomas at other immune-privileged sites such as testis. Perhaps future classifications may consider combining primary DLBCL in the testis together with primary DLBCL of the CNS as a new entity of DLBCL involving immunoprivileged sites.
Herpes-virus (EBV/HHV8)-related large B-cell proliferations
Several large B-cell proliferations related to EBV infection and/or immunodeficiency status have been recognized in the 2008 WHO classification as separate entities or introduced as new ones (Table 1).
The common feature of these processes is the association of an immunodeficiency status with an EBV or HHV-8 related B-cell neoplasm (Table 2). The three newly introduced entities (EBV-positive diffuse large B-cell lymphoma of the elderly, DLBCL associated with chronic inflammation and large B-cell lymphoma arising in HHV-8-associated multicentric Castleman disease) depict mainly the clinical setting in which the lymphoma originated. The recognition of factors such as age and chronic inflammation (not only pyothorax-associated) as causes of immunodeficiency in EBV-associated DLBCL in the absence of any other primary or secondary immune disease prompted the segregation of these into two clinico-pathological categories. Although molecular studies have shown that DLBCL associated with chronic inflammation and nodal diffuse large B-cell lymphoma have significant different gene-expression profiles, EBV-positive diffuse large B-cell lymphoma of the elderly and DLBCL associated with chronic inflammation have no single morphological or phenotypical feature that distinguish them from DLBCL not otherwise specified (NOS). Therefore, the clinico-pathological correlation is the gold standard in the proper diagnosis of these entities. Most cases show a post-germinal center phenotype, and a subset of them have an immunoblast/plasmablast morphology and phenotype, concordantly with the plasma cell differentiation induced by EBV. Similar to post-transplant lymphoproliferative disorders, tumor cells express LMP-1 and EBNA-2 hence a latency pattern III, known as the growth program. Cells expressing the EBV growth program are restricted to the naive B-cell subset in healthy individuals. It might be that in the absence of immune surveillance, EBV-infected germinal center or memory B-cells can switch to latency III and undergo clonal expansion. The expression of the full spectrum of latent EBV genes indicates an important role for the virus in driving proliferation of the infected B-cells [7, 8].
Multicentric Castleman disease is associated with HHV8 infection, and this situation may evolve into a specific type of large B-cell lymphoma, particularly in HIV+ patients. The B-cell lymphoma occurring in this clinical setting was initially defined as a plasmablastic lymphoma because the postulated cell of origin morphologically resembles a plasmablast. However, the tumor cells are often CD20 positive and represent a naive IgM secreting cell without somatic hypermutations [9–11]. These cases must be distinguished from the oral type plasmablastic lymphoma that is EBV positive and HHV8 negative.
Plasmablastic lymphoma of the oral type was regarded in the former WHO classification as a subtype of DLBCL associated with HIV infection. Later descriptions have reported cases of plasmablastic lymphoma outside of this clinical setting and even in patients without any known cause of immunodeficiency [12]. The WHO 2008 classification has broadened the definition of the previous classification to other localizations apart from the oral mucosa and also to other immunodeficiency states other than HIV infection. Plasmablastic lymphoma presents mostly in extranodal sites and in the majority of cases is associated with EBV infection. Tumor cells have immunoblastic morphology with a mature plasma cell phenotype. Clonal IgH rearrangement is demonstrable, and the IgH genes may show evidence of somatic hypermutation or be in an unmutated configuration [13]. The monotonous morphological appearance of the majority of these tumors reflects the lack of a T-cell host response. In these cases, LMP-1 is rarely expressed, and the most frequent latency pattern is type-I, in which EBNA-1 does not serve as a target for CD8 cytotoxic T lymphocytes [7]. The lack of co-stimulatory surface molecules helps the tumor cells to escape from immunological recognition, but also leaves as an open question the role of EBV in the pathogenesis of these lymphomas.
All the entities described above are characterized by a very aggressive behavior and short survival regardless of International Prognostic Index and stage at diagnosis. The immunosuppressed state in the most part of patients contributes to the poor clinical course. Additionally, downregulation of HLA class I expression [14] and activation of the IL-6 [15] receptor signaling pathway have been described in some cases as mechanisms of escape from host cytotoxic T lymphocytes. This is not unlike the mechanisms by which DLBCL of immunoprivileged sites maintain immune-escape.
Previous attempts to segregate different hematolymphoid entities usually started with the observation of morphological features characteristic of a disease and then a later description of a cytogenetic or molecular alteration that now are part of the definition of the entity. In DLBCL, the morphologic, phenotypic, and clinical heterogeneity has led the efforts to segregate different subgroups. In the last WHO classification, several entities with overlapping morphological features have been separated based on clinical features; molecular and cytogenetic studies are required to better understand the biological basis of these entities.
References
Swerdlow S, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (2008) WHO Classification of tumors of hematopoietic and lymphoid tissues. IARC, Lyon
Hans CP, Weisenburger DD, Greiner TC et al (2004) Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 103:275–282
Yamaguchi M, Seto M, Okamoto M et al (2002) De novo CD5+ diffuse large B-cell lymphoma: a clinicopathologic study of 109 patients. Blood 99:815–821
Yamaguchi M, Nakamura N, Suzuki R et al (2008) De novo CD5+ diffuse large B-cell lymphoma: results of a detailed clinicopathological review in 120 patients. Haematologica 93:1195–1202
de JD, Xie W, Rosenwald A et al (2009) Immunohistochemical prognostic markers in diffuse large B-cell lymphoma: validation of tissue microarray as a prerequisite for broad clinical applications (a study from the Lunenburg Lymphoma Biomarker Consortium). J Clin Pathol 62:128–138
Booman M, Douwes J, Glas AM et al (2006) Mechanisms and effects of loss of human leukocyte antigen class II expression in immune-privileged site-associated B-Cell lymphoma. Clin Cancer Res 12:2698–2705
Kuppers R (2003) B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol 3:801–812
Joseph AM, Babcock GJ, Thorley-Lawson DA (2000) Cells expressing the Epstein-Barr virus growth program are present in and restricted to the naive B-cell subset of healthy tonsils. J Virol 74:9964–9971
Dupin N, Diss TL, Kellam P et al (2000) HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma. Blood 95:1406–1412
Du MQ, Liu H, Diss TC et al (2001) Kaposi sarcoma-associated herpesvirus infects monotypic (IgM lambda) but polyclonal naive B cells in Castleman disease and associated lymphoproliferative disorders. Blood 97:2130–2136
Oksenhendler E, Boulanger E, Galicier L et al (2002) High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease. Blood 99:2331–2336
Colomo L, Loong F, Rives S et al (2004) Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities. Am J Surg Pathol 28:736–747
Gaidano G, Cerri M, Capello D et al (2002) Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol 119:622–628
Kanno H, Ohsawa M, Hashimoto M et al (1999) HLA-A alleles of patients with pyothorax-associated lymphoma: anti-Epstein-Barr virus (EBV) host immune responses during the development of EBV latent antigen-positive lymphomas. Int J Cancer 82:630–634
Mauray S, Fuzzati-Armentero MT, Trouillet P et al (2000) Epstein-Barr virus-dependent lymphoproliferative disease: critical role of IL-6. Eur J Immunol 30:2065–2073
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Balague Ponz, O., Ott, G., Hasserjian, R.P. et al. Commentary on the WHO classification of tumors of lymphoid tissues (2008): aggressive B-cell lymphomas. J Hematopathol 2, 83–87 (2009). https://doi.org/10.1007/s12308-009-0038-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-009-0038-8