
Abstract
General anesthesia is an unconscious state induced by anesthetics for surgery. The molecular targets and cellular mechanisms of general anesthetics in the mammalian nervous system have been investigated during past decades. In recent years, K+ channels have been identified as important targets of both volatile and intravenous anesthetics. This review covers achievements that have been made both on the regulatory effect of general anesthetics on the activity of K+ channels and their underlying mechanisms. Advances in research on the modulation of K+ channels by general anesthetics are summarized and categorized according to four large K+ channel families based on their amino-acid sequence homology. In addition, research achievements on the roles of K+ channels in general anesthesia in vivo, especially with regard to studies using mice with K+ channel knockout, are particularly emphasized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the early 19th century, general anesthetics have been used to induce a state of unconsciousness for surgery. Modern anesthesiology defines a complete general anesthetic effect as including unconsciousness, amnesia, analgesia, and muscle relaxation that is indispensable for modern surgery. Hitherto, myriad molecular targets of general anesthetics have been identified, such as gamma-aminobutyric acid receptor, the N-methyl-D-aspartate receptor families, and ion channels [1,2,3,4,5,6]. Among the many kinds of ion channels, potassium (K+) channels are the most diverse and ubiquitous, playing important roles in controlling neuronal excitability and neurotransmitter release in the central nervous system by determining the membrane potential of neurons [7,8,9,10,11,12]. Thus, many studies have been conducted on the regulatory effects of general anesthetics on K+ channel activity. Also, data on these effects have accumulated, and this review summarizes recent research achievements in this area, especially emphasizing studies using K+ channel knockout (KO) mice. K+ channels that have been studied as general anesthetic targets have been assigned to 4 categories according to a standard nomenclature by The International Union of Basic and Clinical Pharmacology (IUPHAR) and the Human Genome Organisation Gene Nomenclature Committee (HGNC) [7]. They are voltage-gated (Kv) channels, the background/leak or tandem 2-pore (K2P) families, inwardly-rectifying (Kir) channels, and Ca2+-activated (KCa) channels. Research advances related to the effects of general anesthetics on these channels are summarized based on the above classification.
Modulation of Kv Channels by General Anesthetics
The opening of Kv channels is regulated by a membrane potential change and triggered by moving of the voltage sensor domain located on the S4 segment. The voltage-gated Kv channel family includes 40 genes encoding pore-forming subunits that are divided into 12 subfamilies (Kv1–Kv12) based on sequence homology (Fig. 1A–C). The HGNC and IUPHAR nomenclature and names based on the homologous channels in Drosophila are shown in Fig. 1A [13].

Phylogenetic trees of the Kv channel family and critical structure of Kv1.2 channel for sevoflurane binding. A Amino acid sequences of Kv1–9 channels were aligned by MEGA software with the Clustal W method and analyzed by the neighbor joining test. B, C Kv7 subfamily and Kv 10–12 subfamilies are shown separately because of the low amino acid sequence similarity with other Kv channels. Names of channel subtypes are labeled in color based on their different responses to volatile anesthetics in experiments performed in vitro with patch clamp. The channels that can be activated by volatile anesthetics are shown in red; channels inhibited by volatile anesthetics are shown in yellow; and channels insensitive to volatile anesthetics are shown in green. Channels with unknown responses to volatile anesthetics are in black color. The protein IDs of Kv channels used in this sequence alignment are as follows: Kv1.1 (NP_000208), Kv1.2 (NP_004965), Kv1.3 (NP_002223), Kv1.4 (NP_002224), Kv1.5 (NP_002225), Kv1.6 (NP_002226), Kv1.7 (NP_114092), Kv1.8 (NP_005540); Kv2.1 (NP_004966), Kv2.2 (NP_004761), Kv3.1 (NP_004967), Kv3.2 (NP_631875), Kv3.3 (NP_004968), Kv3.4 (NP_004969), Kv4.1 (NP_004970), Kv4.2 (NP_036413), Kv4.3 (NP_004971); Kv5.1 (NP_002227), Kv6.1 (NP_002228), Kv6.2 (NP_036415), Kv6.3 (NP_579875), Kv6.4 (NP_758857), Kv7.1 (NP_000209), Kv7.2 (NP_742105), Kv7.3 (NP_004510), Kv7.4 (NP_004691), Kv7.5 (NP_062816), Kv8.1 (NP_055194), Kv8.2 (NP_598004); Kv9.1 (NP_002242), Kv9.2 (NP_065748), Kv9.3 (NP_002243); Kv10.1 (NP_758872) Kv10.2 (NP_647479); Kv11.1 (NP_000229), Kv11.2 (NP_110416), Kv11.3 (NP_150375); Kv12.1 (NP_653234), Kv12.2 (NP_036416), and Kv12.3 (NP_036417). D The local structure of sevoflurane-binding site on Kv1.2 channels. I402 on S6 and the L317 in the S4–S5 linker form a sevoflurane (shown in red)-binding pocket with V265 and A266. Two domains with S5 and S6 linker are shown. The picture was drawn with the VMD software based on crystal structure of Kv1.2 with PDB file 3LUT.
Members of the Shaker-Related K+ Channel Family (Kv1.1–Kv1.6) are Important Targets of Volatile Anesthetics
The Shaker channel was the first cloned voltage-dependent K+ channel from Drosophila [14, 15]. Flies with a mutated Shaker gene shake their legs under ether anesthesia (hence the name). Subsequently, Shaker-related K+ channels were cloned from mammals and named the Kv1.x channel family.
Given the critical role Kv channels play in limiting neuronal excitability, studies of the effects of general anesthetics on the Shaker channel began almost immediately after it was cloned. The earliest study performed on Xenopus oocytes revealed that chloroform and halothane reduce the macroscopic conductance of the Shaker channel while isoflurane increases its macroscopic conductance [16]. Consistent with these results, subsequent studies on Drosophila showed that halothane alters the electroretinogram by reducing its transient component at light-off via inactivation of the Shaker channel [17]. Meanwhile, the sleep state, which differs from anesthesia but shares some of its characteristics, is also shorter in Drosophila with a mutated Shaker channel [18]. These results suggested that Shaker-related K+ channels are important targets of volatile anesthetics (Table 1).
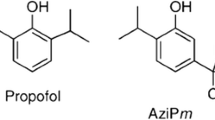
Since then, the effects of general anesthetics on Shaker-related K+ channels in mammals have been widely studied. Barber et al. reported that 1 mmol/L sevoflurane potentiates Drosophila Shaker B, Kv1.2, and Kv1.5 channel currents over the physiological range of membrane potential (−60 to −40 mV) [19]. These results were further confirmed and expanded by a subsequent study by Lioudyno et al., who reported that a sub-surgical dose of sevoflurane (0.2 mmol/L) potentiates the currents of Kv1.1, Kv1.2, Kv1.3, and Kv1.5 channels at low depolarizing potentials (−40 to 0 mV) in a heterologous expression system. But at higher depolarization potentials (30 to 60 mV), the currents of Kv1.1 and Kv1.2 channels are still potentiated, whereas the Kv1.3 and Kv1.5 currents are inhibited [20]. The mechanism of action of sevoflurane on Kv channels can be explained as sevoflurane favoring the open state of Kv1.3 and Kv1.5 channels but accelerating its inactivation at high membrane potentials. By combining mutagenesis and electrophysiology with the use of azisevoflurane, a photoaffinity ligand of sevoflurane, Leu317 within the internal S4-S5 linker of the Kv1.2 channel has been identified as the sevoflurane binding site (Fig. 1D) [21, 22]. Azisevoflurane also labels a second distinct site, Thr384, near the external selectivity filter in the Kv1.2 G329T mutant, and this alters the voltage-dependent gating behavior [21]. These results indicate that the positive allosteric modulation of Kv channels by sevoflurane involves separate processes and multiple sites related to gating modification and the transition of channel state conformation. On the other hand, research has shown that the Kv1.2 channel is resistant to the intravenous anesthetic propofol. But AziPm, a photoactive propofol analog, inhibits the Kv1.2 channel while potentiating the Kv1.2 G329T mutant channel [23]. These results suggest that propofol likely binds to the Kv1.2 channel.
In vivo studies have shown that changing the activity of the Shaker-related K+ channel indeed alters the arousal state of unconscious animals induced by inhaled anesthetics. A microinfusion of an antibody against the Kv1.2 channel into the central medial thalamus (CMT) awakens 17% of rats anesthetized with 3.6% desflurane and 1.2% sevoflurane [24]. In addition, microinfusing a peptide inhibitor of Kv1.1, Kv1.3, and Kv1.6 channels into the CMT restores “consciousness” in rats anesthetized by 1.2% sevoflurane [20]. But the shaker K+ channel is not the target of xenon in short-sleeping D. melanogaster mutants [25].
Taken together, these data confirm that K+ channels in the Kv1.x family are important targets of volatile anesthetics (Table 2) and changing the activity of Kv channels in the CMT plays an important role in altering the arousal state in general anesthesia.
Kv2.1–Kv2.2 (Shab) and Kv3.x (Shaw) Family Channels are Inhibited by Most Inhaled Anesthetics
The Kv2.x family includes two genes that encode the alpha subunit of the voltage-gated K+ channels named Kv2.1 and Kv2.2. They are also called Shab (Shaker cognate B)-related channels because of their homologous channel in Drosophila (Fig. 1A). The effects of anesthetics on these channels have also been studied. Kulkarni et al. reported that ketamine and halothane inhibit the Kv2.1 channel in a dose-dependent manner [26]. Recently, Kv2.1 channel activity was also shown to be inhibited by propofol via a mechanism that decreases its expression level in cortical neurons [27] (Table 3). Similar to Shab-related K+ channels, the voltage-gated Kv3.2 channel, a homolog of the K-Shaw2 channel, is also inhibited by alkanols and halogenated inhaled anesthetics such as halothane, isoflurane, chloroform, and desflurane [28]. Interestingly, the Kv3.2 channel is activated by sevoflurane (Table 1) [29]. Furthermore, many of the intravenous anesthetic drugs, such as thiopental, methohexital, propofol, midazolam, and droperidol (but not barbiturates or ketamine), also inhibit Shaw-like voltage-dependent K+ currents (Table 3) [30]. However, there are no in vivo data to show whether these channels change the arousal state under general anesthesia. The binding sites and inhibitory mechanisms of volatile anesthetics on these Kv channels remain unclear.
Modulation of Other Voltage-Dependent Potassium Channels by Volatile Anesthetics
Among other Kv channels, Kv7.1 channel activity is inhibited to 59% by 190 µmol/L sevoflurane in Kv7.1-expressing cells, but the mechanism is still unclear [31]. Elk1, Kv7.2/7.3, Kv3.4, and Kv4.2 channels are resistant to volatile general anesthetics (Fig. 1A, B, Table 1) [32]. In addition, human ether-a-go-go-related gene (HERG) channels are inhibited by halothane through a mechanism that slows down their activation and accelerates their deactivation and inactivation (Table 1) [33]. Thus far, data to address the roles these channels play in general anesthesia in vivo are lacking. This demonstrates that the effects of general anesthetics on these Kv channels need further work.
Modulation of K2P Channels by Inhaled Anesthetics
The two-pore-domain K+ (K2P) channel family is composed of fifteen KCNK genes, each of which comprises four transmembrane segments and two pore domains in tandem. Two pore-forming subunits form a functional channel as dimers. K2P channels conduct time- and voltage-independent background, or ‘leak’ currents to generate a negative membrane potential in excitable and non-excitable cells. K2P channels can be classified into six subgroups (TREK, TASK, TWIK, TALK, THIK, and TRESK) based on coding genes and biophysical properties (Fig. 2A). The nomenclature provided by HGNC and IUPHAR, and a popular naming scheme are all assigned sequentially in Fig. 2 [34]. Among them, the TASK and TREK-1 channels were the first to be identified and they are potentiated by general anesthetics. Chloroform, diethyl ether, halothane, and isoflurane potentiate the TREK-1 channel, whereas halothane and isoflurane activate the TASK channel (Table 1) [35]. The C-terminus of the TREK-1 and TASK channels is important for activation by general anesthetics [35]. Subsequent studies have revealed many members of this family to be sensitive to volatile anesthetics. Here, we give an introduction according to the subfamilies.

Phylogenetic tree of the K2P channel and local structure of TASK-1 channel for isoflurane binding. A TREK-1, TREK-2, TASK-1, TASK-2, TASK-3 and TRESK channels are activated by volatile anesthetics (red), whereas THIK-1, TALK-1, and TALK-2 channels are inhibited by volatile anesthetics in vitro (yellow). TRAAK channel activity (green) is not modulated by volatile general anesthetics. Responses of others channels (black) to anesthetics remain unknown. The protein IDs used in this alignment are as follows: K2P1.1: NP_002236, K2P2.1: NP_001017425, K2P3.1: NP_002237, K2P4.1: NP_201567, K2P5.1: NP_003731, K2P6.1: NP_004814, K2P7.1: NP_203133, K2P9.1: NP_001269463, K2P10.1: NP_066984, K2P12.1: NP_071338, K2P13.1: NP_071337, K2P15.1: NP_071753, K2P16.1: NP_115491, K2P17.1: NP_113648, and K2P18.1: NP_862823. B Local structure of K2P channel for isoflurane binding. The image was drawn based on the TASK-1 channel structure model by using the Swissport modeling system. The crystal structure of human two-pore-domain potassium ion channel TREK2 (K2P10.1) in complex with a brominated fluoxetine derivative was used as the template (crystal structure PDB file 4xdl.1).
Inhaled Anesthetics Activate TASK-1 and TASK-3 Channels
Both the TASK-1 and TASK-3 channels are activated by clinically relevant concentrations of inhaled anesthetics [36]. Further studies have identified the amino-acid M159 as the critical determinant of the effect of isoflurane on the hTASK-1 channel; it is located between the region of the end of TM2 (transmembrane domain 2) and the middle of TM3 (Fig. 2B) [37]. But the mechanism by which the activity and conformation of the channel change has not been addressed. As a result, TASK-1-KO mice are less sensitive to the anesthetic effects of halothane, isoflurane, and dexmedetomidine than wild-type mice. They require higher concentrations of general anesthetics to induce immobility as reflected by loss of the tail-withdrawal reflex. TASK-3-KO mice also exhibit reduced sensitivity to the inhalation anesthetic halothane, but not isoflurane [36, 38] (Table 2). Although the TASK-3 channel is sensitive to some volatile anesthetics, it is insensitive to clinically-relevant concentrations of the anesthetic gases nitrous oxide, xenon, and cyclopropane (Table 1) [39]. The TASK-1 and TASK-3 channels are also inhibited by etomidate at concentrations used in clinical work but are insensitive to propofol [40] (Table 3). Furthermore, in vivo data suggest that periods of loss of the righting reflex produced by the same concentrations of propofol and pentobarbital in TASK-1-KO mice are longer than such periods in wild-type mice [41] (Table 4).
Activation of TREK Family Channels (TREK-1, TREK-2, and TRAAK) by General Anesthetics
Among the anesthetic-activated neuronal background K+ channels, the TASK and TREK channel subgroups have received the most attention [42]. The TREK channel is highly expressed in the human central nervous system, especially in the subventricular and ventricular zones, hippocampus, striatum, and certain parts of the cortex (layer IV) [43, 44]. The gating mechanisms of TREK channels include stretch, fatty acids, pH, and G protein-coupled receptors [45].
TREK-1 and its functional homologue TREK-2 are sensitive to volatile anesthetics such as chloroform, diethyl ether, halothane, and isoflurane (2–3 fold increase in current amplitude at 1 mmol/L anesthetic) (Table 1) [35]. However, the structurally and functionally related K2P channel TRAAK, which can even form functional heterodimeric channels with TREK-1, is insensitive to volatile anesthetics [35, 46]. This difference may be attributed to the specific structural features of transmembrane helix straightening and buckling of the M4 segment of the TRAAK channel (KCNK4) [47]. Another type of gaseous general anesthetic containing nitrous oxide and xenon has also been reported to open TREK-1 channels [39]. In addition, some intravenous anesthetics (chloral hydrate and trichloroethanol, but not phenobarbital) at pharmacologically relevant concentrations have been shown to open TREK-1 and TRAAK channels [48] (Table 3). Further, several in vivo experiments have demonstrated that the concentrations of some volatile anesthetics, including chloroform, halothane, sevoflurane, and desflurane, required for loss of the righting reflex and the tail clamp withdrawal reflex are significantly increased in TREK-1-KO mice [48, 49] (Table 2). Taken together, all these data suggest that TREK channels are important targets of general anesthetics.
Modulation of the TALK Subfamily (TALK-1, TALK-2, and TASK-2), TWIK Subfamily (TWIK-1, TWIK-2, and KCNK7), THIK Family (THIK-1 and THIK-2), and TRESK Family (TRESK-1 and TRESK-2) Channels by General Anesthetics
The TASK-2 channel has low amino-acid identity with the TASK-1 and TASK-3 channels. The amino-acid identity and high sensitivity to pH suggest that the TASK-2 channel belongs to the TALK subfamily (Fig. 2A) [34]. In heterologous expression systems, a handful of experiments have been done to elucidate the pharmacological effects of volatile anesthetics on the TASK-2 channel. The results demonstrated that volatile anesthetics, among which chloroform was found to be the most potent, activate the TASK-2 channel, but not as potently as the TASK and TREK channels [50]. Thus, it is not surprising that the behavioral effects of inhaled anesthetics are unperturbed in TASK-2-KO mice [51] (Table 2). The minimum alveolar concentration (MAC) values of desflurane, halothane, and isoflurane in TASK-2-KO mice are similar to those of the wild-type [51]. Although in vitro studies have shown that TALK-1 and TALK-2 channel currents are inhibited by chloroform, halothane, and isoflurane, the possibility that they play a role in changing the arousal state in general anesthesia is low because they are mainly expressed in the pancreas [52].
The mRNA expression of TWIK-1 and TWIK-2 channels has been found in many brain regions [53], but their sensitivity to general anesthetics remains unknown (Fig. 2A). KCNK7-KO mice have been used to test the anesthetic effects of varying concentrations of isoflurane, sevoflurane, and desflurane, and the results showed that the MAC values of KCNK7-KO mice defined by the tail clamp withdrawal reflex are indistinguishable from those of the wild-type [54] (Table 2). Thus far, there is no evidence that this channel family is involved in changing the arousal state under general anesthesia.
THIK subfamily channels, including THIK-1 and THIK-2, were first cloned from rat brain and the amino-acid sequences of these two channels are 58% identical [55] (Fig. 2A). THIK-2 is expressed in most brain regions while THIK-1 is expressed in more restricted regions. However, the THIK-2 channel is less functional in vivo probably because its trafficking to the membrane is limited by putative retention/retrieval signals on both the N and C termini [55, 56]. The THIK-1 channel current is inhibited by inhaled anesthetics such as halothane [55]. In addition, in vivo data indicate that isoflurane causes activation of retrotrapezoid nucleus neurons via inhibition of a THIK-1-like background current [57], which suggests that the THIK-1 channel contributes to the maintenance of respiratory motor activity under general anesthesia.
The TRESK channel subfamily contains two members: TRESK-1 and TRESK-2 (Fig. 2A). Protein expression of the TRESK channel has been detected in many regions of the nervous system, including the dorsal root and trigeminal ganglia, cerebrum, cerebellum, brainstem, and sympathetic and parasympathetic ganglia, suggesting potential roles in pain transmission and anesthesia [58].
In the Xenopus oocyte and COS-7 cell expression systems, clinical doses of isoflurane, halothane, sevoflurane, and desflurane strongly increase TRESK currents up to three-fold by increasing the open probability [59]. Isoflurane is the most potent activator with an EC50 of ~ 150 µmol/L. However, some intravenous anesthetics, including etomidate, thiopental, and propofol, have little effect on TRESK channels [60] (Table 3). TRESK-KO mice have also been used to investigate the role of TRESK channels in volatile general anesthesia. The results showed that the MAC value of isoflurane is marginally increased (8%) while those of other volatile anesthetics are not significantly altered [61] (Table 2). These data suggest that either the role of the TRESK channel in general anesthesia is compensated by other channels in KO mice or the TRESK channel plays little role in altering the arousal state in general anesthesia.
Modulation of Inwardly-Rectifying Potassium Channels by General Anesthetics
The mammalian Kir channel family includes 16 genes that encode an α subunit which has two transmembrane segments (M1 and M2) with a pore loop in between (Fig. 3A). Kir channels conduct K+ currents more readily in the inward direction than in the outward direction, and they are activated by ligand-stimulated G protein-coupled receptors. With activation of these receptors, G-protein βγ subunits are released from heterotrimeric receptors to interact with GIRK (G protein-coupled inwardly-rectifying K+) channels and open them. Among these genes, Kir3.x genes (GIRK1–4), such as GIRK1, GIRK2, and GIRK3 are predominantly expressed in neurons while GIRK4 is mainly expressed in the heart [10]. In neurons, GIRK channels either form GIRK2 homotetramers or form heterotetramers of GIRK1, GIRK2, and/or GIRK3 channels [62]. Tetramer channels with different subunits demonstrate different sensitivity to volatile anesthetics. Halothane enhances background currents through hetero-oligomeric GIRK1/GIRK4 channels but not through homo-oligomeric GIRK2 channels. This activation of basal current also requires the presence of G-protein βγ subunits. In contrast to basal GIRK currents, the agonist-induced GIRK currents (via co-expressed M2 muscarinic receptors) are inhibited by halothane (Table 1). In GIRK1/GIRK4 channels, this inhibition is most pronounced at low anesthetic concentrations (0.1 mmol/L–0.3 mmol/L) and occurs also when channels are activated by guanosine-5’-O-(3-thio) triphosphate. However, high concentrations of halothane (0.9 mmol/L) enhance the agonist-induced currents [63] (Table 1). This increase in agonist-induced currents is never seen with GIRK2 homo-oligomeric channels. Subsequent studies using GIRK1 channel (Del 363) and GIRK2 channel (Del 356) mutants that lack the C-terminal ends confirmed that these different modulatory effects produced by halothane are determined by the C terminus [64]. GIRK channels are resistant to intravenous anesthetics and nitrous oxide gas anesthetic (Table 3) [32]. On the other hand, efforts to investigate the roles of GIRK channels in general anesthesia in vivo using GIRK2-KO mice indicated that the MACs for 50% immobility in the tail clamp test for adult wild-type and GIRK2-KO mice are not significantly different [51] (Table 2). This result suggests that there is insufficient evidence to support the hypothesis that GIRK channels are involved in altering the arousal state in general anesthesia.

Phylogenetic trees of the Kir channels and the Ca2+-activated potassium channels and structure of NBD1 domain of Kir6.2 channel that alters the effect of isoflurane activation on the channel. A In Kir channel family, Girk1/Girk4 channels are activated by halothane (red). IRK1, Girk2 and ROMK channels are resistant to volatile anesthetics (green). Protein IDs of the Kir channel family are as follows: Kir1.1: NP_000211, Kir2.1: NP_000882, Kir3.1: NP_002230, Kir2.3: NP_004972, Kir3.4: NP_000881, Kir3.2: NP_002231, Kir6.1: NP_004973, Kir3.3: NP_004974, Kir4.1: NP_002232, Kir6.2: NP_000516, Kir2.2: NP_066292, Kir7.1: NP_002233, Kir2.4: NP_037480, Kir4.2: NP_733933, and Kir5.1: NP_001278554. B Left: Crystal structure of the Kir6.2 channel (based on PDB file 5TWV) with regulatory sulfonylurea receptor (SUR). The NBD1 domain (nucleotide binding domain) located on the SUR receptor is shown in purple color. Right: local structure of NBD1 domain with K708 residue that alters the affinity to isoflurane. C The phylogenetic tree of the Ca2+-activated potassium channels includes large-conductance channels KCa1.1 (BK or Slo1), KCa4.1 (Slack or Slo2.2), KCa4.2 (Slick or Slo2.1), and KCa5.1 (Slo3), intermediate-conductance channel KCa3.1 (IK), and three small-conductance KCa channels are KCa2.1–2.3 (SK). Of particular note, KCa4.1–4.2 are activated by internal Na+ and Cl−; KCa5.1 is activated by internal alkalization (OH). Channels that can be inhibited by volatile anesthetics are shown in yellow. Protein IDs are as follows: KCa1.1: NP_001309764, KCa2.1: NP_002239, KCa2.2: NP_067627, KCa2.3: NP_002240, KCa3.1: NP_002241, KCa4.1: NP_065873, KCa4.2: NP_940905, and KCa5.1: NP_001027006.
Among the Kir channels, Kir6.2 has also been named a KATP channel because it is inhibited by intracellular ATP and is sensitive to intracellular pH. It is composed of the pore-forming inward rectifier Kir6.2 and the regulatory sulfonylurea receptor SUR2A or SUR1. Although isoflurane does not directly interact with the Kir6.2 channel, it interacts with the SUR2A receptor to activate the Kir6.2 channel at low pH (pH 6.8). However, it has no activation effect at pH 7.4. Also, isoflurane cannot activate Kir6.2 channels co-expressed with SUR1 receptors (Table 1). Site-directed mutagenesis in the Walker motif of the SUR2A receptor abolishes the activation of Kir6.2 channels by isoflurane [65, 66]. Based on the recently dissected Kir6.2 channel structure, the nucleotide-binding domain on the SUR receptor is shown in Fig. 3B [67]. This study indicated that isoflurane preferentially enhances opening of the cardiac KATP channel (Kir6.2/SUR2A), suggesting a role of the KATP channel in cardiac protection in the anesthetic state induced by halogenated anesthetics.
Modulation of Calcium-Activated Potassium Channels by General Anesthetics
The KCa family can be divided into three categories: BK (big conductance), IK (intermediate conductance), and SK (small conductance) channels [68]. BK channels are all tetrameric; they are formed by α subunits that have 6/7 transmembrane segments encoded by genes SLO1-3 [69]. But the Slo1 and Slo2 channels are expressed in brain, while the Slo3 channel is specifically expressed in testis and spermatozoa [70]. The Slo2 channel subfamily includes Slo2.1 and Slo2.2, which are activated by Na+ rather than Ca2+ [71]. The Slo1 channel is activated by both voltage and intracellular Ca2+ while the IK and SK channels are gated by intracellular Ca2+ alone. BK channels are diffusely expressed in almost the whole brain, whereas IK channels are mainly expressed in T lymphocytes, red blood cells, and parotid glands [72].
The regulatory effects of general anesthetics on BK, IK, and SK channels have been investigated. The volatile anesthetic halothane, at a clinically relevant dose of 0.5 mmol/L, reduces the open probability of BK channels without altering the single-channel conductance. However, this effect is blocked by increasing the concentration of cytoplasmic free Ca2+ from 1 µmol/L to 100 µmol/L, suggesting that halothane serves as a closed channel blocker [73]. In addition to halothane, isoflurane and enflurane also inhibit BK channels at a clinically relevant dose (Table 1) [74, 75]. Also, the intravenous anesthetic ketamine (at clinically relevant concentrations: 2–500 µmol/L) selectively blocks the BK channel in a dose-dependent manner. Ketamine shifts the open probability vs voltage curve to a higher potential without changing the slope of the voltage-dependence [76]. Interestingly, another intravenous anesthetic (propofol) activates the BK channel to relax coronary arteries [77]. A study using animal models showed that Slo-1-mutated Caenorhabditis elegans are resistant to volatile anesthetics [78]. But the effect of general anesthetics on Slo1-KO mice has not been reported.
IK channel currents are rapidly and reversibly inhibited by many volatile anesthetics such as halothane, isoflurane, enflurane, and sevoflurane with an EC50 from 0.4 to 1 mmol/L, whereas the SK channel is not inhibited by volatile anesthetics [79, 80]. However, the SK channel is blocked by the intravenous anesthetics ketamine, barbital, and methohexital [80]. Experiments using patch clamp in brain slices have further confirmed that propofol enhances the inhibition of spike firing in retrotrapezoid nucleus neurons by blocking SK channels (Table 3) [81].
Although the effects of general anesthetics on Ca2+-activated K+ channels have been investigated, the underlying mechanism and the overall roles played by these channels in general anesthesia remain to be explored.
Concluding Remarks and Future Perspectives
In summary, K+ channel activity is widely regulated by general anesthetics. Among them, the mechanisms by which Kv1.2, Kir6.2, and TASK-1 channels are modulated by general anesthetics have been studied in detail. Local structures and amino-acids critical for anesthetic binding to Kv1.2, Kir6.2, and TASK-1 channels revealed by biophysical studies have further clarified the characteristics of the molecular interactions of general anesthetics with these K+ channels. In addition, recent research has confirmed that activity changes in Kv1.x, TASK-1, TASK-3, and TREK-1 channels are essential for altering the arousal state when volatile anesthetics play their roles (Tables 2 and 4). In addition, the role of the THIK-1 channel in the retrotrapezoid nucleus is to maintain respiratory motor activity under general anesthesia. However, the biophysical mechanisms of the regulatory effects of general anesthetics on other K+ channels remain to be further explored. In the meantime, the roles of many K+ channels in general anesthesia remain elusive, especially the nuclei and neuronal circuits in which they play their roles. In vitro biophysical studies combined with optogenetic electrophysiological studies in vivo using K+ channel KO mice will greatly facilitate further addressing the role of K+ channels in general anesthesia.
References
Covarrubias M, Barber AF, Carnevale V, Treptow W, Eckenhoff RG. Mechanistic insights into the modulation of voltage-gated ion channels by inhalational anesthetics. Biophys J 2015, 109: 2003–2011.
Kopp Lugli A, Yost CS, Kindler CH. Anaesthetic mechanisms: update on the challenge of unravelling the mystery of anaesthesia. Eur J Anaesthesiol 2009, 26: 807–820.
Rudolph U, Antkowiak B. Molecular and neuronal substrates for general anaesthetics. Nat Rev Neurosci 2004, 5: 709–720.
Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med 2003, 348: 2110–2124.
Wang B, Yang X, Sun A, Xu L, Wang S, Lin W, et al. Extracellular signal-regulated kinase in nucleus accumbens mediates propofol self-administration in rats. Neurosci Bull 2016, 32: 531–537.
Moore C, Gupta R, Jordt SE, Chen Y, Liedtke WB. Regulation of pain and itch by TRP channels. Neurosci Bull 2018, 34: 120–142.
Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE, et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev 2003, 55: 583–586.
Huang CG, Chacron MJ. SK channel subtypes enable parallel optimized coding of behaviorally relevant stimulus attributes: A review. Channels (Austin) 2017, 11: 281–304.
Greene DL, Hoshi N. Modulation of Kv7 channels and excitability in the brain. Cell Mol Life Sci 2017, 74: 495–508.
Lujan R, Aguado C. Localization and targeting of GIRK channels in mammalian central neurons. Int Rev Neurobiol 2015, 123: 161–200.
Cooper EC. Made for “anchorin”: Kv7.2/7.3 (KCNQ2/KCNQ3) channels and the modulation of neuronal excitability in vertebrate axons. Semin Cell Dev Biol 2011, 22: 185–192.
Misonou H. Homeostatic regulation of neuronal excitability by K+ channels in normal and diseased brains. Neuroscientist 2010, 16: 51–64.
Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, et al. International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels. Pharmacol Rev 2005, 57: 473–508.
Salkoff L, Wyman R. Genetic modification of potassium channels in Drosophila Shaker mutants. Nature 1981, 293: 228–230.
Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 1987, 237: 770–775.
Correa AM. Gating kinetics of Shaker K+ channels are differentially modified by general anesthetics. Am J Physiol 1998, 275: C1009–1021.
Rajaram S, Nash HA. A specific alteration in the electroretinogram of Drosophila melanogaster is induced by halothane and other volatile general anesthetics. Anesth Analg 2004, 98: 1705–1711.
Cirelli C, Bushey D, Hill S, Huber R, Kreber R, Ganetzky B, et al. Reduced sleep in Drosophila Shaker mutants. Nature 2005, 434: 1087–1092.
Barber AF, Liang Q, Covarrubias M. Novel activation of voltage-gated K(+) channels by sevoflurane. J Biol Chem 2012, 287: 40425–40432.
Lioudyno MI, Birch AM, Tanaka BS, Sokolov Y, Goldin AL, Chandy KG, et al. Shaker-related potassium channels in the central medial nucleus of the thalamus are important molecular targets for arousal suppression by volatile general anesthetics. J Neurosci 2013, 33: 16310–16322.
Woll KA, Peng W, Liang Q, Zhi L, Jacobs JA, Maciunas L, et al. Photoaffinity ligand for the inhalational anesthetic sevoflurane allows mechanistic insight into potassium channel modulation. ACS Chem Biol 2017, 12: 1353–1362.
Liang Q, Anderson WD, Jones ST, Souza CS, Hosoume JM, Treptow W, et al. Positive allosteric modulation of Kv channels by sevoflurane: Insights into the structural basis of inhaled anesthetic action. PLoS One 2015, 10: e0143363.
Bu W, Liang Q, Zhi L, Maciunas L, Loll PJ, Eckenhoff RG, et al. Sites and functional consequence of alkylphenol anesthetic binding to Kv1.2 channels. Mol Neurobiol 2018, 55: 1692–1702.
Alkire MT, Asher CD, Franciscus AM, Hahn EL. Thalamic microinfusion of antibody to a voltage-gated potassium channel restores consciousness during anesthesia. Anesthesiology 2009, 110: 766–773.
Schaper C, Hocker J, Bohm R, Roeder T, Bein B. The shaker potassium channel is no target for xenon anesthesia in short-sleeping Drosophila melanogaster mutants. Sci World J 2012, 2012: 373709.
Kulkarni RS, Zorn LJ, Anantharam V, Bayley H, Treistman SN. Inhibitory effects of ketamine and halothane on recombinant potassium channels from mammalian brain. Anesthesiology 1996, 84: 900–909.
Zhang YZ, Zhang R, Zeng XZ, Song CY. The inhibitory effect of propofol on Kv2.1 potassium channel in rat parietal cortical neurons. Neurosci Lett 2016, 616: 93–97.
Bhattacharji A, Klett N, Go RC, Covarrubias M. Inhalational anaesthetics and n-alcohols share a site of action in the neuronal Shaw2 Kv channel. Br J Pharmacol 2010, 159: 1475–1485.
Barber AF, Liang Q, Amaral C, Treptow W, Covarrubias M. Molecular mapping of general anesthetic sites in a voltage-gated ion channel. Biophys J 2011, 101: 1613–1622.
Friederich P, Urban BW. Interaction of intravenous anesthetics with human neuronal potassium currents in relation to clinical concentrations. Anesthesiology 1999, 91: 1853–1860.
Kim JK, Yoo HY, Kim SJ, Hwang YS, Han J, Kim JA, et al. Effects of sevoflurane on the cAMP-induced short-circuit current in mouse tracheal epithelium and recombinant Cl- (CFTR) and K+ (KCNQ1) channels. Br J Anaesth 2007, 99: 245–251.
Yamakura T, Lewohl JM, Harris RA. Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels. Anesthesiology 2001, 95: 144–153.
Li J, Correa AM. Kinetic modulation of HERG potassium channels by the volatile anesthetic halothane. Anesthesiology 2002, 97: 921–930.
Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 2005, 57: 527–540.
Patel AJ, Honore E, Lesage F, Fink M, Romey G, Lazdunski M. Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci 1999, 2: 422–426.
Linden AM, Aller MI, Leppa E, Vekovischeva O, Aitta-Aho T, Veale EL, et al. The in vivo contributions of TASK-1-containing channels to the actions of inhalation anesthetics, the alpha(2) adrenergic sedative dexmedetomidine, and cannabinoid agonists. J Pharmacol Exp Ther 2006, 317: 615–626.
Andres-Enguix I, Caley A, Yustos R, Schumacher MA, Spanu PD, Dickinson R, et al. Determinants of the anesthetic sensitivity of two-pore domain acid-sensitive potassium channels: molecular cloning of an anesthetic-activated potassium channel from Lymnaea stagnalis. J Biol Chem 2007, 282: 20977–20990.
Linden AM, Sandu C, Aller MI, Vekovischeva OY, Rosenberg PH, Wisden W, et al. TASK-3 knockout mice exhibit exaggerated nocturnal activity, impairments in cognitive functions, and reduced sensitivity to inhalation anesthetics. J Pharmacol Exp Ther 2007, 323: 924–934.
Gruss M, Bushell TJ, Bright DP, Lieb WR, Mathie A, Franks NP. Two-pore-domain K+ channels are a novel target for the anesthetic gases xenon, nitrous oxide, and cyclopropane. Mol Pharmacol 2004, 65: 443–452.
Putzke C, Hanley PJ, Schlichthorl G, Preisig-Muller R, Rinne S, Anetseder M, et al. Differential effects of volatile and intravenous anesthetics on the activity of human TASK-1. Am J Physiol Cell Physiol 2007, 293: C1319–1326.
Linden AM, Aller MI, Leppa E, Rosenberg PH, Wisden W, Korpi ER. K+ channel TASK-1 knockout mice show enhanced sensitivities to ataxic and hypnotic effects of GABA(A) receptor ligands. J Pharmacol Exp Ther 2008, 327: 277–286.
Bayliss DA, Barrett PQ. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol Sci 2008, 29: 566–575.
Aller MI, Wisden W. Changes in expression of some two-pore domain potassium channel genes (KCNK) in selected brain regions of developing mice. Neuroscience 2008, 151: 1154–1172.
Talley EM, Solorzano G, Lei Q, Kim D, Bayliss DA. CNS distribution of members of the two-pore-domain (KCNK) potassium channel family. J Neurosci 2001, 21: 7491–7505.
Lesage F, Terrenoire C, Romey G, Lazdunski M. Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem 2000, 275: 28398–28405.
Patel AJ, Honore E. Anesthetic-sensitive 2P domain K+ channels. Anesthesiology 2001, 95: 1013–1021.
Steinberg EA, Wafford KA, Brickley SG, Franks NP, Wisden W. The role of K(2)p channels in anaesthesia and sleep. Pflugers Arch 2015, 467: 907–916.
Franks NP, Honore E. The TREK K2P channels and their role in general anaesthesia and neuroprotection. Trends Pharmacol Sci 2004, 25: 601–608.
Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, et al. TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J 2004, 23: 2684–2695.
Gray AT, Zhao BB, Kindler CH, Winegar BD, Mazurek MJ, Xu J, et al. Volatile anesthetics activate the human tandem pore domain baseline K+ channel KCNK5. Anesthesiology 2000, 92: 1722–1730.
Gerstin KM, Gong DH, Abdallah M, Winegar BD, Eger EI, 2nd, Gray AT. Mutation of KCNK5 or Kir3.2 potassium channels in mice does not change minimum alveolar anesthetic concentration. Anesth Analg 2003, 96: 1345–1349.
Girard C, Duprat F, Terrenoire C, Tinel N, Fosset M, Romey G, et al. Genomic and functional characteristics of novel human pancreatic 2P domain K+ channels. Biochem Biophys Res Commun 2001, 282: 249–256.
Patel AJ, Honore E. Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 2001, 24: 339–346.
Yost CS, Oh I, Eger EI, 2nd, Sonner JM. Knockout of the gene encoding the K(2P) channel KCNK7 does not alter volatile anesthetic sensitivity. Behav Brain Res 2008, 193: 192–196.
Rajan S, Wischmeyer E, Karschin C, Preisig-Muller R, Grzeschik KH, Daut J, et al. THIK-1 and THIK-2, a novel subfamily of tandem pore domain K+ channels. J Biol Chem 2001, 276: 7302–7311.
Renigunta V, Zou X, Kling S, Schlichthorl G, Daut J. Breaking the silence: functional expression of the two-pore-domain potassium channel THIK-2. Pflugers Arch 2014, 466: 1735–1745.
Lazarenko RM, Fortuna MG, Shi Y, Mulkey DK, Takakura AC, Moreira TS, et al. Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K+ current. J Neurosci 2010, 30: 9324–9334.
Enyedi P, Czirjak G. Properties, regulation, pharmacology, and functions of the K(2)p channel, TRESK. Pflugers Arch 2015, 467: 945-958.
Liu C, Au JD, Zou HL, Cotten JF, Yost CS. Potent activation of the human tandem pore domain K channel TRESK with clinical concentrations of volatile anesthetics. Anesth Analg 2004, 99: 1715–1722.
Huang DY, Yu BW, Fan QW. Roles of TRESK, a novel two-pore domain K+ channel, in pain pathway and general anesthesia. Neurosci Bull 2008, 24: 166–172.
Chae YJ, Zhang J, Au P, Sabbadini M, Xie GX, Yost CS. Discrete change in volatile anesthetic sensitivity in mice with inactivated tandem pore potassium ion channel TRESK. Anesthesiology 2010, 113: 1326–1337.
Herman MA, Sidhu H, Stouffer DG, Kreifeldt M, Le D, Cates-Gatto C, et al. GIRK3 gates activation of the mesolimbic dopaminergic pathway by ethanol. Proc Natl Acad Sci U S A 2015, 112: 7091–7096.
Weigl LG, Schreibmayer W. G protein-gated inwardly rectifying potassium channels are targets for volatile anesthetics. Mol Pharmacol 2001, 60: 282–289.
Milovic S, Steinecker-Frohnwieser B, Schreibmayer W, Weigl LG. The sensitivity of G protein-activated K+ channels toward halothane is essentially determined by the C terminus. J Biol Chem 2004, 279: 34240–34249.
Bienengraeber M, Warltier DC, Bosnjak ZJ, Stadnicka A. Mechanism of cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel activation by isoflurane in a heterologous expression system. Anesthesiology 2006, 105: 534–540.
Marinovic J, Bosnjak ZJ, Stadnicka A. Preconditioning by isoflurane induces lasting sensitization of the cardiac sarcolemmal adenosine triphosphate-sensitive potassium channel by a protein kinase C-delta-mediated mechanism. Anesthesiology 2005, 103: 540–547.
Martin GM, Yoshioka C, Rex EA, Fay JF, Xie Q, Whorton MR, et al. Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife 2017, 6: e24149.
Wei AD, Gutman GA, Aldrich R, Chandy KG, Grissmer S, Wulff H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol Rev 2005, 57: 463–472.
Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci 2006, 7: 921–931.
Schreiber M, Wei A, Yuan A, Gaut J, Saito M, Salkoff L. Slo3, a novel pH-sensitive K+ channel from mammalian spermatocytes. J Biol Chem 1998, 273: 3509–3516.
Zhang Z, Rosenhouse-Dantsker A, Tang QY, Noskov S, Logothetis DE. The RCK2 domain uses a coordination site present in Kir channels to confer sodium sensitivity to Slo2.2 channels. J Neurosci 2010, 30: 7554–7562.
Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, et al. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem 2004, 279: 47681–47687.
Hong Y, Puil E, Mathers DA. Effect of halothane on large-conductance calcium-dependent potassium channels in cerebrovascular smooth muscle cells of the rat. Anesthesiology 1994, 81: 649–656.
Pancrazio JJ, Park WK, Lynch C, 3rd. Effects of enflurane on the voltage-gated membrane currents of bovine adrenal chromaffin cells. Neurosci Lett 1992, 146: 147–151.
Pancrazio JJ, Park WK, Lynch C, 3rd. Inhalational anesthetic actions on voltage-gated ion currents of bovine adrenal chromaffin cells. Mol Pharmacol 1993, 43: 783–794.
Denson DD, Duchatelle P, Eaton DC. The effect of racemic ketamine on the large conductance Ca2+–activated potassium (BK) channels in GH3 cells. Brain Res 1994, 638: 61–68.
Klockgether-Radke AP, Schulze H, Neumann P, Hellige G. Activation of the K+ channel BK(Ca) is involved in the relaxing effect of propofol on coronary arteries. Eur J Anaesthesiol 2004, 21: 226–230.
Hawasli AH, Saifee O, Liu C, Nonet ML, Crowder CM. Resistance to volatile anesthetics by mutations enhancing excitatory neurotransmitter release in Caenorhabditis elegans. Genetics 2004, 168: 831–843.
Namba T, Ishii TM, Ikeda M, Hisano T, Itoh T, Hirota K, et al. Inhibition of the human intermediate conductance Ca2+-activated K+ channel, hIK1, by volatile anesthetics. Eur J Pharmacol 2000, 395: 95–101.
Dreixler JC, Jenkins A, Cao YJ, Roizen JD, Houamed KM. Patch-clamp analysis of anesthetic interactions with recombinant SK2 subtype neuronal calcium-activated potassium channels. Anesth Analg 2000, 90: 727–732.
Ying SW, Goldstein PA. Propofol-block of SK channels in reticular thalamic neurons enhances GABAergic inhibition in relay neurons. J Neurophysiol 2005, 93: 1935–1948.
Acknowledgements
This work was supported by Important Projects of Natural Science in Colleges and Universities in Jiangsu Province (14KJA320002 to ZZ), Jiangsu specially-appointed professorships to ZZ and QYT, the Natural Science Foundation of China (81471314 and 81671090 to ZZ; 31671212 to QYT), the Natural Science Foundation of Jiangsu Province (BK20151170 to ZZ), the Xuzhou Science and Technology Program (KC16SG251 to ZZ; KC16H0230 to QYT), and a General Project of Natural Science in Colleges and Universities in Jiangsu Province (15KJD320001 to XJ). We also appreciate grant support from the Priority Academic Program Development of Jiangsu Higher Education Institutions and from the Jiangsu Provincial Special Program of Medical Science (BL2014029).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors claim that there are no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Y., Xu, J., Xu, Y. et al. Regulatory Effect of General Anesthetics on Activity of Potassium Channels. Neurosci. Bull. 34, 887–900 (2018). https://doi.org/10.1007/s12264-018-0239-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-018-0239-1