
Abstract
Regulatory small RNAs (approximately 20 to 24 nt in length) are produced through pathways that involve several key evolutionarily conserved protein families; the variants of these proteins found in plants are encoded by multigene families and are known as Dicer-like, Argonaute, and RNA-dependent RNA polymerase proteins. Small RNAs include the well-known classes of microRNAs (miRNAs, ~21 nt) and the small-interfering RNAs (siRNAs, ~24 nt). Both of these types of molecules are found across a broad set of eukaryotic species, although the siRNAs are a much larger and more diverse class in plants due to the abundance of heterochromatic siRNAs. Well-studied species such as Arabidopsis have provided a foundation for understanding in rice and other species how small RNAs function as key regulators of gene expression. In this paper, we review the current understanding of plant small RNA pathways, including the biogenesis and function of miRNAs, siRNAs, trans-acting siRNAs, and heterochromatic siRNAs. We also examine the evolutionary relationship among plant species of both their miRNAs and the key enzymatic components of the small RNA pathways. Many of the most recent advances in describing small RNAs have resulted from advances in sequencing technologies used for identifying and measuring small RNAs, and these technologies are discussed. Combined with the plethora of genetic tools available to researchers, we expect that the continued elucidation of the identity and functions of plant small RNAs will be both exciting and rewarding.
Similar content being viewed by others


Introduction to small RNAs in plants
Small RNAs (sRNAs) are short (20 to 30 nt), non-coding RNAs that play important roles in both transcriptional and post-transcriptional gene silencing. These molecules are found across a broad set of eukaryotic species and primarily function through one of several mechanisms, including (1) directing messenger RNA (mRNA) cleavage, (2) translational repression, or (3) triggering modifications that silence genes such as DNA methylation and/or heterochromatic modifications. Data suggest that all of these modes of action result from base pairing to their targets, which may be mRNA, DNA, or even a nascent transcript [1, 2].
Plant small RNAs are generally 20 to 24 nt in length and may be classified based on a series of different criteria. There are two predominant sizes of small RNAs in most plant species, 21 and 24 nt. The 21 nt sRNAs are usually microRNAs (miRNAs), at least in Arabidopsis and rice, that mainly function by cleaving a specific target mRNA in a post-transcriptional manner, based on sequence homology between the miRNA and target mRNA. The 24 nt sRNAs are usually short-interfering RNAs (siRNAs) that predominately control gene expression at the transcriptional level by inducing modifications to silence DNA and histones [3]. These activities take place in heterochromatic regions of the genome. Plant sRNAs may also be categorized as miRNAs or siRNAs based on their origin: miRNAs are derived from imperfectly matched stem-loop structures that are formed from single-stranded RNA (ssRNA) precursors, whereas siRNAs are derived from perfectly—or nearly perfectly—matched double-stranded RNAs (dsRNAs) produced by the activity of RNA-dependent RNA polymerases (with genes named as “RDR1,” “RDR2,” etc.) or from ssRNA transcripts including inverted repeats that fold back to form a dsRNA region [4, 5]. The larger class of siRNAs can be further subdivided into categories including trans-acting siRNAs (ta-siRNAs), natural cis-antisense transcript derived siRNAs (nat-siRNAs) and heterochromatic siRNAs (hc-siRNAs). This division is based on the distinct biogenesis pathway of each subgroup [5].
One key component of plant small RNA biogenesis is a family of RNase III enzymes called Dicer-like (DCL) proteins. These enzymes function to cut or “dice” specific stem-loop structures of ssRNA precursors into miRNA or dsRNA into siRNA duplexes, respectively. There are four DCL proteins in Arabidopsis thaliana and six putative DCL proteins in rice (Oryza sativa), a result of the apparent duplication of DCL3 in rice. All four of the Arabidopsis DCLs have known roles in small RNA biogenesis. DCL1 processes the mature miRNA from the precursor, DCL2 is involved in the production of some 22 and 24 nt viral siRNAs, DCL3 is involved in the accumulation of 24 nt siRNAs from repeat sequences associated with transgenes and heterochromatin, and DCL4 is involved in the processing of 21 nt siRNA from dsRNA precursors, in addition, together with DCL1, participating in the processing of 21 nt ta-siRNAs [6–11]. Functional redundancies and competition among DCL2, DCL3, and DCL4 in small RNA biogenesis have been reported [12, 13]. Similarly, in rice, OsDCL1 and OsDCL4 also function in the biogenesis of miRNAs and siRNAs, respectively [14–16].
Once processed by Dicers, mature small RNAs are incorporated into different Argonaute (AGO) proteins to finally execute their functions. MicroRNAs are mainly bound by AGO1, and ta-siRNAs are bound by AGO6 and AGO2, whereas most of the 24 nt small RNAs are directed to AGO4 and AGO5 [5, 17–19]. During the process of sorting certain classes of small RNAs into their corresponding AGOs, the 5′ terminal nucleotide of the small RNAs plays a significant role, as it was recently reported that different AGOs have a strong bias for a distinct 5′ terminal nucleotide: U for AGO1, A for AGO2, A for AGO4, and C for AGO5 [19, 20]. Interestingly and distinct from most miRNAs associated with AGO1 that have a 5′ U, miR390 with a 5′ A was found to specifically bind to AGO7 and then function at two target sites in the TAS3a transcript [20]. The AGO proteins, like other key elements of the small RNA biogenesis pathway, are conserved across animals, plants, and fungi. Within some of the kingdoms, conservation also extends to include high degrees of similarity among a number of individual miRNAs; there have been some elegant studies recently examining conservation and evolution among plant miRNAs [21–23].
Lessons from Arabidopsis: conserved miRNAs
MicroRNAs have been identified in animals, plants, and viruses. The first miRNA, lin-4, was identified in Caenorhabditis elegans [24, 25], and many more have since been identified in other organisms, as almost all examined multicellular eukaryotes have been found to utilize miRNAs [3]. At present, 6,396 miRNA sequences and annotations have been deposited in the miRBase Sequence Database (release 11.0) [26–29]. Of those, 1,160 are plant miRNAs, of which 184 are from A. thaliana, 269 from rice (O. sativa), 72 from sorghum (Sorghum bicolor), 30 from legume (Medicago truncatula), 234 from cottonwood (Populus trichocarpa), 32 from wheat (Triticum aestivum), 140 from common grape vine (Vitis vinifera), and 96 from maize (Zea mays). The wide variation in numbers is probably due to a lack of intensive study in many of the genomes, as most published studies have focused on Arabidopsis, with rice close but in second place. MicroRNAs are typically identified using experimental approaches, like cloning and sequencing of small RNA libraries, or through computational predictions that are subsequently experimentally validated or even by forward genetics approaches [30–33]. High-throughput sequencing such as massively parallel signature sequencing (MPSS), 454 pyrosequencing, and sequencing-by-synthesis (SBS) of small RNA libraries has substantially increased the rate of identification of miRNAs [34–36]. Deep sequencing across plant lineages has identified evolutionarily conserved miRNA families in gymnosperms, mosses, monocots, and dicots [37, 38]. Notably, miRNA families miR156/157, miR159/319, miR160, miR165/166, miR390, and miR408 are also found in primitive land plants [31, 37–42]. Between Arabidopsis and rice, there are ~20 miRNA families that are evolutionarily conserved (Table 1). A family implies evolutionary relatedness or sequence conservation between the mature miRNAs, and miRNA sequences are typically grouped as a family when the mature miRNAs are identical or there are very few mismatches, i.e., three or fewer nt substitutions and at least one conserved target transcript [22]. For historical reasons, some miRNA families have been annotated with more than one number, i.e., miR156/157, miR159/319, miR165/166, and miR170/171. Sequence conservation has been observed in both the primary and mature miRNAs of plants but is most frequent in the mature sequences and their complementary miRNA* sequences; it is believed that there are generally few selective constraints on the precursor sequences that flank the miRNA-generating stem-loop structure. Some miRNAs are encoded by multiple loci within a genome and demonstrate high levels of sequence conservation in the mature miRNA and miRNA* sequences but are completely unrelated in other parts of the miRNA precursor. The level of conservation of the miRNA precursor varies considerably [43] as does the copy number among miRNAs; the latter point is easily visible in a comparison of Arabidopsis and rice miRNA families (Table 1) [43]. This copy number variation could reflect different expression patterns of each miRNA locus [44]. The evolutionary conservation of miRNAs across plant lineages extends to include the target genes, as sequence changes at the target sites are constrained by the requirement of maintaining close homology to the miRNA. In different plant families, Zhang et al. [43] observed the complementary site of the target to be highly conserved but other regions of the target to have lower nt conservation. The sequence conservation of the miRNAs and their target regions is indicative of the roles of miRNAs in important and conserved physiological processes; this includes a number of important developmental pathways. The difference in the number and size of the miRNA members and families, respectively, is probably shaped by the roles of specific miRNAs, and this could vary somewhat from species to species. It will be interesting to compare across species the expression level differences of miRNA families/members and their targeting efficiencies.
In both Arabidopsis and rice, the conserved miRNAs are usually the most abundantly expressed miRNAs. High-throughput sequencing of rice small RNAs by Sunkar et al. [34] indicated that the relative abundances are high for the conserved miRNAs, with the top ten most-abundant sequence reads coming from conserved miRNAs. For example, miR169 was the most abundantly expressed miRNA family, a family that contains nine members that correspond to 17 rice loci. MiR169 was represented 4,948 times in the small RNA library. Another highly expressed miRNA was miR156. There are three members of the miR156 family that correspond to 12 rice loci and miR156 was represented 1,094 times in the small RNA library. Notably, there were a few conserved miRNA families that were not observed at high frequencies. MicroRNAs with low expression levels included miR394, miR399, and miR408. MicroRNAs miR394 and miR408 are single member families found at a single locus, whereas miR399 has three members clustered in a single locus but, like miR394 and miR408, were sequenced only once in the small RNA library. Overall, the analysis by Sunkar et al. [34] showed that most of the conserved miRNAs are expressed but often with wide variation in the frequency of their expression.
Lessons from Arabidopsis II: non-conserved miRNAs
In contrast to the broad representation of conserved miRNAs, there are some plant miRNAs that are only found in a single species, at least based on the miRNAs and genomes studied to date. These “non-conserved” miRNAs are most often represented by single genes in the genomes in which they are found. Non-conserved miRNAs have had some ambiguities in their identification. This can be illustrated by the case of three small RNAs that were previously annotated as miRNAs, which turned out to be members of the unusual class of ta-siRNAs. The precursors did not have an extensively paired hairpin structure like that of a miRNA [9]. Non-conserved miRNAs require more stringent evidence and proof that they meet the criteria of a real miRNA because they lack one of the strongest pieces of data used to distinguish miRNAs from siRNAs—conservation across species boundaries. Instead, non-conserved miRNAs must be proven using a combination of detailed analyses of their sequence, biogenesis, secondary structure, expression patterns, and silencing functions.
The preponderance of non-conserved miRNAs represented as single gene families suggests a fairly recent evolution for these genes, which may be consistent with the notion that non-conserved miRNAs are evolutionary intermediates between a non-miRNA sequence and a miRNA with an important regulatory role. In some cases, the region of the precursor flanking the mature miRNA has been shown to contain extensive similarity to protein-coding genes [45]. This similarity supports the hypothesis that some of these intermediate miRNAs come from aberrant duplication or transposition events from the expressed gene sequences, such as the inverted duplication of a coding gene. Notably, before the generation of the newly evolved miRNA loci, intermediates may pass through a stage in which heterogeneous populations of siRNA-like sequences are generated [21]. Because DCL1 has insignificant activity on a perfectly paired dsRNA, the duplicated locus would need to accumulate mutations, presumably via genetic drift, to form an imperfect pair in the fold-back structure before the structure is suitable for processing by the DCL1-dependent miRNA biogenesis pathway. There is some evidence indicating that some non-conserved miRNAs can utilize a biogenesis pathway which is DCL4-dependent [23], suggesting that these intermediates have some of the hallmarks of a miRNA but have yet to completely conform to the canonical miRNA-biogenesis pathway.
In rice, many annotated miRNAs have been identified by computational predictions, based on the conservation of sequences with Arabidopsis miRNAs [31]. Despite high levels of homology between Arabidopsis and rice for many genes, there are some highly abundant and well-characterized Arabidopsis miRNAs that have no homologs in rice. These include the Arabidopsis miRNAs miR158, miR161, miR163, miR173 [31], and miR403 [42]. This suggests that each plant lineage, including rice, may evolve a unique set of miRNAs. Direct cloning, traditional sequencing, and deep sequencing approaches have discovered many non-conserved miRNAs in rice, and their predicted target genes encode a broad range of proteins, including some transcription factors (Supplementary Table 1). This set of rice miRNAs and targets is more diverse than the set of conserved miRNAs that mainly target transcription factors. In addition, it is likely that some non-conserved miRNAs have yet to be detected in rice because of their low expression levels or because they are only expressed in specific cells or conditions. The use of mutants in the small RNA biogenesis pathway may yet prove to be helpful in miRNA identification in rice, as some of these mutants are enriched for miRNAs, and analyses with high-throughput sequencing can be quite informative, as demonstrated recently in Arabidopsis [46]. This type of experiment has yet to be done in rice due to the lack of well-characterized small RNA biogenesis mutants. However, deep sequencing in rice has already revealed numerous abundant and consistently expressed non-conserved small RNAs [36]. This method of exploring small RNA profiles in rice has also lead to the identification of natural antisense miRNAs in rice [47].
Lessons from Arabidopsis III: heterochromatic siRNAs
Another type of small RNA molecule with important implications for post-transcriptional gene silencing was discovered in 1999. David Baulcombe’s group demonstrated that, in plants, a type of small RNA molecule triggered by transgenes and viruses is a specificity determinant during the process of post-transcriptional gene silencing [48]. Early estimates suggested that these RNA molecules were a uniform length of 25 nt. This breakthrough discovery provided the conceptual groundwork for the elucidation of RNA interference biochemical pathways. In addition to siRNA-mediated suppression of genes through targeted mRNA degradation, there is another silencing process in some plant systems [49]. This process involves RNA-directed DNA methylation and systemic silencing of specific genomic locations. There are two classes of siRNAs in plants controlling different silencing processes [49]. These two classes of siRNAs were shown to be heterogeneous in both size and function and were referred to as short and long siRNAs. Short siRNAs (like miRNAs) are 21 to 22 nt in length, and they guide the RNA-induced silencing complex (RISC) ribonuclease to target mRNA degradation. Long siRNAs are 24 to 25 nt in length, and they were found to be the signal of systemic RNA silencing, which has been associated with sequence-specific DNA methylation. Due to the chromatin-based events that result in transcriptional silencing, this type of siRNA is often referred to as a “heterochromatic siRNA”. Tang et al. [50] also found that, in wheat germ extracts, exogenous dsRNA can be converted into two distinct length classes of RNAs, which are similar in size. In view of these two classes of RNAs having different preference for the 5′ end nucleotide, they predicted that these RNAs are made by distinct enzymes. Notably, they identified two siRNA-generating DCL activities in wheat germ extracts [50].
A broad and comprehensive analysis of siRNA populations has been carried out in Arabidopsis by a number of laboratories. The first sequencing of small RNAs from inflorescence tissues of Col-0 Arabidopsis indicated that most of the clones corresponded to siRNA-like sequences [51]. These small RNAs ranged in size between 20 and 26 nt, with 24 nt as the most common size. In addition, these data indicated that siRNAs arise more frequently from highly repeated genome sequences such as transposons and retroelements, as well as loci encoding 5S rRNA [8, 52, 53]. Further analysis revealed that DCL3 is the primary enzyme responsible for generating the extensive set of 24 nt siRNAs that match throughout the genome, and DCL3 is particularly specialized in the processing of dsRNA molecules produced by the RNA-dependent RNA polymerase protein known as “RDR2” [8]. Although RDR2 may be unnecessary as a polymerase subunit at some loci like inverted repeats, it still contributes to the formation or stability of a complex that contains active DCL3 [8]. Additional evidence has suggested that, in Arabidopsis, the generation of endogenous heterochromatic siRNAs occurs via an RDR2-DCL3-dependent mechanism [46, 53].
The biological role of the heterochromatic siRNA is performed when one of its strands is loaded into an effector complex. Specifically, AGO4 is required for functionality of heterochromatic siRNAs at a heterochromatic site [54]. It has been proposed that there is a link between small RNA biogenesis and effector programming such that specific siRNAs are loaded into the Argonaute through Dicer–Argonaute interactions. In addition, two non-redundant forms of a nuclear RNA polymerase IV (specific to plants), namely Pol IVa and Pol IVb, are also required at some loci. This has lead to the development of a model for the heterochromatic siRNA pathway in Arabidopsis: Subunits of Pol IVa co-localize with endogenous repeat loci, which are silenced by methylation. It has been proposed that cytosine methylation by a de novo cytosine methyltransferase induces the production of aberrant RNAs, which Pol IVa then uses as templates. Pol IVa transcripts then move to the nucleolar Cajal bodies, where RDR2, DCL3, and AGO4 are located, to form the heterochromatic siRNAs. In the siRNA processing center, the largest subunit of Pol IVb joins the AGO4-containing RISC complex and guides DNA methylation and heterochromatic histone (H3K9) modifications at the endogenous repeats [55–61]. A recent study has uncovered that, in several loci of Arabidopsis, Pol IVb’s role as the effector of RNA silencing is independent of its function in siRNA biogenesis, and the study proposed that some epigenetic marks of chromatin adjacent to the Pol IVb-targeted region could influence the ability of Pol IVb-guided DNA methylation [62]. Although there is evidence showing that heterochromatic siRNAs can trigger epigenetic effects at the target loci, a recent study revealed that some endogenous rice genes, including OsRac, are rarely transcriptionally silenced by promoter-targeted siRNAs, but these genes could be post-transcriptionally suppressed by RNA interference (RNAi) [63]. This discovery led to the proposal that there might be a mechanism that monitors chromatin modifications and may inhibit siRNA-mediated chromatin inactivation [63].
By applying direct cloning methods, in one recent study, a large set of putative endogenous siRNAs were identified from rice root, shoot, and inflorescence small RNA cDNA libraries [64]. The result from this study is consistent with data from Arabidopsis, in that most of the rice siRNAs were from intergenic regions, and they can be sorted into similar sizes and functions for two distinct classes. Both experimental validation and computational predictions indicate that many of these siRNA targets are transposable elements, consistent with the well-described role of plant endogenous siRNAs in genome defense against transposons and viruses [64]. In other studies, high-throughput sequencing has discovered that siRNAs are widely distributed across the rice chromosomes, inconsistent with Arabidopsis in which small RNAs are concentrated in the pericentromeric regions [36]. The difference in small RNA distributions is primarily due to the wider distribution of transposons and related repeats in rice, a phenomenon likely to be reflected in more complex plant genomes as well.
Sequence-based analyses of rice small RNAs
Initially, many miRNAs in rice were sequenced through the traditional Sanger sequencing method, most of which turned out to be the high-abundance miRNAs [16, 33]. However, developments in high-throughput sequencing have enabled more extensive exploration of small RNAs. In 2005, our lab, together with that of Pam Green’s lab, published the first ultrahigh-throughput sequencing-based analysis of small RNAs, resulting in the characterization of more than 1.5 million Arabidopsis small RNAs [65]. This was done using MPSS, and the work greatly expanded our understanding of small RNAs. Subsequently, other next-generation sequencing (NGS) platforms, like 454’s Genome Sequencer, Illumina’s Genome Analyzer (Solexa, also known as SBS for “sequencing by synthesis”) and Applied Biosystems’ (ABI) SOLiD machine have been making sequencing both faster and cheaper (see [66] for a comparison of these techniques). The read length of these NGS platforms is shorter than the original Sanger method (~250 bp for 454 and 35 to 50 bp for Solexa and SOLiD) but ideal for small RNA sequencing: 454 can produce >400,000 reads in one run; Solexa and SOLiD are capable of generating even tens of millions of sequences in parallel [66].
The sequencing of three million reads from three rice libraries by MPSS provided the first overview of the complexity of rice small RNAs. Most of these molecules, as predicted, are low-abundant siRNAs matched to various classes of repeats or genomic regions (Fig. 1) [36]. SBS sequencing of small RNAs from a wild rice relative, Oryza barthii (Fig. 2, an unpublished experiment recently undertaken in our lab) and 454 sequencing of cultivated rice small RNAs (from O. sativa [67]) have both demonstrated the two major sizes of sRNAs, 21 and 24 nt, consistent with prior reports from Arabidopsis and other species. In general, high-throughput analyses have enabled the exploration of rice small RNA populations, and many new miRNAs have been discovered recently in rice [34, 36, 47, 67]. This includes a special class of natural antisense transcript miRNAs (nat-miRNAs), which are derived from natural cis-antisense transcripts with exons primarily located antisense to the introns of their target genes; these nat-miRNAs are DCL1-dependent [47]. Over the next few years, it is likely that there will be an explosion in the breadth of small RNA analyses in rice, leading to more extensive characterization of miRNAs, siRNAs, and other classes of small RNAs (Figs. 3 and 4).

Classes of genomic features matched by rice small RNAs. Bars indicate the total number of small RNAs that were matched to each of the indicated genomic features. The three shaded bars indicate the number of distinct small RNAs matched to each of the three libraries: mixed stage inflorescence, seedling, and stem and exclude small RNAs matching to rRNAs and tRNAs. Modified from Nobuta et al. [36].

Size distribution of Oryza barthii small RNAs from SBS sequencing. A plot comparing the total abundance of small RNA sequences versus their size from an Oryza barthii small RNA library. After trimming adapters, sequences smaller than 18 nt were removed. The SBS read length for these libraries was 35 nt. A total of 2,419,602 small RNAs were successfully sequenced.

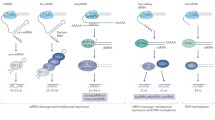
The plant miRNA biogenesis pathway and cellular function. The microRNA gene is transcribed by RNA Polymerase (Pol) II. The miRNA precursor, folded into a hairpin structure, is processed by DCL1 and the associated proteins, HYPONASTIC LEAVES1 (HYL1), a double-stranded RNA binding partner, and SERRATE (SER), a zinc-finger containing protein [79, 80]. The miRNA is processed into a miRNA/MiRNA* duplex that is methylated at the 3′ sugars by HUA ENHANCER1 (HEN1) and exported to the cytoplasm by HASTY (HST) [81, 82]. The mature, methylated miRNA is incorporated into the RNA-induced silencing complex (RISC) that includes ARGONAUTE1 (AGO1), which directs cleavage of the target mRNA. The 5′ cleavage product is thought to be degraded by a 3′ to 5′ exosome [83] and the 3′ fragment is degraded by the 5′ to 3′ EXORIBONUCLEASE4 (XRN4) [84]. An alternative decay pathway has also been proposed [84].
Rice mutants in small RNA biogenesis pathways
Components of the small RNA biogenesis pathway have been characterized functionally in plants (Figs. 3 and 4). While most of this work has been done in Arabidopsis, there is considerable similarity between the key players in Arabidopsis and rice. As mentioned above, in Arabidopsis, there are four DCL proteins, and rice encodes six putative DCL proteins, with duplications in the DCL2 and DCL3 clades (Supplementary Fig. 1a). Redundant, compensatory, and antagonistic roles among members of this multigene family have been described in Arabidopsis. The Arabidopsis loss-of-function mutants dcl1 and dcl4 show pleiotropic developmental defects, which suggests a role for DCLs in plant development. Indeed, the complete knockout of the dcl1 mutant is embryo-lethal, with partial loss-of-function dcl1 mutants demonstrating less severe developmental defects [68]. Information about rice DCLs is limited in comparison to Arabidopsis. However, studies by Liu et al. [15, 16] utilized knock-down and loss-of-function dcl1 and dcl4 RNAi mutants to demonstrate a role for OsDCL1 and OsDCL4 in small RNA biogenesis and plant development. The loss of function of OsDCL1 led to shoot and root abnormalities, such as rolled leaves and reduced root elongation. The plants were also developmentally arrested at the seedling stage. Similarly, loss-of-function of OsDCL4 leads to vegetative growth abnormalities and developmental defects in spikelet organ identity, which results in sterility. This is in contrast to the accelerated vegetative phase change observed in the Arabidopsis DCL mutants [7, 11], which implies that OsDCL4 has a broader role in development than the Arabidopsis DCL4. As previously mentioned, in Arabidopsis, DCL1 is responsible for miRNA accumulation, and DCL1 and DCL4 are necessary for the biogenesis of ta-siRNAs [9, 10]. Similarly, in rice, DCL1 was observed to be essential for miRNA accumulation, but a more prominent role was observed for OsDCL4. Through biochemical and genetic studies, OsDCL4 was observed to be the primary Dicer responsible for the 21 nt siRNAs associated with inverted repeat transgenes and ta-siRNAs that arose from the endogenous TAS3 gene. Clearly, we have much to learn about the nuances of Dicer function, particularly via comparative studies in species other than Arabidopsis (like rice). Much less is known about rice RDR functions (Supplementary Fig. 1b) and Pol IV activities (Supplementary Fig. 1c), although the phylogenetic analysis suggests the possibility of genetic redundancy in rice for each of the three major subunits of Pol IV (Table 2).

The plant heterochromatic and trans-acting siRNA biogenesis pathways. Heterochromatic siRNAs: Pol IV transcribes the genomic DNA into ssRNA [55, 58]. The ssRNA is made into long dsRNA by RNA-dependent RNA polymerase 2 (RDR2) [46]. The long dsRNA is processed by DCL3 to yield siRNA duplexes that are 3′ methylated by HEN1 (as with miRNAs). One strand from the siRNA duplex is incorporated into the RNAi-induced transcriptional silencing (RITS) complex with the aid of AGO4 and the other strand is degraded [60]. Twenty-four-nucleotide heterochromatic siRNAs associated with the RITS complex facilitate chromatin modification and transcriptional silencing. Trans-acting siRNAs: The trans-acting siRNA (TAS) precursor is transcribed by Pol II. AGO1 facilitates miRNA directed cleavage of the TAS precursor. The 5′ cleavage product or 3′ cleavage product is used by RNA-dependent RNA polymerase 6 (RDR6) and suppressor of gene silencing 3 (SGS3) as a template to produce dsRNA [6, 9, 10, 59]. The dsRNA is cleaved in a phased pattern every 21 nt by DCL4 [6, 7, 10, 11]. An interaction between DCL4 and dsRNA binding protein 4 (DRB4) is involved in the processing of the 21 nt ta-siRNAs [11, 85]. The ta-siRNAs are methylated at the 3′ ends by HEN1 and then guided by an AGO protein (AGO1 and possibly another AGO protein) to their targets for cleavage [20, 86]. The cleavage products may be degraded or may serve as a resource for additional ta-siRNAs [87, 88].
Another important component of the small RNA machinery is represented by the set of Argonaute proteins. There are ten conserved members in Arabidopsis and at least 18 members in rice [69, 70]. Phylogenetic analysis of the Argonaute family demonstrates that most of the diversification in rice compared to Arabidopsis took place in the AGO1 and AGO5 clades (Fig. 5). In Arabidopsis, AGO1 facilitates cleavage of mRNAs targeted by miRNAs [71, 72], so it is curious that rice has had a diversification of AGO1 paralogs. The AGO1-associated RNA machinery also functions in determining meristem identity and flower organ identity [73]. It is through posttranscriptional gene silencing that AGO1 mediates vegetative leaf and pollen development [73–75]. Other roles observed for AGO proteins include AGO4-directed DNA methylation and silencing of transposons [76] and the ZIP/AGO7-mediated regulation of developmental timing and proposed ta-siRNA pathway constituent [59, 77, 78]. One AGO protein has been implicated in both rice development and the RNA production pathway. OsAGO7, which is believed to be orthologous to the Arabidopsis ZIP/AGO7 gene (Fig. 5), facilitates upward curling of leaves when over-expressed in rice [69]. In a study performed by Nagasaki et al. [14], the rice genes known as SHOOTLESS2 (SHL2), SHL4/SHOOT ORGANIZATION2 (SHO2), and SHO1, encoding orthologs of the small RNA-associated Arabidopsis proteins RNA-dependent RNA polymerase 6 (RDR6), AGO7, and DCL4, respectively, were shown to play a role in leaf development through the ta-siRNA pathway. Nagasaki et al. [14] were able to show that ectopic expression of SHL4 and mutations in SHL2, SHO2, and SHO1 caused reduced accumulation of miR166 (which regulates the expression of the rice HD-ZIPIII genes OSHB1 and OSHB2), partial adaxialization of leaves, and defects in shoot apical meristem (SAM) formation. Negative regulation of miR166 expression through the SHL/SHO pathway, which contains orthologs of Arabidopsis proteins implicated in ta-siRNA generation [6, 7, 10, 11], suggest that there is a link between RNA-mediated gene regulation and fundamental plant processes such as embryonic SAM formation. The functional role for small RNAs is greatly expanding. As more studies across species are performed, the conservation and evolution of small RNAs will continue to reveal the dependency of plant regulatory pathways on small RNAs and their associated components.

Phylogenetic tree for the Argonaute family of proteins. A phylogenetic tree based on Argonautes (AGOs) from a variety of species, with major clades indicated based on Arabidopsis AGO proteins. Two AGO proteins (AGO716 and AGO710) were removed from the trees because of long branches. The code for each AGO protein is based on the ChromDB identifier plus the species from which the AGO was derived, with plant proteins using the following system of codes: ARATH, Arabidopsis thaliana; POPTR, Populus trichocarpa; ORYSA, Oryza sativa ssp. japonica (from the Nipponbare sequence); ORYSAI, Oryza sativa ssp. indica (from the 93–11 sequence). The phylogenetic methods are described in the legend for Supplementary Fig. 1.
Conclusions and future directions
At this point, many of the major players in small RNA biogenesis have been identified from intensive work in Arabidopsis. The translation of these discoveries to rice and other species, combined with both forward and reverse genetics approaches used directly in those species, is facilitating the elucidation of plant small RNA pathways and activities. With new deep sequencing methods and the prospect of combining these analysis methods with rice mutants in small RNA biogenesis genes, we should soon have a near complete list of rice miRNAs and their targets. This will include characterization of the non-conserved and rice-, grass-, or monocot-specific miRNAs. Identifying miRNAs in rice and more diverse plant species will be important to understand the evolution of miRNAs and the regulation of gene expression by miRNAs. The analysis of rice mutants in genes important for small RNA activities promises to be a particularly exciting area of research. For example, why does rice have nearly twice as many AGO-encoding genes as Arabidopsis, and what are the functions of and levels of redundancy among these proteins? Given the number of mutant populations that are now available for rice, these experiments are now quite feasible. As more components of the plant small RNA machinery are identified by more intricate genetic screens and biochemical methods, the relationship and divergence between plant species and lineages will increasingly be an area of interest.
References
Chapman EJ, Carrington JC. Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet 2007;8:884–96.
Zaratiegui M, Irvine DV, Martienssen RA. Noncoding RNAs and gene silencing. Cell 2007;128:763–76.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97.
Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol 2006;57:19–53.
Vaucheret H. Post-transcriptional small RNA pathways in plants: mechanisms and regulations. Genes Dev 2006;20:759–71.
Allen E, Xie Z, Gustafson AM, Carrington JC. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005;121:207–21.
Xie Z, Allen E, Wilken A, Carrington JC. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc Natl Acad Sci U S A 2005;102:12984–9.
Xie Z, Johansen LK, Gustafson AM, Kasschau KD, Lellis AD, Zilberman D, et al. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2004;2:E104.
Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, et al. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell 2004;16:69–79.
Yoshikawa M, Peragine A, Park MY, Poethig RS. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev 2005;19:2164–75.
Gasciolli V, Mallory AC, Bartel DP, Vaucheret H. Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr Biol 2005;15:1494–500.
Henderson IR, Zhang X, Lu C, Johnson L, Meyers BC, Green PJ, et al. Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 2006;38:721–5.
Bouche N, Lauressergues D, Gasciolli V, Vaucheret H. An antagonistic function for Arabidopsis DCL2 in development and a new function for DCL4 in generating viral siRNAs. EMBO J 2006;25:3347–56.
Nagasaki H, Itoh J, Hayashi K, Hibara K, Satoh-Nagasawa N, Nosaka M, et al. The small interfering RNA production pathway is required for shoot meristem initiation in rice. Proc Natl Acad Sci U S A 2007;104:14867–71.
Liu B, Chen Z, Song X, Liu C, Cui X, Zhao X, et al. Oryza sativa dicer-like4 reveals a key role for small interfering RNA silencing in plant development. Plant Cell 2007;19:2705–18.
Liu B, Li P, Li X, Liu C, Cao S, Chu C, et al. Loss of function of OsDCL1 affects microRNA accumulation and causes developmental defects in rice. Plant Physiol 2005;139:296–305.
Qi Y, He X, Wang XJ, Kohany O, Jurka J, Hannon GJ. Distinct catalytic and non-catalytic roles of ARGONAUTE4 in RNA-directed DNA methylation. Nature 2006;443:1008–12.
Kim VN. Sorting out small RNAs. Cell 2008;133:25–6.
Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell 2008;133:116–27.
Montgomery TA, Howell MD, Cuperus JT, Li D, Hansen JE, Alexander AL, et al. Specificity of ARGONAUTE7-miR390 interaction and dual functionality in TAS3 trans-acting siRNA formation. Cell 2008;133:128–41.
Allen E, Xie Z, Gustafson AM, Sung GH, Spatafora JW, Carrington JC. Evolution of microRNA genes by inverted duplication of target gene sequences in Arabidopsis thaliana. Nat Genet 2004;36:1282–90.
Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PLoS ONE 2007;2:e219.
Rajagopalan R, Vaucheret H, Trejo J, Bartel DP. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 2006;20:3407–25.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993;75:843–54.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993;75:855–62.
Griffiths-Jones S. The microRNA registry. Nucleic Acids Res 2004;32:D109–11.
Griffiths-Jones S. miRBase: the microRNA sequence database. Methods Mol Biol 2006;342:129–38.
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res 2006;34:D140–4.
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008;36:D154–8.
Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol 2003;4:R42.
Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell 2004;14:787–99.
Zhang BH, Pan XP, Wang QL, Cobb GP, Anderson TA. Identification and characterization of new plant microRNAs using EST analysis. Cell Res 2005;15:336–60.
Sunkar R, Girke T, Jain PK, Zhu JK. Cloning and characterization of microRNAs from rice. Plant Cell 2005;17:1397–411.
Sunkar R, Zhou X, Zheng Y, Zhang W, Zhu JK. Identification of novel and candidate miRNAs in rice by high throughput sequencing. BMC Plant Biol 2008;8:25.
Yao Y, Guo G, Ni Z, Sunkar R, Du J, Zhu JK, et al. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.). Genome Biol 2007;8:R96.
Nobuta K, Venu RC, Lu C, Belo A, Vemaraju K, Kulkarni K, et al. An expression atlas of rice mRNAs and small RNAs. Nat Biotechnol 2007;25:473–7.
Wang JF, Zhou H, Chen YQ, Luo QJ, Qu LH. Identification of 20 microRNAs from Oryza sativa. Nucleic Acids Res 2004;32:1688–95.
Adai A, Johnson C, Mlotshwa S, Archer-Evans S, Manocha V, Vance V, et al. Computational prediction of miRNAs in Arabidopsis thaliana. Genome Res 2005;15:78–91.
Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 2002;12:1484–95.
Bonnet E, Wuyts J, Rouze P, Van de Peer Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc Natl Acad Sci U S A 2004;101:11511–6.
Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev 2002;16:1616–26.
Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004;16:2001–19.
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA. Conservation and divergence of plant microRNA genes. Plant J 2006;46:243–59.
Sieber P, Wellmer F, Gheyselinck J, Riechmann JL, Meyerowitz EM. Redundancy and specialization among plant microRNAs: role of the MIR164 family in developmental robustness. Development 2007;134:1051–60.
Wang Y, Hindemitt T, Mayer KF. Significant sequence similarities in promoters and precursors of Arabidopsis thaliana non-conserved microRNAs. Bioinformatics 2006;22:2585–9.
Lu C, Kulkarni K, Souret FF, MuthuValliappan R, Tej SS, Poethig RS, et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res 2006;16:1276–88.
Lu C, Jeong DH, Kulkarni K, Pillay M, Nobuta K, German R, et al. Genome-wide analysis for discovery of rice microRNAs reveals natural antisense microRNAs (nat-miRNAs). Proc Natl Acad Sci U S A 2008;105:4951–6.
Hamilton AJ, Baulcombe DC. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999;286:950–2.
Hamilton A, Voinnet O, Chappell L, Baulcombe D. Two classes of short interfering RNA in RNA silencing. EMBO J 2002;21:4671–9.
Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev 2003;17:49–63.
Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell 2002;14:1605–19.
Lippman Z, Martienssen R. The role of RNA interference in heterochromatic silencing. Nature 2004;431:364–70.
Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, et al. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol 2007;5:e57.
Li CF, Pontes O, El-Shami M, Henderson IR, Bernatavichute YV, Chan SW, et al. An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 2006;126:93–106.
Herr AJ, Jensen MB, Dalmay T, Baulcombe DC. RNA polymerase IV directs silencing of endogenous DNA. Science 2005;308:118–20.
Huettel B, Kanno T, Daxinger L, Aufsatz W, Matzke AJ, Matzke M. Endogenous targets of RNA-directed DNA methylation and Pol IV in Arabidopsis. EMBO J 2006;25:2828–36.
Kanno T, Huettel B, Mette MF, Aufsatz W, Jaligot E, Daxinger L, et al. Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat Genet 2005;37:761–5.
Onodera Y, Haag JR, Ream T, Nunes PC, Pontes O, Pikaard CS. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 2005;120:613–22.
Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev 2004;18:2368–79.
Pontes O, Li CF, Nunes PC, Haag J, Ream T, Vitins A, et al. The Arabidopsis chromatin-modifying nuclear siRNA pathway involves a nucleolar RNA processing center. Cell 2006;126:79–92.
Zhang X, Henderson IR, Lu C, Green PJ, Jacobsen SE. Role of RNA polymerase IV in plant small RNA metabolism. Proc Natl Acad Sci U S A 2007;104:4536–41.
Mosher RA, Schwach F, Studholme D, Baulcombe DC. PolIVb influences RNA-directed DNA methylation independently of its role in siRNA biogenesis. Proc Natl Acad Sci U S A 2008;105:3145–50.
Okano Y, Miki D, Shimamoto K. Small interfering RNA (siRNA) targeting of endogenous promoters induces DNA methylation, but not necessarily gene silencing, in rice. Plant J 2008;53:65–77.
Sunkar R, Girke T, Zhu JK. Identification and characterization of endogenous small interfering RNAs from rice. Nucleic Acids Res 2005;33:4443–54.
Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ. Elucidation of the small RNA component of the transcriptome. Science 2005;309:1567–9.
von Bubnoff A. Next-generation sequencing: the race is on. Cell 2008;132:721–3.
Morin RD, Aksay G, Dolgosheina E, Ebhardt HA, Magrini V, Mardis ER, et al. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res 2008;18:571–84.
Schauer SE, Jacobsen SE, Meinke DW, Ray A. DICER-LIKE1: blind men and elephants in Arabidopsis development. Trends Plant Sci 2002;7:487–91.
Shi ZWJ, Wan X, Shen G, Wang X, Zhang J. Over-expression of rice OsAGO7 gene induces upward curling of the leaf blade that enhanced erect-leaf habit. Planta 2007;226:99–108.
Carmell MA, Xuan Z, Zhang MQ, Hannon GJ. The Argonaute family: tentacles that reach into RNAi, developmental control, stem cell maintenance, and tumorigenesis. Genes Dev 2002;16:2733–42.
Vaucheret H, Vazquez F, Crete P, Bartel DP. The action of ARGONAUTE1 in the miRNA pathway and its regulation by the miRNA pathway are crucial for plant development. Genes Dev 2004;18:1187–97.
Baumberger N, Baulcombe DC. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci U S A 2005;102:11928–33.
Kidner CA, Martienssen RA. The role of ARGONAUTE1 (AGO1) in meristem formation and identity. Dev Biol 2005;280:504–17.
Goodrich J, Puangsomlee P, Martin M, Long D, Meyerowitz EM, Coupland G. A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 1997;386:44–51.
Katz A, Oliva M, Mosquna A, Hakim O, Ohad N. FIE and CURLY LEAF polycomb proteins interact in the regulation of homeobox gene expression during sporophyte development. Plant J 2004;37:707–19.
Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 2003;299:716–9.
Garcia D, Collier SA, Byrne ME, Martienssen RA. Specification of leaf polarity in Arabidopsis via the trans-acting siRNA pathway. Curr Biol 2006;16:933–8.
Hunter C, Sun H, Poethig RS. The Arabidopsis heterochronic gene ZIPPY is an ARGONAUTE family member. Curr Biol 2003;13:1734–9.
Yang L, Liu Z, Lu F, Dong A, Huang H. SERRATE is a novel nuclear regulator in primary microRNA processing in Arabidopsis. Plant J 2006;47:841–50.
Chen X. MicroRNA biogenesis and function in plants. FEBS Lett 2005;579:5923–31.
Bollman KM, Aukerman MJ, Park MY, Hunter C, Berardini TZ, Poethig RS. HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development 2003;130:1493–504.
Han MH, Goud S, Song L, Fedoroff N. The Arabidopsis double-stranded RNA-binding protein HYL1 plays a role in microRNA-mediated gene regulation. Proc Natl Acad Sci U S A 2004;101:1093–8.
Houseley J, LaCava J, Tollervey D. RNA-quality control by the exosome. Nat Rev Mol Cell Biol 2006;7:529–39.
Souret FF, Kastenmayer JP, Green PJ. AtXRN4 degrades mRNA in Arabidopsis and its substrates include selected miRNA targets. Mol Cell 2004;15:173–83.
Adenot X, Elmayan T, Lauressergues D, Boutet S, Bouche N, Gasciolli V, et al. DRB4-dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr Biol 2006;16:927–32.
Vaucheret H. MicroRNA-dependent trans-acting siRNA production. Sci STKE 2005;2005:pe43.
Howell MD, Fahlgren N, Chapman EJ, Cumbie JS, Sullivan CM, Givan SA, et al. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell 2007;19:926–42.
Chen HM, Li YH, Wu SH. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc Natl Acad Sci U S A 2007;104:3318–23.
Wassenegger M, Krczal G. Nomenclature and functions of RNA-directed RNA polymerases. Trends Plant Sci 2006;11:142–51.
Margis R, Fusaro AF, Smith NA, Curtin SJ, Watson JM, Finnegan EJ, Waterhouse PM. The evolution and diversification of Dicers in plants. FEBS Lett. 2006;580:2442–50
Hirochika H, Guiderdoni E, An G, Hsing YI, Eun MY, Han CD, et al. Rice mutant resources for gene discovery. Plant Mol Biol 2004;54:325–34.
Acknowledgments
We are particularly indebted to Dr. Carolyn Napoli of ChromDB for her work in generating the phylogenetic trees. We gratefully acknowledge the assistance of Dr. Kan Nobuta for assistance with handling the O. barthii data and Xiang Song and Dr. Rod Wing for providing the material for this library. This work was supported primarily by NSF Plant Genome Research Program award 0701745.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Table 1
Non-conserved miRNAs identified in rice. (DOC 76.5 KB)
Supplementary Fig. 1
Phylogenetic trees for Dicers, RNA dependent RNA polymerases, and Pol IV subunits. (DOC 225 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Simon, S.A., Zhai, J., Zeng, J. et al. The Cornucopia of Small RNAs in Plant Genomes. Rice 1, 52–62 (2008). https://doi.org/10.1007/s12284-008-9008-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12284-008-9008-5