
Abstract
The chronic nature of rheumatoid arthritis (RA) suggests immune dysfunction, including persistent systemic activation. Therefore, we evaluated the activatory and inhibitory potential as well as proliferative activity of peripheral blood (PB) CD4+ T cells from RA patients in different stages of the disease and after different therapeutic interventions. We found that CD4+ T cells from RA patients were activated in vivo concerning decreased CD28 expression and increase of CD40L, CD69, and CTLA-4 expression; however, the extent of stimulation was suboptimal when compared to healthy controls. Consequently, impaired proliferative activities of these cells were found in all patients irrespective of the active disease duration. Treatment with methotrexate (MTX) and/or inhibitors of TNF-alpha (iTNF) did not significantly influence systemic activation in RA patients, which corresponded with the maintenance of inflammation markers; however, partial restoration of CD28 and CTLA-4 expression as well as clinical improvement were observed. In patients with early disease (the MTX group), we noted higher capacity of CD4+ T cells for restoration of T cell function, whereas cells from the iTNF group with progressive disease remained with a proliferative defect after the treatment. In conclusion, our study demonstrates that the dysregulated expression of molecules interfering with CD4+ T cell signaling may result in functional impairment of the effector T cells and correlates with disease progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by swollen and tender joints, cartilage damage, the production of autoantibodies, and systemic inflammation as a hallmark of disease progression. The pathogenesis of RA includes T cells’ contribution to initiation and maintenance of the disease. The persistent inflammatory process in RA is a result of disturbed B or T cell stimulation including autoreactive T cells, which initiate a stimulating cascade of events [1].
Immune homeostasis requires optimal T cell activation. A major role in the maintenance of balanced T cell responses is played by CD28, CD40L, and CTLA-4 molecules. CD28 is an antigen constitutively expressed on T cells transducing a costimulatory signal after ligand B7 binding, thus promoting full T cell activation [2], whereas both CD40L and CTLA-4 are inducible upon activation, thus being transiently expressed on the cell surface of T lymphocytes [3, 4]. The cross-linking of CTLA-4 during T cell activation results in suppression of cytokine production and cell proliferation [4]; hence CTLA-4 is suggested to be an antagonist of CD28 [5]. They both share common B7 ligands with obviously higher affinity displayed by CTLA-4. Reciprocal regulation of CD28 and CTLA-4 at the mRNA level has been previously demonstrated; in particular, transient down-regulation of CD28 mRNA seen early after stimulation is a stronger inducer of CTLA-4 gene transcription [6]. CD40L, one of the earliest and the most specific marker of T cell activation, is crucially involved in the positive cell signaling process after binding to CD40 expressed on B cells [7]. Thus, CD40-CD40L interactions are involved in both humoral and cellular immune responses, including autoimmune activity and inflammation [3].
Several studies showing genetic associations of CD28 and CTLA-4, their increased soluble form as well as clinical improvement of RA after CTLA-4Ig administration clearly emphasize the importance of both signaling molecules in the pathogenesis of RA [8–10]. Also, CD40L expression has been found to be significantly higher in the circulation of RA patients [11, 12]. Furthermore, a long-lasting remission of experimental autoimmune diseases has been achieved by blocking CD40L, suggesting its role in the pathological mechanisms of RA as well [13].
Previous studies, including observations from our laboratory, have shown that progression/severity of RA might be accompanied by systemic CD4+ T cell subtypes imbalance [14, 15, unpublished data]. The present study was undertaken to assess whether disease evolution may affect the activatory and inhibitory potential of CD4+ T cells. Therefore, we evaluated the expression of CD28, CD40L, and CTLA-4 molecules in the population of peripheral blood (PB) helper T cells from RA patients in the different stages of the disease. Furthermore, we performed a proliferation assay to find out if the state of activation of CD4+ T cells may influence their function. We also examined the impact of different therapeutic interventions on the studied parameters.
Materials and Methods
Study Populations
The study was approved by the Ethics Committee of Wroclaw Medical University. Peripheral blood was taken from 36 patients (26 women and 10 men) diagnosed with RA based on the 1987 revised classification criteria of the American College of Rheumatology (ACR). Patients were divided into 2 groups depending on the severity and duration of the disease; 19 patients, including 12 women and 7 men (mean age: 54.7 ± 16.4 years), with a mean disease duration of 15.2 months were assigned to methotrexate therapy (MTX group). Seventeen patients, including 14 women and 3 men (mean age: 50.1 ± 6.9 years), with a mean RA duration of 123.7 months were assigned to therapy with inhibitors of TNF-alpha (iTNF group). Blood samples were collected just before and after 6 months of the therapy.
All the patients had active disease and were consistent with the criteria of the activity: for methotrexate (MTX) treatment, erythrocyte sedimentation rate (ESR) > 30 mm/h and/or C-reactive protein (CRP) > 1.5 mg/dL, disease activity score DAS28 > 3.2; for treatment with inhibitors of the human tumor necrosis factor alpha (iTNF), ESR > 30 mm/h and/or CRP > 1.5 mg/dL, DAS28 > 5.1. Treatment failure with at least two traditional disease-modifying antirheumatic drugs (DMARDs), including MTX, was required for biologics to be used. Therefore, based on the statistically significant differences in the active disease duration (p = 0.0000001) and a mean DAS28 score either before (5.6 ± 0.9 vs. 6.2 ± 0.8, p = 0.02) or after the treatment (3.3 ± 1.2 vs. 4.8 ± 1.1, p = 0.0001) between the two groups, iTNF patients were assigned to the progressive form of the disease.
All patients from the MTX group were receiving a stable dose of MTX (10–15 mg once a week orally) alone; in the biologic treatment group, patients received recommended doses of the following inhibitors of TNF-alpha: infliximab, etanercept, and adalimumab. Due to the persistently active disease, iTNF patients were also treated with DMARDs, including MTX (n = 15), sulphasalazine (n = 9), non-steroidal anti-inflammatory drugs (n = 17), and glucocorticoids at a daily dose of less than 10 mg of prednisone (n = 16), if the treatment regimens were not modified 4 weeks before the study; hence, 13 and 3 out of 17 patients were still receiving MTX and sulphasalazine, respectively, when iTNF therapy was started. With regard to the MTX group, patients were treated with unmodified doses of steroids (n = 13) and/or anti-inflammatory drugs (n = 16) for more than 4 weeks before MTX administration.
Also the clinical and biochemical parameters allowed determination of the improvement according to the criteria suggested by the European League Against Rheumatism (EULAR): no response if reduction of DAS28 < 0.6; moderate efficacy of the therapy if reduction is 0.6 < DAS28 < 1.2; and good efficacy of the therapy if reduction of DAS28 > 1.2. Although at least partial improvement was achieved in 14 (82.35 %) and 19 (100 %) patients administered with iTNF and MTX, respectively, only 3 out of 19 MTX patients and none of 17 iTNF patients reached clinical remission (DAS28 < 2.6) at 6 month of the therapy. Eighteen age- and sex-matched healthy controls, including 14 women and 4 men (mean age: 51.8 ± 8.4 years), free of inflammatory syndrome (CRP < 0.5 mg/dL), infectious diseases, steroid or immunosuppressive therapy, and cancers were also included in the study. Written informed consent was obtained from each patient and healthy donor after a full explanation of the procedure.
Cell Preparation, Culture Conditions, and Flow Cytometry
Peripheral blood mononuclear cells (PBMCs) were obtained by buoyant density-gradient centrifugation on Lymphoflot (Biotest, Germany). The expression of CD28, CD40L (CD154), CD69, and CTLA-4 molecules was studied on the freshly isolated and cultured for 48 h CD4+ T cells by a triple immunostaining method. After isolation the cells were stained with anti-CD3/FITC, anti-CD4/PerCP, and anti-CD28/RPE, anti-CD40L(CD154)/RPE, anti-CD69/RPE or anti-CTLA-4(CD152)/RPE monoclonal antibodies (MoAbs) (Becton Dickinson). For intracellular detection of CTLA-4, the cells were first incubated with pure anti-CTLA-4 MoAb to block surface CTLA-4 molecules with subsequent fixation, permeabilization, and staining with anti-CTLA-4/RPE MoAb as described above.
For estimation of the optimal range of CD4+ T cells activation, the PBMCs from healthy individuals were suspended in 1 × 106 PBMCs/ml in RPMI-1640 medium (Gibco, Paisley, UK) supplemented with 10 % fetal calf serum (Flow Laboratories, UK), 2 mmol/l L-glutamine and 50 μg/ml gentamycin (Gibco), and cultured with 10 ng/ml of anti-CD3 monoclonal antibody (MoAb) (clone OKT3) (eBioscience). The cultures were incubated at 37 °C in a humidified atmosphere containing 5 % CO2 for 48 h. Then, immunostaining with markers of activation and inhibition was performed as described above. For each immunostaining procedure, negative controls were always done by omitting the MoAb and by incubating the cells with mouse Ig of the same isotype as the MoAbs conjugated with PerCP, FITC or RPE. The cells were analyzed by flow cytometric analysis using a FACScan cytometer equipped with Cell Quest software (BD Bioscience Pharmingen). Of the acquired 10,000 events, at least 1,500–2,000 were analyzed in the final histograms.
Proliferation Assays
Cell proliferation was analyzed using the colorimetric methyl thiazol tetrazoliumbromide (MTT) method. After negative separation by magnetic cell sorting (Miltenyi Biotec), CD4+ T cells were cultured at 1 × 105 cells/200 μl in 96-well plates in medium alone or with anti-CD3 monoclonal antibody (10 ng/ml) for the induction of cell proliferation. The plates were incubated at 37 °C in a humidified atmosphere containing 5 % CO2 for 48 h. After 48 h, 25 μl of MTT (5 mg/ml stock solution) was added to each well and incubated for 3 h. Next, 90 μl of the extraction buffer (20 % sodium dodecyl sulfate with 50 % dimethylformamide, pH 4.7) was added. After 4 h of incubation, the optical density (OD) was measured at 550 nm using the microplate reader (Dynatech 5000). The experiments were repeated 3 times.
Statistical Analysis
One-way ANOVA test was used to determine significant differences between groups. The Wilcoxon signed rank test was used to compare paired patients before and after the treatment. Results were considered statistically significant when p ≤ 0.05. Statistica’99 Edition was used in the statistical calculations.
Results
Before Therapy
Expression of Co-Stimulatory CD28 Molecule and Activation Markers in PB CD4+ T Cells
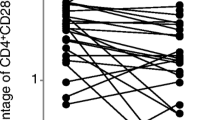
The frequencies of CD4+ T cells co-expressing CD28, CD40L, CD69, and both membrane/surface and cytoplasmic CTLA-4 molecule (mCTLA-4 and cCTLA-4, respectively) were estimated to define the activation status of the PB CD4+ T cells. The mean frequencies of CD28+, CD40L+, CD69+ Th cells in all groups examined are shown in Table 1 and Fig. 1.

Expression of CD4+ T cells co-expressing CD28, CD40L, and CD69 molecules in RA patients before and after 6 months of therapy (mean proportion ± SD)
Although the differences were of borderline significance (p ≤ 0.06), the proportions of CD4+CD28+ T cells within PBMCs were obviously lower in RA patients in comparison with corresponding cells from controls. OKT3 stimulation decreased the CD28+ population in all examined individuals, but only in the iTNF patients was it compromised to significantly lower values compared to controls (p = 0.001).
The population of in vivo activated T cells with the respective phenotype CD4+CD40L+ and CD4+CD69+ was significantly increased in early (p ≤ 0.00001) as well as in progressive disease (p < 0.002) compared to controls; remarkably, among the patients, the activated CD4+ T cell population was higher in MTX patients (p ≤ 0.04). The expression of antigen CD69 on PB cells did not reach the physiological values observed in optimally ex vivo stimulated T cells from controls, remaining at significantly lower levels in all patients (p < 0.002). Stimulation with OKT3 revealed no impact on the frequency of CD40L+ T cells only in iTNF patients, whereas a decrease in MTX patients (p = 0.006) and an increase in controls were found (p = 0.001). OKT3-stimulated CD40L expression on Th cells was comparable between all individuals studied. As for CD69 expression, we observed a further significant increment after ex vivo stimulating culture in all studied groups (p < 0.004), although its level in iTNF patients did not reach values observed in controls (p = 0.02).
The expression of inhibitory CTLA-4 molecule in the CD4+ T cell population is shown in Table 1 and Fig. 2. Only in MTX patients was the mCTLA-4 molecule expressed on a significantly higher proportion of CD4+ T cells compared to controls (p = 0.02). In contrast, the expression of the cCTLA-4 molecule was diminished in the iTNF group as compared to either corresponding cells from MTX patients (p = 0.008) or to the optimally ex vivo stimulated cells from healthy controls (p = 0.04). OKT3 stimulation resulted in no changes in the mCTLA-4+ T cell population in MTX patients, and its marked increase in others (p ≤ 0.04). Thus, there were no differences in ex vivo stimulated mCTLA-4 expression between studied groups. As regards cCTLA-4 expression, stimulating culture led to significant increase in controls only (p = 0.003), whereas it decreased, however non-significantly, in the MTX group. In iTNF patients, OKT3 stimulation did not significantly change the frequency of cCTLA-4+ Th cells; thus its defective proportion in RA was sustained in comparison with controls (p ≤ 0.02).

Expression of CD4+ T cells co-expressing membrane/surface CTLA-4 (mCTLA-4) and cytoplasmic CTLA-4 (cCTLA-4) in RA patients before and after 6 months of therapy (mean proportion ± SD)
Proliferative Capacity of PB CD4+ T Cells
In order to verify whether the affected capacity of reaching the optimal level of T cell activation in circulation of RA patients may result in subsequent functional impairment of these cells, we performed a proliferation assay. Compared to controls, we found that both groups of patients exhibited defective proliferative activity of CD4+ T cells under either unstimulated or OKT3-stimulated conditions (p ≤ 0.01 and p ≤ 0.006, respectively). Although the differences between patients were non-significant, the functional impairment of T cells was evident in the iTNF group. The results are provided in Table 2.
After 6 Months of Therapy
Expression of Co-Stimulating CD28 Molecule and Activation Markers in PB CD4+ T Cells
The influence of the therapy on CD28, CD40L, and CD69 expression is shown in Table 1 and Fig. 1. After MTX and iTNF therapy, we noted a marked increase in the frequency of PB CD28+ Th cells (p = 0.006 and p = 0.04, respectively); thus a similarly expanded CD28+ cell population was seen in all examined groups. OKT3 stimulation markedly diminished the CD28+ T cell population to comparable values in all groups (p ≤ 0.02).
Similarly to pre-treatment observations, the proportions of activated PB CD4+ T cells with positive expression for CD40L and CD69 were significantly enhanced in both groups of RA patients compared to PB healthy cells (p ≤ 0.0005). Remarkably, the occurrence of these activated cells in PB from the iTNF group remained lower than in MTX patients (p ≤ 0.07). Likewise, the frequency of circulating CD4+ T cells co-expressing CD69 in both groups of treated RA patients did not reach the physiological values found in healthy T cells cultured under optimally stimulating conditions (p ≤ 0.00002). Compared to pre-stimulation values, only in controls was an increase in CD40L+ cell frequency after culture with OKT3 noted (p = 0.001). In contrast, a diminished proportion of cells from RA patients expressed CD40L (p ≤ 0.05); nevertheless, the differences between all groups were non-significant. Regarding CD69 antigen expression, the post-treatment response of T cells to OKT3 did not differ as compared with those observed in untreated patients; thus the cells expressed a significantly higher frequency of CD69 antigen after stimulation than corresponding cells in PB (p ≤ 0.001), and reached similar levels in all studied groups.
The impact of the treatment on CTLA-4 expression within the CD4+ T cell population is shown in Table 1 and Fig. 2. After the therapy, a decrease with subsequent normalization of the frequency of mCTLA-4+ T cells in the MTX group was demonstrated (p = 0.031). Therefore, there were no significant differences in PB mCTLA-4 and cCTLA-4 expression in CD4+ T cells between studied groups of treated patients and controls. OKT3 stimulation led to a marked increment in the frequency of mCTLA-4+ Th cells to similar values in all individuals (p ≤ 0.01). As for cCTLA-4, after 48 h of culture with OKT3, the cells from RA patients responded to stimulation in the same way, their cells expressing the cCTLA-4 molecule in an unchanged proportion of CD4+ T cells; in contrast, the frequency of healthy cCTLA-4+ Th cells significantly increased under ex vivo stimulating conditions (p = 0.003) to obviously higher values compared to those seen in RA (p ≤ 0.002).
Proliferative Activity of PB CD4+ T Cells
The effect of the therapy on proliferation of CD4+ T lymphocytes is shown in Table 2. Treatment with MTX resulted in either spontaneous or stimulated proliferation improvement of CD4+ T cells (p = 0.05 and p = 0.03, respectively), thus leading to normalization of both. In contrast, iTNF administration influenced neither spontaneous nor stimulated proliferative capacity of CD4+ T cells; therefore they remained with a functional defect compared to healthy T cells (p = 0.05). Nevertheless, there were no significant differences in either the unstimulated or the stimulated proliferation rate of CD4+ T cells from treated patients, although it seemed that the cells obtained from the iTNF group underwent weaker cell division.
Discussion
In the current study we report dysregulated expression of the activator and suppressor molecules within the population of PB CD4+ T cells in RA patients before and after 6 months of different therapeutic interventions. Furthermore, the observed alterations seem to be pronounced during disease progression. In particular, we observed that in the peripheral circulation in the course of RA, there is an expansion of CD4+ T cells lacking the co-stimulatory CD28 molecule. It is noteworthy that the magnitude of the CD4+CD28- T cell population was similar in both groups of studied patients with different duration of the active disease. In many autoimmune diseases, such as Crohn’s disease [16], juvenile idiopathic arthritis [17], ankylosing spondylitis [18], and multiple sclerosis [19], an unusually high proportion of CD28- T cells in the periphery was also found. Also, normal aging is related to increased loss of both the CD28 molecule and immunocompetence. Our finding parallels several studies demonstrating a decreased CD4+CD28+ T cell population in RA, which is disproportionally diminished with patients’ age [20, 21]. In fact, the RA T cell pool is characterized by premature aging correlated with the loss of CD28 [22].
CD28 expression on T cells is normally modulated following T cell activation with transient down-regulation early after stimulation [23]. Thus the loss of CD28 expression on PB CD4+ T cells in RA patients could reflect chronic immune stimulation by arthrogenic antigens or continuous activation in a cytokine-rich microenvironment with the prime position of TNF-alpha [23, 24]. This cytokine plays a role as the dominant repressor of CD28 gene transcription and has been found up-regulated in RA sera [25]. The impact of the in vivo activation on CD28 expression decline is consistent with studies showing that repeated stimulation of T lymphocytes in long-term culture may lead to progressive loss of the CD28 molecule [26]. Consistently with the above suggestion, our data also show significant down-regulation of the CD28 T cell pool when T cells are cultured with additional stimulation with OKT3, more pronounced in patients with progressive RA. Also, the extensive TNF-alpha half-life in the culture and post-culture estimation cannot be neglected due to its known interaction with membrane proteins [27], resulting in the maintenance of TNF-alpha biologic action for a longer period. Thus, membrane-bound TNF-alpha might reveal its prolonged inhibitory effect on CD28 expression in the culture. The OKT3-mediated CD28 defect found in the most advanced RA is in agreement with the previous demonstration that the frequency of CD28- T cells is correlated with severity of RA [28]. Nonetheless, this alteration is reversible irrespective of the disease duration and type of therapeutic intervention. The reversal of CD28 deficit seems to be associated more with the clinical outcome of RA therapy [16], which is in line with our observation of post-treatment clinical improvement in almost all patients even if RA remission usually did not occur.
The suggestion on chronic in vivo stimulation of CD4+ T cells is consistent with other findings of the significantly increased expression of both the activation markers CD40L and CD69 in all RA patients as well as inducible antigen CTLA-4 in those in the early phase of RA. Furthermore, CD40L and CD69 antigens were found to be strongly interrelated in all patients at every time point tested, as was also previously demonstrated [12]. Based on the observation that CD40L expression is closely correlated with CD39, a lymphoid activation marker with prolonged expression after activation [29], we can speculate that CD40L+ cells represent a subset of CD4+ T cells that have undergone prolonged and increased activation. The variability of CD40L and CD69 antigen expression found between both groups of patients favors the interpretation that at distinct phases of the disease, patients’ lymphocytes undergo activation with different intensity. Therefore, CD4+ T cells from patients with early RA exhibited the highest extent of systemic activation compared to those obtained from progressive disease. This could be caused by the fact that patients qualified for iTNF administration were under intensified immunosuppressive treatment, including MTX and other DMARD medications. The second explanation is related to possible CD4+ T cell exhaustion in advanced RA due to prolonged in vivo activation. The observation that OKT3-stimulated CD69 expression in this group of patients did not reach the level of activation observed in corresponding cells from controls seemed to confirm this suggestion. Given also that DNA demethylation of the X-linked gene CD40L in CD4+ T cells from women with RA corresponded with increased CD40L expression, together with our observation of female preponderance in the group of RA patients examined, a role of genetic factors in CD40L overexpression in RA cannot be excluded [30].
In addition, we did not observe post-treatment normalization in the expression of activation markers in PB. The percentages of CD40L+ and CD69+ cells within CD4+ T cells remained at a markedly higher level in PB of RA patients even after 6 months of therapy with MTX and/or iTNF. Yet, an increase of the CD40L+ T helper cell pool in the circulation of MTX patients was seen, which may result from CD28 post-treatment increase, as the correlated dynamics of their expressions have been previously demonstrated in RA [31]. We suggest that reversal of systemic activation in RA could be related to clinical outcome after therapeutic interventions. In fact, almost all patients enrolled in the current study did not achieve clinical remission, which corresponded with the maintenance of increased clinical and laboratory markers of inflammation.
The variability between both groups of patients also concerned the fraction of CD4+ T cells co-expressing inhibitory CTLA-4 molecule inducible upon activation. Its increased expression on the surface of T cells from the MTX group strengthens the suggestion of the highest extent of in vivo activation in untreated patients in early disease. On the other hand, CTLA-4 plays a key role in T cell inhibition, thus indicating a potency for sufficient termination of the ongoing T cell activation in early RA. OKT3 stimulation did not change surface CTLA-4 expression in the same group despite the intact cytoplasmic compartment of CTLA-4 molecules, suggesting that the activation in vivo had occurred to a maximal extent. In contrast, patients with progressive RA exhibited impaired suppressor potential in PB CD4+ T cells, considering affected cytoplasmic CTLA-4 expression, probably due to T cell exhaustion related to disease progression. In addition, this also could be a result of immunosuppressive agent administration in the past. Nevertheless, ex vivo re-stimulation revealed an intrinsic defect in CTLA-4-mediated suppression in the CD4+ T cell pool in all RA patients either before or after the therapeutic intervention, suggesting that the inhibitory potential declined progressively during prolonged activation in RA, which is in line with the previous observations in other autoimmune diseases [32].
We suggest that the dysregulated expression of the examined molecules with capacity for interfering with cell signaling might have functional implications. To verify this hypothesis, we extended our recent functional study assessing the capacity of RA CD4+ T cells for secretion of cytokines (unpublished data) and performed the proliferation assay. At entry to the study, all RA patients exhibited profoundly defective proliferative activity of T cells under either unstimulated or stimulated conditions. This observation is in line with previous studies [33–35]. The impaired overall proliferative capacity of T cells in RA has been attributed to premature senescence of PB T cells in established disease. Rheumatoid arthritis-related aging of CD4+ T cells is characterized e.g. by shorter telomerase length resulting from repeated in vivo stimulation and the loss of CD28 expression especially in the most severe RA [34, 36]. Accordingly, in the present study, the most evident proliferative defect was observed in progressive RA. Our recent finding that within the CD4+ T cell compartment from the same RA patients, only those with advanced and long-lasting disease exhibited a profoundly and irreversibly defected Th1 cell population, other subsets having similarly expanded, clearly shows quantitative disturbances within the existing pool of lymphocytes during RA evolution (unpublished data). Furthermore, we demonstrated that patients with early and short-term active disease treated with MTX had a higher potency for restoration of T cell proliferative capacity. In contrast, those with advanced RA possessed a profound proliferative defect even after therapy with iTNF. Consistently, the previous reports showed that iTNF addition into the culture delayed loss of CD28 and led to only transient proliferation improvement, resulting nonetheless in an overall decrease in the ex vivo rate of CD4+ T cell proliferation [37]. Remarkably, after multiple rounds of cell division, the cells did reach the non-proliferative end stage of replicative senescence, thus being arrested in the G0/G1 phase of the cell cycle [35, 37, 38]. In addition, RA-related aging T cells showed both a significant reduction in the production of IL-2 and an increase in IL-6 secretion, thus supporting the shift in the cytokine balance towards pro-inflammatory conditions. Similarly disturbed imbalance in serum IL-2/IL-6 concentration, confirming the functional impairment of PB CD4+ T lymphocytes more pronounced in progressive RA, was observed by our group recently (unpublished data). The post-treatment maintenance of affected proliferative capacity in RA related to disease evolution has also been demonstrated [22], which is consistent with our present findings.
In summary, our data demonstrate that patients with varying severity of RA exhibit several abnormalities of the activatory and inhibitory potential of PB CD4+ T cells corresponding with the impaired proliferative capacity. The observed alterations of the immune system develop progressively and might facilitate progression of the disease by affecting the effector functions of CD4+ T lymphocytes.
Abbreviations
- ACR:
-
American College of Rheumatology
- CRP:
-
C-reactive protein
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen-4
- cCTLA-4:
-
Cytoplasmic CTLA-4
- DAS28:
-
Disease activity score
- ESR:
-
Erythrocyte sedimentation rate
- EULAR:
-
European league against rheumatism
- iTNF:
-
Inhibitors of the human tumor necrosis factor alpha
- mCTLA-4:
-
Membrane/surface CTLA-4
- MTX:
-
Methotrexate
- MoAb:
-
Monoclonal antibody
- mRNA:
-
Messenger ribonucleic acid
- MTT:
-
Methyl thiazol tetrazoliumbromide
- OD:
-
Optical density
- OKT3:
-
Ortho kung t3
- PB:
-
Peripheral blood
- PBMC:
-
Peripheral blood mononuclear cells
- RA:
-
Rheumatoid arthritis
- Th:
-
Helper T cell
References
Weyand CM (2000) New insights into the pathogenesis of rheumatoid arthritis. Rheumatology 39:13–18
Lenschow DJ, Walunas TL, Bluestone JA (1996) CD28/B7 system of T cell costimulation. Annu Rev Immunol 14:233–258
Elgueta R, Benson MJ, Vries VC, Wasiuk A, Guo YX, Noelle RJ (2009) Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev 229:152–171
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Greene JM et al (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1:405–413
Krummel MF, Allison JP (1995) CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med 182:459–465
Lindsten T, Lee KP, Harris ES, Petryniak B, Craihead N, Reynolds PJ et al (1993) Characterization of CTLA-4 structure and expression on human T cells. J Immunol 151:3489–3499
Noelle RJ, Roy M, Shepherd DM, Stamenkovic I, Ledbetter JA, Aruffo A (1992) A 39-kDa protein signal for an activated helper T cells binds CD40 and transduces the signal for cognate activation of B cell. Proc Natl Acad Sci 89:6550–6554
Kim YO, Kim HJ, Kim SK, Chung JH, Hong SJ (2010) Association of the CD28/CTLA4/ICOS polymorphisms with susceptibility to rheumatoid arthritis. Clin Chem Lab Med 48:345–353
Kremer JM, Westhovens R, Leon M, Di Giorgio E, Alten R, Steinfeld S et al (2003) Treatment of rheumatoid arthritis by selective inhibition of T-cell activation with fusion protein CTLA-4Ig. N Engl J Med 349:1907–1915
Cao J, Zou L, Luo P, Chen P, Zhang L (2012) Increased production of circulating soluble co-stimulatory molecules CTLA-4, CD28 and CD80 in patients with rheumatoid arthritis. Int Immunopharmacol 14:585–592
Goules A, Tzioufas AG, Manousakis MN, Kirou KA, Crow MK, Routsias JG (2006) Elevated levels of soluble CD40 ligand (sCD40L) in serum of patients with systemic autoimmune diseases. J Autoimmun 26:165–171
Berner B, Wolf G, Hummel KM, Muller GA, Reuss-Borst MA (2000) Increased expression of CD40 ligand (CD154) on CD4+ T cells as a marker of disease activity in rheumatoid arthritis. Ann Rheum Dis 59:190–195
Early GS, Zhao W, Burns CM (1996) Anti-CD40 ligand antibody treatment prevents the development of lupus-like nephritis in a subset of a New Zealand black x New Zealand white mice. Response correlates with the absence of an anti-antibody response. J Immunol 157:3159–3164
Kawashima M, Miossec P (2005) mRNA quantification of T-bet, GATA-3, IFN-gamma, and IL-4 shows a defective Th1 immune response in the peripheral blood from rheumatoid arthritis patients: link with disease activity. J Clin Immunol 25:209–214
van Roon JAG, Verhoef CM, van Roy JLAM et al (1997) Decrease in peripheral type 1 over type 2 T cell cytokine production in patients with rheumatoid arthritis correlates with an increase in severity of disease. Ann Rheum Dis 56:656–660
Garcia de Tena J, Manzano L, Leal JC, San Antonio E, Sualdea V, Alvarez-Mon M (2004) Active Crohn’s disease patients show a distinctive expansion of circulating memory CD4+CD45RO+CD28null T cells. J Clin Immunol 24:185–196
Martens PB, Goronzy JJ, Schaid D, Weyand CM (1997) Expansion or unusual CD4+ T cells in severe rheumatoid arthritis. Arthritis Rheum 40:1106–1114
Duftner C, Goldberger C, Falkenbach A, Wurzner R, Falkensammer B, Pfeiffer FP et al (2003) Prevalence, clinical relevance and characterization of circulating cytotoxic CD4+CD28- T cells in ankylosing spondylitis. Arthritis Res Ther 5:R292–R300
Markovic-Plese S, Cortese I, Wandinger KP, McFarland HF, Martin R (2001) CD4+CD28- costimulation-independent T cells in multiple sclerosis. J Clin Invest 108:1185–1194
Bryl E, Vallejo AN, Matteson EL, Witkowski JM, Weyand CM, Goronzy JJ (2005) Modulation of CD28 expression with anti-tumor necrosis factor alpha therapy in rheumatoid arthritis. Arthritis Rheum 52:2996–3003
Schmidt D, Goronzy JJ, Weyand CM (1996) CD4+CD7-CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J Clin Invest 97:2027–2037
Pawlowska J, Smolenska Z, Zdrojewski Z, Witkowski JM, Bryl E (2012) Changes in proliferation kinetics of T cells: a new predictive cellular biomarkers for early rheumatoid arthritis? J Clin Immunol. doi:10.1007/s10875-012-9692-1
Bryl E, Vellejo AN, Weyand CM, Goronzy JJ (2001) Down-regulation of CD28 expression by TNF-alpha. J Immunol 167:3231–3238
Vallejo AN, Weyand CM, Goronzy JJ (2004) T-cell senescence: a culprit of immune abnormalities in chronic inflammation and persistent infection. Trends Mol Med 10:119–124
Feldmann M, Maini RN (2001) Anti-TNF-alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol 19:163–196
Valenzuela HF, Effros RB (2002) Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol 105:117–125
Yamashita K, Fukushima K (2004) The carbohydrate recognition by cytokines modulates their physiological activities. Glycoconj J 21:31–34
Pawlik A, Ostanek L, Brzosko I, Brzosko M, Masiuk M, Machalinski B et al (2003) The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res Ther 5:R210–R213
Kansas GS, Wood GS, Tedder TF (1991) Expression, distribution, and chemistry of human CD39. Role in activation-associated homotypic adhesion of lymphocytes. J Immunol 146:2235–2244
Liao J, Liang G, Xie S, Zhao H, Zuo X, Li F et al (2012) CD40L demethylation in CD4+ T cells from women with rheumatoid arthritis. Clin Immunol 145:13–18
Warrington KJ, Vallejo AN, Weyand CM, Goronzy JJ (2003) CD28 loss in senescent CD4+ T cells; reversal by interleukin-12 stimulation. Blood 101:3543–3549
Kosmaczewska A, Bilinska M, Ciszak L, Noga L, Pawlak E, Szteblich A et al (2007) Different patterns of activation markers expression and CD4+ T-cell responses to ex vivo stimulation in patients with clinically quiescent multiple sclerosis. J Neuroimmunol 186:137–146
Cope AP, Londei M, Chu NR, Cohen SB, Elliott MJ, Brennan FM, Maini RN, Feldmann M (1994) Chronic exposure to tumor necrosis factor (TNF) in vitro impairs the activation of T cells through the T cell receptor/CD3 complex; reversal in vivo by anti-TNF antibodies in patients with rheumatoid arthritis. J Clin Invest 94:749–760
Koetz K, Bryl E, Spickschen K, O’Fallon WM, Goronzy JJ, Weyand CM (2000) T cell homeostasis in patients with rheumatoid arthritis. Proc Natl Acad Sci U S A 97:9203–9208
Wagner U, Pierer M, Wahle M, Moritz F, Kaltenhauser S, Hantzschel H (2004) Ex vivo homeostatic proliferation of CD4+ T cells in rheumatoid arthritis is dysregulated and driven by membrane-anchored TNF-alpha. J Immunol 173:2825–2833
Hodes RJ, Hathcock KS, Weng NP (2002) Telomeres in B and T cells. Nat Rev Immunol 2:699–706
Parish ST, Wu JE, Effros RB (2009) Modulation of T lymphocyte replicative senescence via TNF-alpha inhibition: role of caspase-3. J Immunol 182:4237–4243
Fujii H, Shao L, Colmegna I, Goronzy JJ, Weyand CM (2009) Telomerase insufficiency in rheumatoid arthritis. Proc Natl Acad Sci U S A 106:4360–4365
Acknowledgments
This work was supported by a grant from the National Science Centre (Poland): grant no. N N402 244935.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Kosmaczewska, A., Ciszak, L., Swierkot, J. et al. Alterations in Both the Activatory and Inhibitory Potential of Peripheral Blood CD4+ T Cells in Rheumatoid Arthritis Patients Correlate with Disease Progression. Pathol. Oncol. Res. 20, 235–243 (2014). https://doi.org/10.1007/s12253-013-9687-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12253-013-9687-0