
Abstract
One person on three will receive a diagnostic of cancer during his life. About one third of them will die of the disease. In most cases, death will result from the formation of distal secondary sites called metastases. Several events that lead to cancer are under genetic control. In particular, cancer initiation is tightly associated with specific mutations that affect proto-oncogenes and tumour suppressor genes. These mutations lead to unrestrained growth of the primary neoplasm and a propensity to detach and to progress through the subsequent steps of metastatic dissemination. This process depends tightly on the surrounding microenvironment. In fact, several studies support the point that tumour development relies on a continuous cross-talk between cancer cells and their cellular and extracellular microenvironments. This signaling cross-talk is mediated by transmembrane receptors expressed on cancer cells and stromal cells. The aim of this manuscript is to review how the cancer microenvironment influences the journey of a metastatic cell taking liver invasion by colorectal cancer cells as a model.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carcinogenesis is a multistep process that results from genetic alterations that underlie the transformation of normal cells into malignant derivatives. In fact, the genome of tumour cells is altered at several sites as a result of point mutations or changes in chromosome integrity. It is now accepted that cancer arises from a succession of genetic alterations that confer growth advantages leading to the progressive conversion of normal cells into cancer cells [1]. The genetic modifications that contribute to cancer initiation and progression are regrouped within six phenotypic traits that constitute the essential hallmarks of cancer. These have been described in detail in a didactic review by Hanahan and Weinberg and are summarized in Fig. 1 [1]. Firstly, cancer cell have acquired self-sufficiency of growth through mutations that activate proto-oncogenes allowing cancer cells to generate their own growth signals such as secretion of transforming growth factor-α (TGFα) or platelet-derived growth factor (PDGF). Secondly, cancer cells are characterized by their insensitivity to antigrowth signals, in part through disruption of the Rb pathway. Thirdly, cancer cells have gained the capability to escape apoptosis through mutations that inactivate tumour suppressor genes such as p53 or PTEN, or through activation of survival pathways involving insulin-like growth factor-1/2 (IGF1/2) or interleukin-3 (IL-3). Fourthly, cancer cells have lost the cell-autonomous program that limits their multiplication. This results mainly from turning on of telomerase, an enzyme that maintains the telomeres at the end of chromosomes, thereby impairing senescence and death. Fifthly, the growing neoplasm becomes capable to sustain its oxygen and nutritive supplies by promoting angiogenesis. This neovascularization process is turned on by the expression of pro-angiogenic agents such as vascular endothelial growth factor (VEGF) produced by cancer cells or by the repression of anti-angiogenic processes such as a fall in thrombospondin levels. Sixthly, cells from the primary neoplasm detach from this site to invade adjacent tissues and travel to distant sites that they will colonize to form metastases. Recent studies have indicated that during the acquisition of these inherent hallmarks, the cancer cells need to interact synergistically with their surrounding microenvironment to form a neoplasm and to progress further to colonize distant organs. This review will focus on the major cancer cell-microenvironment interactions that pave the journey of a metastatic cell from the primary site to the metastatic site, taking the invasion of the liver by colorectal cancer cells as a model.

The hallmarks of cancer cells. Cancer cells from most human cancers share six acquired traits that collectively dictate malignant growth: (a) self-sufficiency with respect to growth signals, (b) insensitivity to growth-inhibitory signals, (c) evasion of programmed cell death, (d) limitless replicative potential, (e) sustained angiogenesis, and (f) tissue invasion and metastasis. All these traits contribute to growth, detachment and invading potential of cancer cells (adapted from [1])
Interaction of the Primary Neoplasm with its Microenvironment
Colorectal cancer arises mostly from dysplastic adenomatous polyps. As with other types of cancers, it involves a multistep process characterized by the inactivation of a variety of genes that suppress tumours (e.g.: APC, DCC, p53) and repair DNA (e.g.:hMSH2, hMSLH1) and the simultaneous activation of proto-oncogenes (e.g.: K-ras, N-Ras and c-Myc). This confers a selective growth advantage to colonic epithelial cells and triggers the transformation of the normal epithelium into pre-cancerous adenomatous polyps and subsequently leads to local invasion and metastasis. There are several distinct neoplastic syndromes in which individuals display a marked predisposition to the development of colorectal cancer. In particular, genetic lesions in the familial form of adenomatous polyposis (FAP) and hereditary non-polyposis colorectal cancer (HNPC) are now well delineated and their study has contributed enormously to our understanding of the molecular pathology of sporadic colorectal cancer [2]. Beyond the intrinsic genetic alterations that affect the colon cancer cells, a dynamic interaction occurs between cancer cells and the host stromal microenvironment to support cancerous growth and dissemination [3, 4]. The stroma is constituted mainly of cellular elements such as fibroblasts and non-cellular elements such as the extracellular matrix (ECM). These stromal elements act in a synergistic cross-talk with cancer cells from the primary sites to sustain cancer growth and metastasis. The importance of the stroma in cancer is highlighted by histological observations indicating that some types of neoplasms such as pancreatic cancer are composed mostly of stromal cells [5, 6]. We will first review the influence played by the stroma on the development of the primary neoplasm and thereafter on the formation of metastases.
Influence of Stromal Cells on the Primary Neoplasm
Cancer cells of the primary neoplasm constitute a histological lesion that is embedded in the stromal microenvironment of a given tissue. The tumour stroma plays an essential role in the development of colorectal cancer and is referred to as a “reactive stroma” [7]. This reactive stroma is associated with an increased number of fibroblasts, enhanced capillary density and deposition of a new ECM rich in type-1-collagen and fibrin. Macrophages are also recruited in the reactive stroma in response to the healing process generated by the tumour. There is evidence indicating that the primary neoplasm and the reactive stroma communicate in a reciprocal way via the basement membrane [7]. Moreover, several studies show that the reactive stroma influences the initiation and promotion of cancer by secreting cytokines and growth factors or by expressing their receptors (Fig. 2). For example, stromal cells within colon carcinoma express high level of PDGF receptor (PDGFR). In turn, this has a marked impact in colon cancer progression since chemical inhibition of PDGFR signaling in the stromal cells inhibits cancer progression [8]. On the other hand, several studies indicate that fibroblasts within a tumour harbor mutations that activate them into myofibroblasts or cancer associated fibroblasts (CAFs). These genetically altered fibroblasts may directly be involved in cancer initiation as demonstrated in mouse models. For example, when immortalized prostate epithelial cells are grafted in mice together with normal fibroblasts or CAFs taken from the primary site, intraepithelial neoplasia of the prostate emerge only in the presence of CAFs [9]. This clearly indicates that CAFs have the potential to initiate tumour formation. Along these lines, CAFs produce cytokines such as TGFβ that activate cancer cells and trigger their detachment from the primary neoplasm. Interestingly, a recent study showed that TGFβ also leads to the transformation of lung endothelial cells into CAFs, which favors progression of lung cancer in a mouse model. Overall, this study indicates that endothelial cells contribute to expand the pool of CAFs that synergize with cancer cells to favor their progression [10]. This important study further highlights the intricate signaling network that exists between a tumour and its environment.

Interaction of cancer cells with the microenvironment. Beyond genetic alterations that affect cancer cells, a dynamic interaction occurs between cancer cells and the host stromal microenvironment to support cancerous growth and dissemination. The stroma is constituted mainly of cellular elements such as fibroblasts and immune cells, and non-cellular elements such as ECM. These stromal elements act in a synergistic cross-talk with the cancer cells from the primary sites to sustain cancer growth and metastasis. For example, cancer-associated macrophages and fibroblasts influence cancer initiation/promotion by secreting cytokines, growth factors and chemokines
Macrophages are another important type of cells recruited to the primary neoplasm. These tumour-associated macrophages (TAMs) can constitute a large proportion of the tumour mass [11]. Incidentally, their recruitment to the tumours is a response to cancer-associated inflammation and it may explain why some cancers respond to anti-inflammatory agents [12, 13]. The importance of these TAMs in cancer is supported by the fact that their presence within a tumour correlates with a poor prognosis. As a corollary, removal of macrophages in mice (via a homozygous null mutation of the genes encoding macrophage growth factor, colony stimulating factor-1) reduces the rate of tumour progression and impairs metastasis in a mouse model of breast cancer [14]. In an excellent review, Condeelis and Pollard proposed six extrinsic traits by which macrophages enhance tumour incidence, progression and metastasis. These are chronic inflammation, matrix remodeling, tumour cell invasion, intravasation, angiogenesis and seeding at distant sites [15]. In the case of colon cancer, it seems that TAMs favor metastasis by secreting VEGF, thereby promoting angiogenesis at the primary site [16]. Intriguingly, a recent study shows that TAMs promote removal of apoptotic colon cancer cells that express the sulfoglycolipids SM4s. During the process, the phenotype of TAMs is modified, as it is associated with an increased expression of TGF-β1 and secretion of IL-6. This phenotypic modification may putatively contribute to further activate the angiogenic process [17].
Interestingly, the recruitment of macrophages and other immune cells to a tumour contributes to develop adaptive immune response against this tumour. Along these lines, certain subsets of lymphocytes within a tumour are associated with a favorable prognostic. In particular, the presence of intra-tumoural T cells correlates with a better clinical outcome in advanced colorectal carcinoma [18, 19]. Tumours also develop mechanisms that allow evading immunosurveillance [20]. Tumour-infiltrating macrophages may generate arginine metabolites that lead to T-cell death [20]. More insidiously, tumour cells or their microenvironment can produce molecules that kill effector T cells. For example, colon cancer cells show an increased production of Fas ligand that binds to its receptor on immune cells to trigger their apoptosis and decrease immunosurveillance [21]. Another mechanism developed by cancer cells to escape the immune system is to increase the expression of carbohydrates at their surface. This masks surface antigens on the cancer cells, thereby impeding their recognition by immune cells. In this context, galectin-1 expression by head and neck cancer cells negatively correlates with patient survival [22]. Moreover, it is now apparent that in tumour draining lymph nodes of advanced cancers, dendritic cells are actively immunosuppressive. These dendritic cells do not activate T cells but rather induce tolerance to tumour antigens [23].
Influence of the Extracellular Matrix on the Primary Neoplasm
Most mammalian cells are in contact with an ECM whose composition and structure vary with development and diseases such as cancer as well as with cell types and their location. The ECM is composed of five classes of macromolecules (collagens, laminins, fibronectins, proteoglycans and hyaluronans). Each class of macromolecules exists as various isoforms depending on the cell type within the ECM. By binding to integrins at the cell surface, the ECM provides mechanical and physicochemical support for cells. It also provides pathways for cell migration and sequesters growth factors that influence cellular behaviour. The basement membrane (BM), a specialized sheet produced through cooperation of epithelial and stromal cells, is a particular type of ECM with a significant role in cancer. It is a dense meshwork of ECM molecules such as Type IV collagen, laminins, Type VII collagen, and heparan sulfate proteoglycans. Type IV collagen is a major structural component of BMs that forms a network enabling the binding of other BM components. Inappropriate synthesis or degradation of any ECM molecules alters cell physiology and causes disease. Accordingly, a loss of BM components due to degradation by proteolytic enzymes and/or a lack of biosynthesis are correlated with tumour progression. For example, Type VII collagen is lost early in the development of malignant melanoma [24], breast cancers [25], and prostate carcinomas [26]. Similarly, laminin-5 is commonly lost in colon carcinomas but not in pre-malignant tumours. On the other hand, de novo synthesis and/or deposition of BM components are associated with different types of tumours, which suggests an active role for BM molecules in tumour invasion. In gastric, liver, and pancreatic carcinomas, laminins are produced and deposited at the invasive front of the tumour clusters. The expression of laminin-1 in gastric carcinoma is a risk factor for liver metastasis. Moreover, in human gastric and pancreatic carcinomas, laminin-5 is expressed by invasive malignant cells, and is considered as a marker of invasiveness [27–30]. These modifications of the BM occur simultaneously to changes in transmembrane adhesive receptors expressed by tumour cells in response to the “new” ECM ligands. In this sense, the best-characterized example is the “switch” of function of the α6β4 integrin. The usual role of this integrin is to provide stable adhesion of epithelial cells to BM. However, this integrin has been identified as a tumour antigen also designated as TSP-180 or A9 [31, 32]. Furthermore, increased expression of α6β4 and changes in its distribution correlate with increased aggressiveness of tumours and poor prognosis [33, 34]. In colon carcinoma, the α6β4 integrin promotes cell migration on laminin-1, since it induces and stabilizes actin-containing motility structures, instead of being associated with hemidesmosomes to support stable adhesive structures [35].
Metabolism of ECM molecules is an important aspect of tissue homeostasis and determines how cells respond to acute and chronic stresses. Various types of proteinases participate in ECM turnover, but matrix metalloproteinases (MMPs) are the principal ECM-degrading enzymes. These enzymes have a central role in cancer progression given that ECM degradation products can influence stroma–cancer cell interactions. In particular, metastatic colon cancer cells are able to induce the expression and/or secretion of MMP-2 and MMP-9 in stromal cells, either via direct contact or via a paracrine regulation [36]. Because of the multi-functionality of MMPs, it is conceivable that they influence several stages of cancer development.
Epithelial–Mesenchymal Transition
Cancer cell detachment from the primary site is one of the key initial events required for metastasis. This is tightly associated with epithelial–mesenchymal transition (EMT), a morphogenetic process in which epithelial cells loose their characteristics and gain mesenchymal properties during embryogenesis [37] (Fig. 3). There is now accumulating evidence indicating that carcinoma cells usurp normal developmental EMT to detach from the primary neoplasm and migrate to distant sites. In this context, histochemical studies reveal that adenocarcinoma is accompanied by the release of single cells through EMT [38]. In particular, it appears that EMT is an integral component of colorectal cancer progression [39, 40]. One of the major elements that characterize EMT of carcinoma cells is the loss of E-cadherin-mediated cell–cell adhesion [41]. This latter event results from mutations in the E-cadherin gene, proteolytic degradation of E-cadherin, IGF1-mediated internalization of E-cadherin and disruption of the function of E-cadherin involving β-catenin [41]. Loss of E-cadherin further results from a decrease in its expression that is subsequent to promoter hypermethylation and activation of transcriptional repressors such as Snail and FOXC2 [42, 43]. In colon cancer cells, deregulation of E-cadherin and EMT is associated with a peripheral accumulation of Src and phospho-myosin and with an increased expression of the guanine nucleotide exchange factor TIAM 1 [44, 45]. Moreover, the level of RhoC varies widely in colon cancer cells and an elevated RhoC expression correlates with a poor prognostic and with an aberrant expression and localization of E-cadherin [46]. Interestingly, in melanoma, breast cancer and prostate cancer, the disruption of the E-cadherin-mediated cancer cell adhesion is associated with the so-called cadherin-switch in which the E-cadherin loss is accompanied by de novo expression of mesenchymal cadherin, such as N-cadherin. This shift is important since the loss of E-cadherin by cancer cells is associated with their inability to adhere with themselves and with normal epithelial cells. Furthermore, by up-regulating the expression of N-cadherin, cancer cells will interact with stromal cells, thereby changing their location and favoring the invasion of the surrounding stroma. Moreover, N-cadherin confers motility and migration to cancer cells [47]. Hence, the cadherin switch is an important component of the EMT that characterizes the early step of cancer progression toward an invasive and metastatic phenotype (Fig. 3).

The epithelial–mesenchymal transition. Epithelial-to-Mesenchymal Transition (EMT) is a morphogenetic process in which epithelial cells loose their characteristics and gain mesenchymal properties during embryogenesis and during progression of cancer. Carcinoma cells acquire a mesenchymal-like state in order to facilitate their migration and invasion. The EMT process is induced and regulated by effectors such as growth factors (TGFβ, PDGF, EGF), cytokines (Il-8) and ECM components. It is characterized by loss of epithelial markers such as E-cadherin and cytokeratins and gain of mesenchymal markers such as N-cadherin and vimentin
The Tumour Microenvironment in Epithelial–Mesenchymal Transition Signaling
The tumour microenvironment is a potent factor that may facilitate and even initiate EMT. In this regard, several arguments support the concept that cancer EMT is importantly regulated by the surrounding stroma via the activation of the same signaling pathways as those regulating EMT during embryogenesis. Among others, embryonic and cancer EMTs are characterized by fluctuating levels of TGFβ, EGF and PDGF [3]. Notably, TGFβ signaling is associated with the induction and maintenance of EMT and its effect may be mediated through autocrine and paracrine pathways [48]. For example, TGFβ plays a crucial role in EMT of colon cancer cells and it has been proposed that its role is triggered by TNFα produced by infiltrating macrophages [49, 50]. Typically, the binding of TGFβ with serine–threonine kinase receptors TGFβR1/TGFβR2 triggers the phosphorylation of the Smad2/Smad3 dimers that dissociate from the receptors to interact with Smad4 before entering the nucleus to regulate transcriptional modulation of EMT. In particular, in kidney tubular epithelia and NMuMG breast epithelial cells, TGFβ-induced EMT is dependent on the down-regulation of E-cadherin via Smad3 [51]. Moreover, the high mobility group A2 (HMGA2) gene is induced by the TGFβ–Smad pathway, which is necessary and sufficient for TGFβ-induced EMT. HMGA2 is a nuclear factor that links TGFβ signaling with the EMT-inducing transcription factors Snail1, Snail2 and Twist [52]. Signaling pathways that mediate the activity of β-catenin and LEF may also cooperate with TGFβ/Smads to form new transcriptional complexes inducing EMT [3]. For example, PDGF and PDGFR signaling is essential for TGFβ-induced EMT in mammary epithelial cells. It is possible that these pathways interact at the transcriptional level. Along these lines, TGFβ/Smads may interact with the PDGFR-dependent phosphorylation of nuclear p68 RNA helicase to trigger nuclear translocation of β-catenin via a Wnt-independent pathway [53]. Several other signaling pathways activated by TGFβ are also involved in modulating EMT. In NMuMG mouse mammary epithelial cells, TGFβ, produced via an autocrine pathway, induces EMT in conjunction with integrin β1, upstream of RhoA and the p38 MAP kinase [54, 55]. Interestingly, it has been proposed that the propensity of colon cancer to undergo EMT relies on a synergistic cooperativity between continuous TGFβ signaling and an activated Ras pathway [39, 56]. In mice, Ras-transformed EpH4 cells progressively acquire a mesenchymal phenotype in association with an autocrine production of TGFβ [3]. On the other hand, Ras-transformed hepatocytes and MDCK cells undergo TGFβ-induced EMT in contrast to their parental counterpart and this effect transits through both ERK and PI3 kinase pathways [57]. The precise events that modulate TGFβ-mediated EMT via these pathways are unclear. However, it is likely that they are associated with cytoskeletal remodeling and increased cell motility. In this context, the expression of integrin α6β4 as a consequence of the EMT enables motile and invasive colorectal cancer cells to interact with interstitial matrices and to sustain activation of TGFβ [39].
Overall, a lot of observations converge on the fact (1) that cancer EMT “highjacks” the same signaling pathways as those leading to embryonic EMT, and (2) that tumour microenvironment may act as an initiator of these signaling cascades.
Angiogenesis
Tumour growth and progression are both dependent on angiogenesis to provide cancer cells with oxygen and nutriments required for their growth. In fact, cancer cells cannot grow when they are located at a distance greater than 110 μm from a blood vessel. At a later stage during the process, angiogenesis is further involved as an important step of metastasis, given that the cancer cells that detach from the primary site will use the new vessels as a road to reach and colonize new sites. Accordingly, cancer cells begin to promote angiogenesis early in tumourigenesis in order to sustain their growth. The process of angiogenesis is regulated by a tight balance between pro- and anti-angiogenic agents. Typically, the “angiogenic switch” is initiated by tumour-associated hypoxic conditions, which contributes to activate the transcription of hypoxia-inducible factor-1 (HIF-1). In turn, HIF-1 promotes the expression of several angiogenic factors by the cancer cells including VEGF, basic fibroblast growth factor (bFGF) and placenta-like growth factor (PLGF) [58]. Interestingly, there is accumulating evidence indicating that the expression of these angiogenic agents by cancer cells is also increased through HIF-1-independent pathways [59]. In this context, tumour angiogenesis is preserved in HIF-1-deficient xenografts [60]. It seems that oncogenic ras and the intracellular levels of calcium play a key role in regulating angiogenic gene expression in response to hypoxia [59]. Once they are expressed, the angiogenic growth factors interact with their respective receptors at the surface of endothelial cells activating the angiogenic program. In particular, VEGF binds to VEGFR2, which triggers signaling cascades involving eNOS, PI3 kinase, p38 and ERK MAP kinases and leading to the induction of vasodilatation, endothelial cell migration/proliferation and vessel assembly [61]. Fibroblasts in or near the tumour bed also produce pro-angiogenic factors, and tumours recruit progenitor endothelial cells from bone marrow. The angiogenic switch also involves down-regulation of angiogenesis suppressor proteins, such as thrombospondin [62]. We will not further review the mechanisms of angiogenesis, as the topic has been covered in recent reviews [61, 63, 64].
Angiogenesis is importantly involved in the growth and dissemination of colorectal cancer, and the level of angiogenesis is a predictor of survival in patients with this disease [65]. Along these lines, a color doppler vascularity index (ratio of colored pixels/total pixels per tumour section) predicts distant metastasis and survival in colon cancer patients. More precisely, patients who have a doppler vascularity index at the primary site greater than 15% have a poorer overall survival than the patients with an index <15% [66]. As a corollary, the doppler hepatic perfusion index (ratio of hepatic arterial flow to total liver flow) is increased even in patients with occult micro-metastases and is predictive of subsequent hepatic relapse [67]. Similarly, the correlation existing between angiogenesis of the primary colon cancer site and distal hepatic sites is supported by several imaging approaches including dynamic contrast-enhanced magnetic resonance imaging and contrast-enhanced computed tomography [65]. The importance of angiogenesis in colon cancer metastasis to the liver is further highlighted by the finding that anti-angiogenic agents such as the anti-VEGF-directed antibody bevacizumab in combination with standard chemotherapy offers stimulating promises in blocking the metastatic process and prolonging patient survival [68].
Lymphangiogenesis
In solid cancers including colorectal cancer, the status of the lymph node is the most important prognostic indicator for the clinical outcome of patients [69–71]. However, the mechanistic basis underlying the role played by the lymphatic system in metastasis remained rather elusive until the pioneered work of Kari Alitalo’s group who reported the existence of lymphangiogenesis and showed that the process is used by cancer cells to disseminate [72]. VEGF-C is the main regulator of lymphangiogenesis, and its levels correlate with lymph node metastases. The VEGF-C lymphangiogenic effect is mediated through its binding to VEGFR-3 present on lymphatic endothelial cells. VEGF-D also binds to this receptor and contributes to lymphangiogenesis. The activation of this receptor in association with β1 integrin sub-unit initiates different signaling pathways in the lymphatic endothelial cells, including ERK and JNK MAP kinases, Pyk2, NF-κB and AKT, that govern proliferation, survival and sprouting [73]. As for blood vessel angiogenesis, several other growth factors participate to lymphangiogenesis. In particular, angiopoietin-1, by binding to its receptor Tie2, promotes lymphatic vessel sprouting, hyperplasia and growth in experimental animal models [74]. At present, it is not clear how lymphangiogenesis is induced but it is likely that it follows similar steps as blood vessel angiogenesis. Moreover, lymphangiogenesis may be initiated by the incorporation of circulating endothelial progenitors (CEPs) into existing lymphatic vessels. CEPs are a subpopulation of circulating endothelial cells (CECs) derived from the bone marrow [71]. These CEPs are more abundant in the circulation during pathological conditions including cancer. Interestingly, a subset of CD34+ cells co-express VEGFR-3 and CD133 and may represent a subpopulation of lymphatic endothelial progenitors that may migrate from the circulation and integrate into expanding tumour-related lymphatic vessels [75]. Other studies also show that peri-tumoural lymphangiogenesis occurs without incorporation of donor-derived CEPs. In this context, tumour lymphangiogenesis in a mouse xenograft model can occur by sprouting from pre-existing local lymphatic networks without contribution of CEP [76]. An alternative contribution to tumour lymphangiogenesis is the transdifferentiation of other cell types especially macrophages and host stromal cells into lymphatic endothelial cells [77, 78].
Lymphangiogenesis is present in colorectal cancer and the lymphatic vessel density of the tumour correlates with depth of invasion and metastasis not only to regional lymph nodes but also to the liver. The reason for the close correlation between hepatic and lymph node metastasis of colorectal cancer is still unclear. One possibility relies on the fact that colorectal cancer cells express the CXCR5 chemokine receptor and that both lymph endothelial cells and the liver express its ligand BCA-1/CXCL13 [79]. Hence, it is possible that cancer cells may be directed via lymphangiogenesis to both lymph nodes and liver, which would explain the correlation between lymph node and liver metastasis of colon cancer. On the other hand, lymph node metastases are found at the site of lymph node drainage of the liver namely, celiac, portal and mediastinal lymph nodes. This further raises the possibility that these lymph node metastases are derived from the liver metastases during a process of colon cancer re-metastasis [80]. Here again, this finding could explain the correlation between lymph node and liver metastasis of colorectal cancer.
Obviously, tumour blood vessel angiogenesis and lymphangiogenesis are among the best examples of the insidious cross-talk that occurs between colon cancer cells and their surrounding stroma in tumour development and dissemination.
Intravasation of Cancer Cells
Following their detachment from the primary neoplasm, cancer cells enter or intravasate into the existing or newly-formed blood or lymphatic vessels to disseminate. The process of intravasation is not very well understood in the case of colon cancer. Several molecular steps involving matrix metalloproteinases and interaction between cancer cells and endothelial adhesion molecules have been described. For example, studies in patients with familial adenomatous polyposis, indicate that urokinase plasminogen activator (u-PA) levels increase during the normal epithelia transition to dysplastic lesions, thus implicating u-PA in the early stages of tumour development [81, 82]. In fact, the u-PA system plays a determinant role in the intravasation of colorectal cancers [83]. Following its binding to receptor (u-PAR), u-PA activates plasminogen and other proteases such as matrix metalloproteinases-2 (MMP-2) and MMP-9. In turn, this contributes to breakdown the ECM, a step that favors intravasation. A recent study has shown that u-PAR gene expression is induced downstream of Src-mediated activation of the AP-1 promoter and by c-Jun phosphorylation at Ser73 and Ser63. Src activity increases during the transition from benign colonic polyps to primary colon cancers and to colon cancer metastases [84]. In addition, Src activity is associated with an increased invasion in vitro of colon cancer cells [85]. Moreover, an elevated Src activity is a poor prognostic factor in colorectal cancer patients [86]. Hence, it is possible that Src by regulating the u-PA/u-PAR system may contribute to ECM breakdown, thereby enabling the access of the detached cancer cells to the vessels and favoring their intravasation. By degrading the ECM, motile colon cancer cells create their path to the vasculature. Moreover, the ability of cancer cells to survive in the presence of broken ECM fragments constitutes an inherent property of metastatic cells. Accordingly, selection for anoikis-resistant cells increases the in vivo metastatic capacity of melanoma cells in mouse model systems [87]. In the case of colon cancer cells, this might possibly be due to the fact that they overexpress focal adhesion kinase (FAK), suggesting that they can by-pass integrin signaling and thereby survive in the absence of contact with the ECM [88]. Reciprocally, specific experimental deletion of FAK in a mouse model of skin tumour is associated with the induction of cell death and the inhibition of tumour progression [89]. FAK may contribute to confer survival by activating survival signal pathways, such as ERK or AKT [90, 91]. Following their survival and migration in the degraded ECM, cancer cells gain access to the blood vessel. At this step, it seems that microvessel diameter is the dominant parameter underlying colon cancer intravasation via passive entry [92, 93]. Once again, the cancer cells must acquire further capacity to survive the process of intravasation. Indeed, a large number of non-metastatic cells undergo fragmentation when they interact with blood vessels, whereas metastatic cells demonstrate an increased survival during this process [94].
As described in the previous sections, colon cancer cells can also disseminate via the lymphatic vessels, which is supported by the close correlation existing between liver metastasis and lymphangiogenesis. The mechanisms by which lymphangiogenesis contributes to colon cancer and cancer metastasis in general are still unclear. It is possible that VEGF-C stimulated lymphatic endothelial cells secrete chemotactic or mitogenic factors that will attract cancer cells to the newly growing lymph vessels enabling their adhesion and intravasation on and through the lymphatic endothelium [72]. In this context, expression of CXCR4 and CCR7 by human breast cancer cells facilitate their migration to the lymph nodes [95]. Once they have interacted with the endothelium of the lymphangiogenic vessels, the entry of cancer cells in these vessels will be greatly facilitated by the fact that they are leaky and tortuous (Fig. 4). As mentioned earlier, colorectal cancer cells express the CXCR5 chemokine receptor and both lymph endothelial cells and the liver express its ligand BCA-1/CXCL13 [79]. It is, then, possible that cancer cells may be directed via lymphangiogenesis to both lymph nodes and liver.

Mechanisms of VEGF-C-induced intravasation of cancer cells across lymph vessels. In several human cancers, increased expression of VEGF-C in primary tumours correlates with regional lymph node metastases. It is possible that a reciprocal cross-talk exists between tumour cells and lymphatic endothelial cells to induce tumour lymphangiogenesis and formation of lymph node metastases. Notably, VEGF-C activates lymphatic endothelial cells (1) that in turn may secrete chemotactic factors (2). This will contribute to attract cancer cells bearing appropriate chemokine receptors to the growing lymph vessels (3), and enable their adhesion and their intra-lymphatic intravasation
Circulation of Cancer Cells
As described above, tumour cells enter the blood circulation directly or indirectly via the lymphatic system. In the case of colon cancer cells, they use the hepatic–portal circulatory system to enter the liver. Interestingly, a large number of cancer cells can be detected in the blood of cancer patients. However, the number of circulating cancer cells do not correlate with the level of metastasis. This suggests that circulation is deleterious for most cancer cells. In fact, several experimental evidences have shown that, only 0.01% of metastatic clonal cancer cells injected into the circulation are able to generate metastases. A major question is then to understand what the fate of cancer cells that enter the circulation is. This question has been addressed in reviews by Ann Chambers, Patrick Mehlen and their colleagues [91, 96]. They report that death of solitary cells in the circulation occurs mostly through tumour immune surveillance or destruction by mechanical stresses. In fact, studies using radio-labeled or fluorescent-labeled cancer cell lines injected into the circulation and intravital videomicroscopy support the hypothesis that solitary cancer cells in the circulation, or soon after extravasation, are sensitive to apoptosis and die [97–99]. In line with this concept, there is a tenfold decrease in the number of apoptotic cancer cells that are found at the secondary site 1 hour following the injection of cells rendered resistant to apoptosis via overexpression of Bcl2 [98]. As reported above, cancer cell death might occur as a result of two major mechanisms: cell destruction by mechanical stress and cell destruction by the immune system.
The mechanical stress imposed by the circulation is a major stress that cancer cells must survive to form metastases. In fact, mechanical destruction of circulating cancer cells is the first line of defence in the host microenvironment that acts against hematogenous cancer dissemination [100]. Cancer cells circulating in the blood are submitted to strong mechanical forces caused by blood flow. This is especially important in narrow capillaries and within the microvasculature of contracting skeletal and heart muscle, in which sphere-to-cylinder shape transformation is lethal to most cancer cells [101–103]. Moreover, NO and ROS are known to trigger apoptosis of cancer cells, and their production is activated by mechanical forces including shear and pulsatile stresses and contraction of the endothelium [91]. The production of these oxyradicals is required for apoptosis of intravenously injected melanoma cells or colorectal cancer cells in hepatic sinusoids [104, 105]. Cancer cells have developed several ways to survive the mechanical stresses associated with circulation. Notably, they express high levels of stress proteins such as HSP70. Moreover, they associate with platelets and with themselves in order to form a shield that protects them against external aggression [106, 107]. In addition, shear stress can induce pathways in the cancer cells that involve activation of integrins leading to adhesion of cancer cells to endothelial cells, thereby allowing them to escape anoikis. Nevertheless, these surviving processes are effective to only a small percentage of circulatory cancer cells since only 0.01% of these form metastases. In summary, the balanced response of tumour cells to mechanical destruction and adhesion initiation by circulating stress is an important mechanism that regulates the spread of hematogenous cancer.
Immunosurveillance is also involved in detecting and eliminating circulating tumour cells [108]. Accordingly, interleukin-2 (IL-2) is effective in treating patients with metastatic melanoma and kidney cancers, and its anti-tumoural activity is closely related to its ability to expand and activate specific subsets of immune cells, such as T cells and natural killer (NK) cells [109]. Other cytokines, such as IL-12 and IL-18, stimulate NK cells to suppress metastasis [110]. Mechanisms of tumour-cell killing by NK cells seem to be mediated either by the NKG2/perforin or Trail pathways [111, 112]. Circulating cancer cells have developed several mechanisms to escape immunosurveillance. In some cases, they can interact with platelets and fibrinogen to form clots that protect them against immune cells and facilitate their arrest and their adhesion on the endothelium [113, 114]. Furthermore, cancer cells may use inflammation-associated mechanisms to further protect themselves against apoptosis. For example acute-phase glycoproteins reach abnormally high levels in patients with cancer, which correlate with the extent of disease. It has been shown that these glycoproteins, synthesized by the liver in response to an inflammatory stimulus, may act as ‘non-specific blocking factors’ protecting tumours against the host’s immunological attack [115]. Intriguingly, there is no evidence that human immunodeficiencies are associated with increased development of metastasis from solid tumours. Hence, it is still difficult to understand the specific contribution of tumour-cell death induced by the immune system in limiting metastasis. Nevertheless, manipulating the immune system to induce tumour regression still has a great therapeutic interest.
Extravasation of Cancer Cells
The tumour microenvironment encountered by the invading cancer cells is a critical component of metastasis and should be permissive for the metastatic growth of incoming tumour cells [116]. Notably, the adhesive interactions between the cancer cells and the endothelial cells of the target organs are determining components of metastasis. These interactions determine the arrest of the circulatory cells in the capillaries and initiate the cascade of events that culminate in extravasation or diapedesis of the cancer cells in the colonized organs. The extravasation of circulating tumour cells in the host organ requires successive adhesive interactions between endothelial cells and their ligands or counter-receptors present on the cancer cells [117]. Typically, the colon cancer cell/endothelial cell interactions in the liver imply first a selectin-mediated initial attachment and rolling of the circulating cancer cells on the endothelium. The rolling cancer cells then become activated by locally released chemokines present at the surface of endothelial cells. This triggers the activation of integrins from the cancer cells allowing their firmer adhesion to members of the Ig-CAM family such as ICAM and VCAM, initiating the transendothelial migration and extravasation processes [118] (Fig. 5). Interestingly, the expression of endothelial adhesion receptors may be induced by the cancer cells via a paracrine pathway. This is supported by studies showing that culture medium supernatants of cancer cells can trigger the expression of E-selectin on endothelial cells suggesting that cancer cells may release cytokines such as TNF-α, IL-β or INF-γ that will directly activate endothelial cells to express E-selectin and by extension P-selectin, ICAM-2 or VCAM [119, 120]. On the other hand, other studies show that cancer cells may initiate the expression of endothelial adhesion molecules in more indirect ways. In particular, highly metastatic human colorectal and mouse lung carcinoma cells, on their entry into the hepatic microcirculation, initiate a rapid host inflammatory response by inducing TNF-α production in resident Kupffer cells. In turn, this event triggers the expression of E-selectin and VCAM by endothelial cells and enhances the binding and extravasation of the cancer cells across sinusoidal endothelial cells [121, 122]. Moreover, the kinetics of the host inflammatory response are tumour-type specific and rely on interactions with hepatic Kupffer cells, which supports the concept that the process is local and liver-specific. In this sense, the process of E-selectin- and other endothelial adhesion receptor-mediated metastasis appears to be local. In particular, increased hepatic local metastasis of B16F1 melanoma cells is observed following the administration of exogenous IL-1α, which induces the expression of vascular adhesion receptor expression, including E-selectin, VCAM-1 and ICAM-1 enabling cell arrest in terminal portal venules [123]. Moreover, syngenic colon adenocarcinoma C-26 cells trigger an endogenous inflammatory cascade upon entry into the liver. The cascade is absent in TNFR1-deficient mice, which is associated with reduced formation of liver metastases [124]. Overall, these findings indicate that, at least in the case of the formation of colorectal metastases in the liver, the process results from a local remodeling of the hepatic microenvironment that enables adhesion and extravasation of the cancer cells. In corollary, these results further suggest that hepatic remodeling involving E-selectin-mediated adhesion is an important component of the hepatic homing of colon cancer metastases.

Extravasation of cancer cells is a multi-step process. The first step consists in the transient adhesion of cancer cells to the endothelium. It involves endothelial adhesion molecules such as E-selectin and P-selectin and their counter-receptors present on cancer cells. This step is associated with the rolling of the cancer cells on the endothelium (1). The second step consists in a firmer adhesion of cancer cells to endothelial cells (2). It is mediated through chemoattractants and cell adhesion molecules on the endothelium and integrins on the cancer cells. The third step is characterized by the extravasation of cancer cells through endothelial cell–cell junctions (3). EC Endothelial cells, TC tumour cell (adapted from [107])
The binding of cancer cells to E-selectin requires a molecular scaffold composed of oligosaccharides that are borne by a carrier protein and possibly a lipid on cancer cells. This macromolecular scaffolding complex is known as the selectin counter-receptor and is essential for cell binding [120]. We previously reported that Death receptor 3 (DR3) is a typical sialylated-a/-x protein that binds E-selectin and transmits its signal within cancer cells [125]. Other E-selectin ligands expressed by cancer cells involve ESL-1 and CD44 [107]. Interestingly, the binding of DR3 to E-selectin triggers the reciprocal activation of E-selectin, which induces a forward signaling in endothelial cells that contributes to increase endothelial permeability enabling transendothelial migration of cancer cells [126].
Since the adhesion of colon cancer cells to liver endothelium requires the presence of endothelial selectins and their appropriate receptors on the cancer cells, the degree of expression of selectins on the vascular wall and the presence of the appropriate ligand on cancer cells are determinant for their adhesion and extravasation into a specific organ. Along these lines, the homing of B16F10 melanoma cells to the liver requires the expression of E-selectin ligands by melanoma cells and the expression of soluble E-selectin in the liver [127]. On the other hand, endothelial selectins may be expressed differentially in blood vessels from different tissues. For example, LPS and TNFα strongly induce the expression of P-selectin on endothelial cells of the leptomeninges, but at a weaker level on some blood vessels of the brain parenchyma [128]. Based on these findings, it can be proposed that the differential specific interactions of selectins expressed by endothelial cells of potential target organs and their ligands expressed on cancer cells are major determinants that underlie metastasis and organ-specific distribution of metastases. Along these lines, lung is a secondary metastatic site for colon cancer and lung endothelial cells also express E-selectin upon appropriate stimulation [129, 130]. As mentioned previously in this section, it is clear that adhesion molecules other than E-selectin and its ligands are also involved in the early adhesion and extravasation of cancer cells. Notably, direct integrin (α2, α6, β1, β4)-mediated cell adhesion to the ECM in the space of Disse of liver sinusoids is required for the successful formation of liver metastases by colon cancer cells [131]. In addition, αvβ5 integrin expressed by metastatic colon cancer cells is required to enable their adhesion to an appropriate counter-receptor on hepatic microvessels [132]. Moreover, ICAM1 has been shown to dictate the binding of tumour cells to the pulmonary endothelial cells during metastasis [133].
Colonization
Once circulating cancer cells have gained access to the secondary site, their subsequent growth will depend on the compatibility of the cancer cells (seed) with the “soil” that they encounter in the new organ. Their growth will then be tightly regulated by molecular interactions between cancer cells and the new environment. Most cancer cells will die by apoptosis initiated by an adverse environment [99]. Only a small subset of them initiate cell division to form micrometastases, and only a small proportion of these micrometastases become vascularized and grow to form macroscopic metastases. In the case of colon cancer cells, it has been proposed that they release soluble CD44 which acts as a decoy receptor impairing the interaction of colon cancer cells with its hyaluronate ligand within the ECM, thereby conferring resistance to apoptosis [134]. On the other hand, several lines of evidence indicate that ECM, by modulating the expression of growth factors and growth-factor receptors in colon cancer cells, also modulates the proliferation of these cancer cells in the liver. For example, hepatocyte-derived ECM, in particular heparin proteoglycan, stimulate the proliferation of colon cancer cell lines via induction of autocrine growth factors and their receptors [135]. Another factor that may limit the proliferation of all the incoming cancer cells at the secondary site is the fact that the latter is poorly vascularized at the onset, being devoid of angiogenesis. This will yield dormant micrometastases that may eventually develop in macrometastases following appropriate remodeling of the microenvironment. Incidentally, dormant micrometastases that overexpress Ras will later be more prone to develop metastases that depend or not on angiogenesis. Moreover, the expression of the EGF receptor coupled to the expression of growth factors like TGF-α in the tissue are among the well-known molecular factors that influence the ability of colon and other cancer cells to grow in the liver [136, 137].
During the last 10 years, cancer researches have focussed on the role of chemokines in cancer progression. Chemokines are chemotactic cytokines that cause the directed migration of leukocytes along a chemical gradient leading to the “homing” of leukocytes to specific organs [138]. These chemokines are induced by inflammatory cytokines, growth factors and pathogenic stimuli. Immune cells, endothelial cells and cancer cells express chemokine receptors and respond to chemokine gradients. Many cancers have a complex chemokine network that influence the immune-cell infiltration of the primary and secondary tumour, as well as tumour cell growth, survival, migration and angiogenesis [139]. It is now well accepted that chemokines and their receptors influence the metastatic potential and site-specific spread of tumour cells [140]. For example, breast cancer cells and primary breast tumours express patterns of chemokine receptors that “match” chemokines specifically expressed in organs to which these cancers commonly metastasize, namely the lymph nodes, lung, bone marrow and the liver. Furthermore, blocking one of the chemokine receptors was found to inhibit metastasis of breast cancer cells in experimental animal models [95]. Similarly, the chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases in the liver [141].
As described in earlier sections of this review, progression of solid tumours involves transition of epithelial transformed cells into mesenchymal cells (EMT), a process by which cancer cells acquire a more invasive and metastatic phenotype. Subsequently, the disseminated mesenchymal cancer cells must undergo the reverse transition, MET, at the metastatic site to allow micrometastases to give rise to a secondary neoplasm corresponding to the primary one. In this regard, cancer cells from the secondary site re-express markers of epithelial cells such as E-cadherin [142]. Since initiation of tumour growth at the secondary site is the rate-limiting step in metastasis, this suggests that the ability of mesenchymal cells to undergo MET in the appropriate microenvironments, is a key feature of metastasis [143]. Curiously, despite the importance of MET, little is known of the mechanisms that are involved. In the case of colon cancer cell metastasis, it may be speculated that the liver microenvironment, especially a subpopulation of CAFs, is involved in altering the microenvironment that will contribute to activate the MET process. This will be achieved by promoting the cross-talk between autocrine/paracrine growth factors and cell adhesion molecules expressed by cancer cells and stromal cells. For instance, CAFs taken from liver of patients having colon metastases overexpress COX2 and TGF-β2 and they enhance the proliferation of colon cancer cells in in vitro co-culture systems [144].]. It is possible that the role of CAFs in colorectal metastasis is modulated through the production of IL-8, a chemokine related to invasion and angiogenesis [145, 146]. In addition, a recent study shows that the kinase profile of colorectal cells greatly differs whether they are taken from the primary or the secondary hepatic metastatic sites, which further indicates that the hepatic microenvironment has remodeled the signaling network of the colon cancer cells [147].
In summary, the ability of cancer cells to grow in a specific site depends on features that are inherent to the cancer cell and to the invaded organ, and on the active interplay between these factors. Much remains to be learned about the detailed molecular interactions between cancer cells and specific secondary site, and probably many organ-specific growth factor pathways remain to be identified. However, there is strong evidence indicating that these interactions are important in determining the survival and growing potential of a cancer cell in a specific organ.
Concluding Remarks and Clinical Outlook
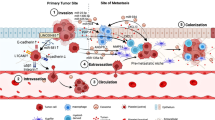
The journey of colon cancer cells from the primary site to the secondary sites is paved by obstacles that render the process destructive and ineffective (Fig. 6). Yet, some cells succeed and traverse all these impediments to generate metastases that will become fatal. In each case, the molecular events that lead to the massive destruction of the cancer cells or allow their survival during their journey is governed by an intricate interacting cross-talk between the cancer cells and its microenvironment. We have briefly reviewed some of these molecular interactions that characterize the journey of colon cancer cells to the liver. Much remains to be elucidated to fully understand the whole process. Nevertheless, little by little our knowledge increases, which contributes to improve our therapeutic intervention. The discovery that the anti-VEGF monoclonal antibody bevacuzimab increases the survival of colon cancer patient by impairing angiogenesis is one the best example. In fact, any therapy that interferes with the interaction of cancer cells with its microenvironment has promising clinical outcome. For example, one may envision targeting CAFs, EMT or MET as therapeutic purposes. Targeting the growth of metastases at the secondary site is particularly attractive since this event occurs often before the clinical detection of the primary neoplasm. In this context, components of the TGFβ pathway, a major player of EMT, are considered to be important new therapeutic targets. In line with this, new inhibitors of this pathway are already being tested in pre-clinical and clinical trials. Notably, four main strategies have been designed for disrupting TGFβ signaling: inhibition or sequestration of TGFβ, inhibition of TGFβ receptor kinase activity, inhibition of SMAD signaling downstream of TGFβ and restoration of anti-tumour immunity upon TGFβ inhibition [148]. In this regard, an antisense-based therapy against TGF-β2 is presently in a phase I/II study investigating treatment of colorectal cancer [149]. Moreover, approaches to increase immunosurveillance are under investigation and should lead to important therapeutic breakthroughs. Accordingly, interesting results have been obtained in mice using dendritic cell vaccination [150]. In our opinion, we believe that the next few years will be more fruitful in identifying new therapeutic targets in this direction.

The journey of a metastatic cell: from the primary site to the secondary site. Cancer cells have a “long and difficult” journey to go on before colonizing a secondary site and form metastases. This journey comprises several milestones that are summarized as follows: a Proliferation of primary tumour, b Local invasion of detached cells. c Intravasation in a capillary, and tumour cell survival in blood circulation: 1, interaction with leukocytes that may be destructive or not; 2, aggregation with platelets, which protects cancer cells against mechanical stress and leukocytes; 3, tumour cell–cell aggregation, which protects against stress and formation of intra capillary thrombosis. d Arrest and extravasation in a target organ. e Metastatic growth in the new appropriate environment and formation of a secondary neoplasm
References
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100:57–70
Ballinger AB, Anggiansah C (2007) Colorectal cancer. BMJ 335:715–718
Tse JC, Kalluri R (2007) Mechanisms of metastasis: epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J Cell Biochem 101:816–829
Li H, Fan X, Houghton J (2007) Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem 101:805–815
Mareel M, Leroy A (2003) Clinical, cellular, and molecular aspects of cancer invasion. Physiol Rev 83:337–376
Mahadevan D, Von Hoff DD (2007) Tumor–stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther 6:1186–1197
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6:392–401
Kitadai Y, Sasaki T, Kuwai T et al (2006) Targeting the expression of platelet-derived growth factor receptor by reactive stroma inhibits growth and metastasis of human colon carcinoma. Am J Pathol 169:2054–2065
Olumi AF, Grossfeld GD, Hayward SW et al (1999) Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 59:5002–5011
Zeisberg EM, Potenta S, Xie L et al (2007) Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67:10123–10128
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4:71–78
Balkwill F, Charles KA, Mantovani A (2005) Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 7:211–217
Bingle L, Brown NJ, Lewis CE (2002) The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol 196:254–265
Lin EY, Nguyen AV, Russell RG et al (2001) Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med 193:727–740
Condeelis J, Pollard JW (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266
Barbera-Guillem E, Nyhus JK, Wolford CC et al (2002) Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res 62:7042–7049
Popovic ZV, Sandhoff R, Sijmonsma TP et al (2007) Sulfated glycosphingolipid as mediator of phagocytosis: SM4s enhances apoptotic cell clearance and modulates macrophage activity. J Immunol 179:6770–6782
Pages F, Berger A, Camus M et al (2005) Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med 353:2654–2666
Galon J, Costes A, Sanchez-Cabo F et al (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Lu B, Finn OJ (2008) T-cell death and cancer immune tolerance. Cell Death Differ 15:70–79
O’Connell J, O’Sullivan GC, Collins JK et al (1996) The Fas counterattack: Fas-mediated T cell killing by colon cancer cells expressing Fas ligand. J Exp Med 184:1075–1082
Le QT, Shi G, Cao H et al (2005) Galectin-1: a link between tumor hypoxia and tumor immune privilege. J Clin Oncol 23:8932–8941
Shurin MR, Shurin GV, Lokshin A et al (2006) Intratumoral cytokines/chemokines/growth factors and tumor infiltrating dendritic cells: friends or enemies? Cancer Metastasis Rev 25:333–356
Kirkham N, Price ML, Gibson B et al (1989) Type VII collagen antibody LH 7.2 identifies basement membrane characteristics of thin malignant melanomas. J Pathol 157:243–247
Wetzels RH, Kuijpers HJ, Lane EB et al (1991) Basal cell-specific and hyperproliferation-related keratins in human breast cancer. Am J Pathol 138:751–763
Nagle RB, Hao J, Knox JD et al (1995) Expression of hemidesmosomal and extracellular matrix proteins by normal and malignant human prostate tissue. Am J Pathol 146:1498–1507
Pyke C, Romer J, Kallunki P et al (1994) The gamma 2 chain of kalinin/laminin 5 is preferentially expressed in invading malignant cells in human cancers. Am J Pathol 145:782–791
Pyke C, Salo S, Ralfkiaer E et al (1995) Laminin-5 is a marker of invading cancer cells in some human carcinomas and is coexpressed with the receptor for urokinase plasminogen activator in budding cancer cells in colon adenocarcinomas. Cancer Res 55:4132–4139
Tani T, Karttunen T, Kiviluoto T et al (1996) Alpha 6 beta 4 integrin and newly deposited laminin-1 and laminin-5 form the adhesion mechanism of gastric carcinoma. Continuous expression of laminins but not that of collagen VII is preserved in invasive parts of the carcinomas: implications for acquisition of the invading phenotype. Am J Pathol 149:781–793
Tani T, Lumme A, Linnala A et al (1997) Pancreatic carcinomas deposit laminin-5, preferably adhere to laminin-5, and migrate on the newly deposited basement membrane. Am J Pathol 151:1289–302
Kennel SJ, Foote LJ, Falcioni R et al (1989) Analysis of the tumor-associated antigen TSP-180. Identity with alpha 6-beta 4 in the integrin superfamily. J Biol Chem 264:15515–15521
Van Waes C, Kozarsky KF, Warren AB et al (1991) The A9 antigen associated with aggressive human squamous carcinoma is structurally and functionally similar to the newly defined integrin alpha 6 beta 4. Cancer Res 51:2395–2402
Falcioni R, Kennel SJ, Giacomini P et al (1986) Expression of tumor antigen correlated with metastatic potential of Lewis lung carcinoma and B16 melanoma clones in mice. Cancer Res 46:5772–5778
Wolff WL, Shinya H, Cwern M et al (1990) Cancerous colonic polyps. “Hands on” or “hands off?”. Am Surg 56:148–152
Rabinovitz I, Mercurio AM (1997) The integrin alpha6beta4 functions in carcinoma cell migration on laminin-1 by mediating the formation and stabilization of actin-containing motility structures. J Cell Biol 139:1873–1884
Mook OR, Frederiks WM, Van Noorden CJ (2004) The role of gelatinases in colorectal cancer progression and metastasis. Biochim Biophys Acta 1705:69–89
Berx G, Raspe E, Christofori G et al (2007) Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis 24(8):587–597
Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2:442–454
Bates RC, Mercurio AM (2005) The epithelial–mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther 4:365–370
Bates RC, Pursell BM, Mercurio AM (2007) Epithelial–mesenchymal transition and colorectal cancer: gaining insights into tumor progression using LIM 1863 cells. Cells Tissues Organs 185:29–39
Cavallaro U, Christofori G (2004) Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer 4:118–132
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol 7:131–142
Cano A, Perez-Moreno MA, Rodrigo I et al (2000) The transcription factor snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2:76–83
Avizienyte E, Fincham VJ, Brunton VG et al (2004) Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial–mesenchymal transition. Mol Biol Cell 15:2794–2803
Minard ME, Ellis LM Gallick GE (2006) Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin Exp Metastasis 23:301–313
Bellovin DI, Simpson KJ, Danilov T et al (2006) Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene 25:6959–6967
Hazan RB, Phillips GR, Qiao RF et al (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 148:779–790
Zavadil J, Bottinger EP (2005) TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774
Bates RC, Mercurio AM (2003) Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell 14:1790–1800
Bates RC, DeLeo MJ 3rd, Mercurio AM (2004) The epithelial–mesenchymal transition of colon carcinoma involves expression of IL-8 and CXCR-1-mediated chemotaxis. Exp Cell Res 299:315–324
Zeisberg M, Hanai J, Sugimoto H et al (2003) BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 9:964–968
Thuault S, Valcourt U, Petersen M et al (2006) Transforming growth factor-beta employs HMGA2 to elicit epithelial–mesenchymal transition. J Cell Biol 174:175–183
Yang L, Lin C, Zhao S et al (2007) Phosphorylation of p68 RNA helicase plays a role in platelet-derived growth factor-induced cell proliferation by up-regulating cyclin D1 and c-Myc expression. J Biol Chem 282:16811–16819
Bhowmick NA, Ghiassi M, Bakin A et al (2001) Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 12:27–36
Bhowmick NA, Zent R, Ghiassi M et al (2001) Integrin beta 1 signaling is necessary for transforming growth factor-beta activation of p38MAPK and epithelial plasticity. J Biol Chem 276:46707–46713
Calvert PM, Frucht H (2002) The genetics of colorectal cancer. Ann Intern Med 137:603–612
Janda E, Lehmann K, Killisch I et al (2002) Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol 156:299–313
Liao D, Johnson RS (2007) Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 26:281–290
Mizukami Y, Kohgo Y, Chung DC (2007) Hypoxia inducible factor-1 independent pathways in tumor angiogenesis. Clin Cancer Res 13:5670–5674
Mizukami Y, Jo WS, Duerr EM et al (2005) Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med 11:992–997
Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100:782–794
Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2:727–739
Hillen F, Griffioen AW (2007) Tumour vascularization: sprouting angiogenesis and beyond. Cancer Metastasis Rev 26:489–502
Shchors K, Evan G (2007) Tumor angiogenesis: cause or consequence of cancer? Cancer Res 67:7059–7061
Goh V, Padhani AR, Rasheed S (2007) Functional imaging of colorectal cancer angiogenesis. Lancet Oncol 8:245–255
Chen CN, Cheng YM, Liang JT et al (2000) Color Doppler vascularity index can predict distant metastasis and survival in colon cancer patients. Cancer Res 60:2892–2897
Leen E, Goldberg JA, Angerson WJ et al (2000) Potential role of Doppler perfusion index in selection of patients with colorectal cancer for adjuvant chemotherapy. Lancet 355:34–37
Prat A, Casado E, Cortes J (2007) New approaches in angiogenic targeting for colorectal cancer. World J Gastroenterol 13:5857–5866
Wilting J, Hawighorst T, Hecht M et al (2005) Development of lymphatic vessels: tumour lymphangiogenesis and lymphatic invasion. Curr Med Chem 12:3043–3053
Sundar SS, Ganesan TS (2007) Role of lymphangiogenesis in cancer. J Clin Oncol 25:4298–4307
Sundlisaeter E, Dicko A, Sakariassen PO et al (2007) Lymphangiogenesis in colorectal cancer—prognostic and therapeutic aspects. Int J Cancer 121:1401–1409
Karkkainen MJ, Makinen T, Alitalo K (2002) Lymphatic endothelium: a new frontier of metastasis research. Nat Cell Biol 4:E2–E5
Alitalo K, Tammela T, Petrova TV (2005) Lymphangiogenesis in development and human disease. Nature 438:946–953
Tammela T, Saaristo A, Lohela M et al (2005) Angiopoietin-1 promotes lymphatic sprouting and hyperplasia. Blood 105:4642–4648
Salven P, Mustjoki S, Alitalo R et al (2003) VEGFR-3 and CD133 identify a population of CD34+ lymphatic/vascular endothelial precursor cells. Blood 101:168–172
He Y, Rajantie I, Ilmonen M et al (2004) Preexisting lymphatic endothelium but not endothelial progenitor cells are essential for tumor lymphangiogenesis and lymphatic metastasis. Cancer Res 64:3737–3740
Kerjaschki D (2005) The crucial role of macrophages in lymphangiogenesis. J Clin Invest 115:2316–2319
Kerbel RS (1991) Inhibition of tumor angiogenesis as a strategy to circumvent acquired resistance to anti-cancer therapeutic agents. Bioessays 13:31–36
Meijer J, Zeelenberg IS, Sipos B et al (2006) The CXCR5 chemokine receptor is expressed by carcinoma cells and promotes growth of colon carcinoma in the liver. Cancer Res 66:9576–9582
Rashidi B, Sun FX, Jiang P et al (2000) A nude mouse model of massive liver and lymph node metastasis of human colon cancer. Anticancer Res 20:715–722
Sier CF, Vloedgraven HJ, Griffioen G et al (1995) Plasminogen activators and inhibitor type 1 in neoplastic colonic tissue from patients with familial adenomatous polyposis. Br J Cancer 71:393–396
Ploplis VA, Tipton H, Menchen H et al (2007) A urokinase-type plasminogen activator deficiency diminishes the frequency of intestinal adenomas in ApcMin/+ mice. J Pathol 213:266–274
Leupold JH, Asangani I, Maurer GD et al (2007) Src induces urokinase receptor gene expression and invasion/intravasation via activator protein-1/p-c-Jun in colorectal cancer. Mol Cancer Res 5:485–496
Iravani S, Mao W, Fu L et al (1998) Elevated c-Src protein expression is an early event in colonic neoplasia. Lab Invest 78:365–371
Boyd DD, Wang H, Avila H et al (2004) Combination of an SRC kinase inhibitor with a novel pharmacological antagonist of the urokinase receptor diminishes in vitro colon cancer invasiveness. Clin Cancer Res 10:1545–1555
Allgayer H, Wang H, Gallick GE et al (1999) Transcriptional induction of the urokinase receptor gene by a constitutively active Src. Requirement of an upstream motif (−152/−135) bound with Sp1. J Biol Chem 274:18428–18437
Zhu Z, Sanchez-Sweatman O, Huang X et al (2001) Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res 61:1707–1716
Owens LV, Xu L, Craven RJ et al (1995) Overexpression of the focal adhesion kinase (p125FAK) in invasive human tumors. Cancer Res 55:2752–2755
McLean GW, Komiyama NH, Serrels B et al (2004) Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev 18:2998–3003
Zeng Q, Chen S, You Z et al (2002) Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J Biol Chem 277:25203–25208
Mehlen P, Puisieux A (2006) Metastasis: a question of life or death. Nat Rev Cancer 6:449–458
Tsuji T, Sasaki Y, Tanaka M et al (2002) Microvessel morphology and vascular endothelial growth factor expression in human colonic carcinoma with or without metastasis. Lab Invest 82:555–562
Tien YW, Jeng YM, Hu RH et al (2004) Intravasation-related metastatic factors in colorectal cancer. Tumour Biol 25:48–55
Wyckoff JB, Jones JG, Condeelis JS et al (2000) A critical step in metastasis: in vivo analysis of intravasation at the primary tumor. Cancer Res 60:2504–2511
Muller A, Homey B, Soto H et al (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–56
Chambers AF, Naumov GN, Varghese HJ et al (2001) Critical steps in hematogenous metastasis: an overview. Surg Oncol Clin N Am 10:243–255, vii
Fidler IJ (1970) Metastasis: quantitative analysis of distribution and fate of tumor embolilabeled with 125 I-5-iodo-2¢-deoxyuridine. J Natl Cancer Inst 45:773–782
Wong CW, Lee A, Shientag L et al (2001) Apoptosis: an early event in metastatic inefficiency. Cancer Res 61:333–338
Luzzi KJ, MacDonald IC, Schmidt EE et al (1998) Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153:865–873
Gassmann P, Haier J (2007) The tumor cell–host organ interface in the early onset of metastatic organ colonisation. Clin Exp Metastasis (in press) DOI 10.1007/s10585-007-9130-6
Weiss L, Nannmark U, Johansson BR et al (1992) Lethal deformation of cancer cells in the microcirculation: a potential rate regulator of hematogenous metastasis. Int J Cancer 50:103–107
Weiss L (1988) Biomechanical destruction of cancer cells in the heart: a rate regulator for hematogenous metastasis. Invasion Metastasis 8:228–237
Weiss L (1989) Biomechanical destruction of cancer cells in skeletal muscle: a rate-regulator for hematogenous metastasis. Clin Exp Metastasis 7:483–491
Wang HH, McIntosh AR, Hasinoff BB et al (2000) B16 melanoma cell arrest in the mouse liver induces nitric oxide release and sinusoidal cytotoxicity: a natural hepatic defense against metastasis. Cancer Res 60:5862–5869
Edmiston KH, Shoji Y, Mizoi T et al (1998) Role of nitric oxide and superoxide anion in elimination of low metastatic human colorectal carcinomas by unstimulated hepatic sinusoidal endothelial cells. Cancer Res 58:1524–1531
Kluger HM, Chelouche Lev D, Kluger Y et al (2005) Using a xenograft model of human breast cancer metastasis to find genes associated with clinically aggressive disease. Cancer Res 65:5578–5587
Gout S, Tremblay PL, Huot J (2007) Selectins and selectin ligands in extravasation of cancer cells and organ selectivity of metastasis. Clin Exp Metastasis (in press) DOI 10.1007/s10585-007-9096-4
Malmberg KJ, Ljunggren HG (2006) Escape from immune- and nonimmune-mediated tumor surveillance. Semin Cancer Biol 16:16–31
Rosenberg SA (2001) Progress in human tumour immunology and immunotherapy. Nature 411:380–384
Kim S, Iizuka K, Aguila HL et al (2000) In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci U S A 97:2731–2736
Smyth MJ, Swann J, Kelly JM et al (2004) NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J Exp Med 200:1325–1335
Takeda K, Smyth MJ, Cretney E et al (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol 214:194–200
Im JH, Fu W, Wang H et al (2004) Coagulation facilitates tumor cell spreading in the pulmonary vasculature during early metastatic colony formation. Cancer Res 64:8613–8619
Biggerstaff JP, Seth N, Amirkhosravi A et al (1999) Soluble fibrin augments platelet/tumor cell adherence in vitro and in vivo, and enhances experimental metastasis. Clin Exp Metastasis 17:723–730
Samak R, Israel L (1982) [Extraction and identification of circulating immune complexes from the serum of cancer patient by affinity chromatography followed by high pressure steric exclusion chromatography. Demonstration of their effect on the mitogenesis of normal lymphocytes]. Ann Med Interne (Paris) 133:362–366
Kaplan RN, Rafii S, Lyden D (2006) Preparing the “soil”: the premetastatic niche. Cancer Res 66:11089–11093
Nicolson GL (1988) Cancer metastasis: tumor cell and host organ properties important in metastasis to specific secondary sites. Biochim Biophys Acta 948:175–224
Walzog B, Gaehtgens P (2000) Adhesion molecules: the path to a new understanding of acute inflammation. News Physiol Sci 15:107–113
Narita T, Kawakami-Kimura N, Kasai Y et al (1996) Induction of E-selectin expression on vascular endothelium by digestive system cancer cells. J Gastroenterol 31:299–301
Kannagi R, Izawa M, Koike T et al (2004) Carbohydrate-mediated cell adhesion in cancer metastasis and angiogenesis. Cancer Sci 95:377–384
Khatib AM, Auguste P, Fallavollita L et al (2005) Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am J Pathol 167:749–759
Auguste P, Fallavollita L, Wang N et al (2007) The host inflammatory response promotes liver metastasis by increasing tumor cell arrest and extravasation. Am J Pathol 170:1781–1792
Wang HH, Qiu H, Qi K et al (2005) Current views concerning the influences of murine hepatic endothelial adhesive and cytotoxic properties on interactions between metastatic tumor cells and the liver. Comp Hepatol 4:8
Kitakata H, Nemoto-Sasaki Y, Takahashi Y et al (2002) Essential roles of tumor necrosis factor receptor p55 in liver metastasis of intrasplenic administration of colon 26 cells. Cancer Res 62:6682–6687
Gout S, Morin C, Houle F et al (2006) Death receptor-3, a new E-Selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res 66:9117–9124
Tremblay PL, Auger FA, Huot J (2006) Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene 25:6563–6573
Araki M, Araki K, Biancone L et al (1997) The role of E-selectin for neutrophil activation and tumor metastasis in vivo. Leukemia 11(Suppl 3):209–212
Gotsch U, Jager U, Dominis M et al (1994) Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes Commun 2:7–14
Hanna WC, Ponsky TA, Trachiotis GD et al (2006) Colon cancer metastatic to the lung and the thyroid gland. Arch Surg 141:93–96
Esposito V, Groeger AM, De Luca L et al (2002) Expression of surface protein receptors in lung cancer. Anticancer Res 22:4039–4043
Enns A, Gassmann P, Schluter K et al (2004) Integrins can directly mediate metastatic tumor cell adhesion within the liver sinusoids. J Gastrointest Surg 8:1049–1059, discussion 1060
Enns A, Korb T, Schluter K et al (2005) Alphavbeta5-integrins mediate early steps of metastasis formation. Eur J Cancer 41:1065–1072
Fingar VH, Taber SW, Buschemeyer WC et al (1997) Constitutive and stimulated expression of ICAM-1 protein on pulmonary endothelial cells in vivo. Microvasc Res 54:135–144
Subramaniam V, Gardner H, Jothy S (2007) Soluble CD44 secretion contributes to the acquisition of aggressive tumor phenotype in human colon cancer cells. Exp Mol Pathol 83:341–346
Zvibel I, Halpern Z, Papa M (1998) Extracellular matrix modulates expression of growth factors and growth-factor receptors in liver-colonizing colon-cancer cell lines. Int J Cancer 77:295–301
Radinsky R (1995) Molecular mechanisms for organ-specific colon carcinoma metastasis. Eur J Cancer 31A:1091–1095
Radinsky R, Ellis LM (1996) Molecular determinants in the biology of liver metastasis. Surg Oncol Clin N Am 5:215–229
Baggiolini M (1998) Chemokines and leukocyte traffic. Nature 392:565–568
Balkwill F, Coussens LM (2004) Cancer: an inflammatory link. Nature 431:405–406
Murphy PM (2001) Chemokines and the molecular basis of cancer metastasis. N Engl J Med 345:833–835
Zeelenberg IS, Ruuls-Van Stalle L, Roos E (2003) The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res 63:3833–3839
Yates CC, Shepard CR, Stolz DB et al (2007) Co-culturing human prostate carcinoma cells with hepatocytes leads to increased expression of E-cadherin. Br J Cancer 96:1246–1252
Hugo H, Ackland ML, Blick T et al (2007) Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol 213:374–383
Nakagawa H, Liyanarachchi S, Davuluri RV et al (2004) Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene 23:7366–7377
Mueller L, Goumas FA, Affeldt M et al (2007) Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol 171:1608–1618
Mueller L, Goumas FA, Himpel S et al (2007) Imatinib mesylate inhibits proliferation and modulates cytokine expression of human cancer-associated stromal fibroblasts from colorectal metastases. Cancer Lett 250:329–338
Belluco C, Mammano E, Petricoin E et al (2005) Kinase substrate protein microarray analysis of human colon cancer and hepatic metastasis. Clin Chim Acta 357:180–183
Pennison M, Pasche B (2007) Targeting transforming growth factor-beta signaling. Curr Opin Oncol 19:579–585
Schlingensiepen KH, Schlingensiepen R, Steinbrecher A et al (2006) Targeted tumor therapy with the TGF-beta2 antisense compound AP 12009. Cytokine Growth Factor Rev 17:129–139
Proudfoot O, Pouniotis D, Sheng KC et al (2007) Dendritic cell vaccination. Expert Rev Vaccines 6:617–633
Acknowledgments
This work was supported by The Canadian Cancer Society/NCI. Stéphanie Gout holds a Post-Doctoral Fellowship from Le Fonds de la Recherche en Santé du Québec (FRSQ).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Gout, S., Huot, J. Role of Cancer Microenvironment in Metastasis: Focus on Colon Cancer. Cancer Microenvironment 1, 69–83 (2008). https://doi.org/10.1007/s12307-008-0007-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-008-0007-2