
Abstract
In this study, we investigated the lipolytic effects of eicosapentaenoic acid (EPA) in 3T3-L1 adipocytes. The differentiated 3T3-L1 adipocytes were treated in a serum-free medium with 300 μM of EPA for 3, 6, 12, and 24 h. In comparison with the control, intracellular lipid accumulation was significantly decreased by 24% at 24 h following the addition of EPA (P < 0.05). Under the same experimental conditions, there was an increase of glycerol and free fatty acids (FFAs). The mRNA level of carnitine palmitoyltransferase I-a, a component of the fatty-acid shuttle system involved in the mitochondrial oxidation of long-chain fatty acids, was also significantly elevated by EPA (P < 0.05). However, the expression of peroxisome proliferator-activated receptor-γ and acetyl-CoA carboxylase (ACC), which are involved in adipogenesis, was significantly down-regulated by EPA (P < 0.05). These results suggest that EPA may modulate lipid metabolism by stimulation of lipolysis, which was associated with induction of lipolytic gene expression and suppression of adipogenic gene expression in 3T3-L1 adipocytes.
Similar content being viewed by others

Introduction
Adipose tissue stores energy in the form of lipids and releases fatty acids in response to nutritional signals or energy insufficiencies [1]. Excessive fat accumulation in the white adipose tissue causes obesity and leads to in an increased risk of various disorders, such as type II diabetes, hypertension and coronary heart disease [2, 3]. Consequently, there has been considerable interest in the role of dietary fat in the development of adiposity. Compared to lard or corn oil-fed groups, total body fat or abdominal fat mass were considerably reduced in rodents fed with fish oil or perilla oil containing plenty of n-3 polyunsaturated fatty acids (PUFA) [4, 5]. The polyunsaturated fatty acids, especially those in the class of n-3 fatty acids, are now known to affect all four of the metabolic nuclear receptors that modulate triglyceride (TG) levels. These include liver × receptor (L × R), farnesoid × receptor (F × R), and hepatocyte nuclear factor-4α (HNF-4α), and peroxisome proliferator-activated receptors (PPARs) controlling the expression of genes involved in lipid and glucose metabolism [6, 7]. However, the direct effect of n-3 fatty acids on lipolysis in vitro remains largely unresolved.
In the present study, we investigated the effects of eicosapentaenoic acid (EPA), known as one of the most critical components of n-3 PUFA, on lipid metabolism and the underlying mechanisms in 3T3-L1 adipocytes. We hypothesize that EPA stimulates lipolysis through induction of lipolytic gene expression and suppression of adipogenic gene expression. We measured the lipid accumulation and release of free fatty acids (FFAs) and glycerol into medium followed by EPA treatment. In addition, the mRNA levels of the adipogenic genes including peroxisome proliferator-activated receptor-γ (PPAR-γ) and acetyl-CoA carboxylase (ACC), and carnitine palmitoyltransferase I-a (CPT-Iα), one of lipolytic genes, were measured.
Materials and methods
Materials
EPA (Sigma, St. Louis, MO; purity >99%) was dissolved in ethanol and stored in the dark as stock solution at −20°C. The EPA were freshly prepared from the stock solution and diluted with growth medium. An acyl-CoA oxidase-based colorimetric kit (NEFA-C) was obtained from Wako (Tokyo, Japan), and a BCA protein assay kit was obtained from Pierce (Rockford, USA). The 3T3-L1 cell line was obtained from American Type Culture Collection (Manassas, VA).
Cell culture
3T3-L1 fibroblasts were initially maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM/l glutamine, 100 U/l penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 95% air/5% CO2 at 37°C. To induce adipocytic differentiation, 3T3-L1 cells were allowed to grow to confluence and cultured with differentiation medium containing 0.5 mM/l isobutylmethylxanthine, 1 μM/l dexamethasone, and 5 μg/ml insulin. After 48 h exposure to the differentiation medium, cells were maintained for an additional 5–7 days in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Cells were then treated with 300 μM/l of EPA for 3, 6, 12, or 24 h in serum-free media.
Oil Red O staining
The 3T3-L1 adipocytes were washed with phosphate-buffered saline (pH 7.4) and then fixed with 10% formalin in phosphate-buffered saline. Cells were stained with Oil Red O dye (saturated Oil Red O dye in six parts of isopropanol and four parts of water). Spectrophotometric quantification of the stain was performed by dissolving the stained oil droplets in the cell monolayer with 4% Nonidet P-40 in isopropanol and measuring absorbance at 520 nm [8]. The values were calculated as percentages of the control cells treated without EPA and expressed as the mean ± SD.
Lipolysis assays
The amounts of glycerol and FFA released from cells into the medium were measured to analyze the lipolytic effect of EPA on the accumulated triacylglycerol in adipocytes. Medium was collected from the culture plate and heated at 65°C for 8 min to inactivate any enzymes released from the cells. The amounts of glycerol and FFA were measured by using a commercial glycerol analysis kit (Roche) and an ACO-based colorimetric kit (Wako), respectively. Cellular protein content was analyzed using a BCA protein assay kit (Pierce). Data were expressed as the mean ± SD.
Quantitative real-time reverse transcription-polymerase chain reaction (PCR)
Total RNA was extracted from 3T3-L1 adipocytes using TRIzol® Reagent (Promega, Madison, WI). The cDNAs were synthesized from 5 μg of RNA using M-MLV reverse transcriptase (Promega). After cDNA synthesis, quantitative real-time PCR was performed in 25 μl of Universal SYBR Green PCR Master Mix (Qiagen, Chatsworth, CA) using a fluorometric thermal cycler (Rotor-Gene™ 2000; Corbett Research, Mortlake, NSW, Australia). Reaction mixtures were incubated for an initial denaturation at 95°C for 10 min, followed by 50 PCR cycles. Each cycle consisted of 95°C for 10 s, 55°C for 20 s, and 72°C for 20 s. Primers were designed using an on-line program (primer3_http://www.cgivo.2) [9]. The sequences of the sense and antisense primers used for amplification were as follows: PPARγ, 5′-TTGATTTCTCCAGCATTTCT-3′ and 5′-TGTTGTAGAGCTGGGTCTTT-3′; ACC, 5′-CTGTGAGGTGGATCAGAGAT-3′ and 5′-TTCAGCTCTAACTGGAAAGC-3′; CPT-Iα, 5′-GTGTT GGAGGTGACAGACTT-3′ and 5′-CACTTTCTCTTTCCACAAGG-3′; β-actin, 5′-GTTGCCAATAGTGATGACCT-3′ and 5′-GGACCTGACAGACTACCTCA-3′. The ΔC T method was used to measure relative quantification. Values were expressed as fold change over control and expressed as the mean ± SD.
Statistical analysis
The results are expressed as mean ± SD. The statistical significance of differences between groups was determined by the Student’s t-test (two-tailed) using the SPSS package program version 11.0 (SPSS, Chicago, IL). Also, the significant differences among groups were determined by one-way analysis of variance. The results were considered to be significant if the value of P was <0.05, and Tukey’s multiple range test was performed if differences were identified between groups at α = 0.05.
Results and discussion
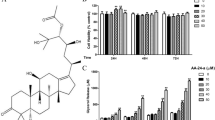
The differentiated 3T3-L1 adipocytes were treated in a serum-free medium with 300 μM/l of EPA for 3, 6, 12, and 24 h. The intracellular oil droplets were stained with Oil Red O dye and quantified. In comparison with the control, lipid accumulation was significantly decreased by 24% at 24 h following the addition of EPA, but had no effect during early stages 3 to 12 h of the experiment (Table 1). The amounts of glycerol released into the medium was notably increased by 61% at 24 h following the addition of EPA, but had no effect during the early stages 3 to 12 h of the experiment (Fig. 1a). Additionally, the amounts of FFA in the medium were increased compared to the control by 38.5 and 44.6% at 12 and 24 h, respectively (Fig. 1b). These results led to the conclusion that EPA stimulated lipolysis in adipocytes as evidenced by the decreased lipid content in cells and the increased concentrations of glycerol and FFA in the medium. Lending further support to our findings, other investigators reported that the plasma TG levels were decreased in animals given n-3 PUFA diet, compared with the control diet [10, 11], in order to investigate the underlying mechanisms of inhibitory effects of EPA on lipid accumulation, the mRNA levels of the lipolytic gene, such as CPT-Iα (and adipogenic genes such as PPAR-γ and ACC were analyzed by real-time PCR). CPT-Iα, which is located on the outer mitochondrial membrane, is the rate-limiting enzyme for fatty acid oxidation. Following the activation of fatty acids to produce acyl-CoA thioesters, CPT-Iα catalyzes the transfer of acyl groups from acyl-CoA to carnitine, producing acylcarnitine. This reaction is the initial step in the transport of long-chain fatty acids from the cytosol into the mitochondrial matrix for fatty acid oxidation [12, 13]. In this study, the mRNA level of CPT-Iα was significantly increased by 2.3-fold compared to the control (P < 0.05) following EPA supplementation (Fig. 2). This finding strongly indicats that EPA may promote CPT-Ia gene expression for mitochondrial fatty acid β-oxidation. PPAR-γ is the adipocyte-specific transcription factor that appears to promote adipocyte differentiation and control the expression of several fat-specific genes [14, 15]. ACC is known as the rate-limiting enzyme in the de novo synthesis of fatty acids [16, 17]. In our study, the mRNA level of PPAR-γ was found to be decreased after EPA supplementation by 53.4% compared to the control (P < 0.05). In addition, the mRNA level of ACC was decreased by 63.7% (Fig. 2). These results imply that EPA attenuates lipid accumulation in adipocytes, possibly via down-regulation of PPAR-γ and induction of ACC gene expression. In conclusion, EPA may modulate lipid metabolism by stimulation of lipolysis, which likely is mediated by induction of lipolytic gene expression and suppression of adipogenic gene expression in adipocytes.

Time course of EPA-induced lipolysis in 3T3-L1 adipocytes. The 3T3-L1 adipocytes were treated in serum-free medium with 300 μM/l of EPA for 3, 6, 12, or 24 h
The medium was collected and assayed for glycerol (a) and FFA content (b). Data are mean ± SD values (n = 5). Values with different superscript letters are significantly different at P < 0.05 by Tukey’s test

Effect of EPA on the mRNA levels of CPT1-α, PPAR-γ, and ACC. The 3T3-L1 adipocytes were incubated for 24 h in serum-free medium with or without 300 μM of EPA. The amounts of mRNA were measured by quantitative real-time PCR. Data are mean ± SD values (n = 9). * P < 0.05 and ** P < 0.01 versus control (no treatment)
References
Spiegelman BM, Flier JS (1996) Adipogenesis and obesity: rounding out the big picture. Cell 87:377–389
Kopelman PG (2000) Obesity as a medical problem. Nature 404:635–643
Visscher TL (2001) The public health impact of obesity. Annu rev public health 22:355–375
Hainault I, Carlotti M, Hajduch E, Guichard C, Lavau M (1993) Fish oil in a high lard diet prevents obesity, hyperlipidemia, and adipocyte insulin resistance in rats. Annu N Y Acad Sci 683:98–101
Parrish CC, Pathy DA, Angel A (1990) Dietary fish oils limit adipose tissue hypertrophy in rats. Metabolism 39:217–219
Davidson MH (2006) Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am J Cardiol 98:27–33
Jump DB (2002) The biochemistry of n-3 polyunsaturated fatty acids. J Biol Chem 277:8755–8758
Moreno-Aliaga MJ, Matsumura F (1999) Endrin inhibits adipocyte differentiation by selectively altering expression pattern of CCAAT/enhancer binding protein-alpha in 3T3-L1 cells. Mol Pharmacol 56:91–101
Rozen S and Skaletsky HJ (1998) Primer3. Code available at http://www-/genome.wi.mit.edu/genome_software/otherprimer3.html
Bravo E, Napolitano M, Lopez-Soldado I, Valeri M, Botham KM, Stefanutti C (2006) Hypercholesterolaemia alters the responses of the plasma lipid profile and inflammatory markers to supplementation of the diet with n-3 polyunsaturated fatty acids from fish oil. Eur J Clin Invest 36:788–795
Peyron-Caso E, Quignard-Boulang A, Laromiguiere M, Feing-Kwon-Chan S, Veronese A, Ardoun B, Slama G, Rixkalla SW (2003) Dietary fish oil increases lipid mobilization but does not decrease lipid storage-related enzyme activities in adipose tissue of insulin-resistant, sucrose-fed rats. J Nutr 133:2239–2243
McGarry JD, Foster DW (1980) Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem 49:395–420
McGarry JD, Brown NF (1997) The mitochondrial carnitine palmitoyltransferase system: from concept to molecular analysis. Eur J Biochem 244:1–14
Rousseau V, Becker DJ, Ongemba LN, Rahier J, Henquin JC, Brichard SM (1997) Developmental and nutritional changes of ob and PPAR gamma 2 gene expression in rat white adipose tissue. Biochem J 321:451–456
Schoonjans K, Staels B, Auwerx J (1996) The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta 1302:93–109
Kim KH (1997) Regulation of mammalian acetyl-coenzyme A carboxylase. Annu Rev Nutr 17:77–99
Wakil SJ, Stoops JK, Joshi VC (1983) Fatty acid synthesis and its regulation. Annu Rev Biochem 52:537–579
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MOST) (no. M10510130005-07N1013-00510).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, MS., Kwun, IS. & Kim, Y. Eicosapentaenoic acid increases lipolysis through up-regulation of the lipolytic gene expression and down-regulation of the adipogenic gene expression in 3T3-L1 adipocytes. Genes Nutr 2, 327–330 (2008). https://doi.org/10.1007/s12263-007-0068-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-007-0068-8