
Abstract
Estuaries are important conduits between terrestrial and marine aquatic systems and function as hot spots in the aquatic methane cycle. Eutrophication and climate change may accelerate methane emissions from estuaries, causing positive feedbacks with global warming. Boreal regions will warm rapidly in the coming decades, increasing the need to understand methane cycling in these systems. In this 3-year study, we investigated seasonal and spatial variability of methane dynamics in a eutrophied boreal estuary, both in the water column and underlying sediments. The estuary and the connected archipelago were consistently a source of methane to the atmosphere, although the origin of emitted methane varied with distance offshore. In the estuary, the river was the primary source of atmospheric methane. In contrast, in the adjacent archipelago, sedimentary methanogenesis fueled by eutrophication over previous decades was the main source. Methane emissions to the atmosphere from the study area were highly variable and dependent on local hydrodynamics and environmental conditions. Despite evidence of highly active methanogenesis in the studied sediments, the vast majority of the upwards diffusive flux of methane was removed before it could escape to the atmosphere, indicating that oxidative filters are presently still functioning regardless of previous eutrophication and ongoing climate change.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Methane (CH4) is an important greenhouse gas that contributes significantly to global warming (IPCC 2014) and influences atmospheric chemistry through a complex chain of oxidation reactions (Cicerone and Oremland 1988). The majority of CH4 on Earth is produced by microbial methanogenesis, which is the ultimate pathway of anaerobic fermentation of organic matter occurring in a multitude of environments (Knittel and Boetius 2009). Of the natural sources of atmospheric CH4, wetlands and freshwater systems are among the most significant, while agriculture, fossil fuels, waste treatment, and other anthropogenic sources make up between 46 and 67% of global emissions (Kirschke et al. 2013). As a consequence of human activities, the concentration of CH4 in the atmosphere has more than doubled since pre-industrial times (Blasing 2016).
Coastal regions globally are experiencing intensified anthropogenic influence. In 2010, 1.9 billion people lived within a 100 km of a coastline; this figure is expected to rise to 2.4 billion by 2050 (Kummu et al. 2016). Agriculture, wastewater, industrial activities, and transport all contribute to nutrient and organic carbon loading to coastal aquatic systems (Syvitski et al. 2005; Lotze 2006; Paerl et al. 2006). Due to their role as conduits of land-to-sea transfer, estuaries are hot spots for biogeochemical cycling and are among the most productive aquatic systems in the world (Bianchi 2007). Eutrophication has increased the organic matter loading to estuarine sediments, leading to expanded areas of oxygen stress (Diaz and Rosenberg 2008; Middelburg and Levin 2009).
An important consequence of the eutrophication-driven expansion of low-oxygen conditions in estuaries is the increased production of CH4 in the underlying sediments, as remineralization of organic matter increasing proceeds by anaerobic pathways (Naqvi et al. 2010; Gelesh et al. 2016). Estuaries have long been considered a potential source of atmospheric CH4 emissions (Reeburgh 1969), due to outgassing of allochthonous inflowing CH4 from the terrestrial environment, as well as autochthonous production within the estuary itself (Upstill-Goddard et al. 2000). However, the relative importance of these two main sources is not always easy to quantify (Reeburgh 2007). Large spatial and temporal variability in CH4 concentrations is observed within and between estuaries, hampering efforts to construct methane mass balances for estuarine systems or to quantify the extent of human impact. Yet, typical estuarine surface water CH4 concentrations are well above atmospheric equilibrium (de Angelis and Scranton 1993; Abril and Borges 2004), implying that estuarine systems are indeed a source for atmospheric CH4.
The anthropogenic impact on methane cycling in estuaries is potentially exacerbated by climate change. The combined effects of eutrophication and warmer temperatures have been shown to increase atmospheric fluxes of CH4 from lakes (Davidson et al. 2018). Boreal regions are excepted to be especially vulnerable to the warming climate and the changes introduced might not be linear in nature (Soja et al. 2007). Furthermore, other effects of climate change such as increased or more variable runoff, decreased solubility of oxygen, and increased water mass stratification (Gelesh et al. 2016) can all potentially contribute to the methanogenic potential of estuaries.
Despite their productivity and potential for greenhouse gas emissions, the exact contribution of estuaries to the global CH4 budget is still relatively poorly constrained and they have often been excluded from global carbon budgets (Kirschke et al. 2013). In estuaries, as in other methanogenic environments, the emissions reaching the atmosphere are dependent on the balance of methanogenic and methanotrophic processes. The microbial processes and communities involved in the production and consumption of CH4 are both directly and indirectly affected by environmental conditions. Hence, it is important to understand how different environmental conditions may lead to cascading effects in the microbial communities mediating CH4 processes, and impact directly on process rates, for example through influencing metabolic activity or substrate availability (Dean et al. 2018).
Of the biogeochemical processes occurring in estuarine sediments, anaerobic oxidation of methane (AOM) is the most important limiting the escape of CH4 to the water column and eventually to the atmosphere. AOM is capable of removing up to 90% of all CH4 produced by methanogenesis (Knittel and Boetius 2009). It is typically most active in sediments (Iversen and Blackburn 1981), though it has been also shown to be active in the water column in strongly stratified systems (Jakobs et al. 2014). In the water column, aerobic oxidation of methane (MOX) is also a major process removing significant amounts of methane (Fenchel et al. 1995). Although these processes are very effective at preventing methane from escaping to the atmosphere, the combined effects of climate change and eutrophication have the potential to drastically increase CH4 emissions from estuaries (Davidson et al. 2018), and it remains unknown whether it is possible for these filters to be overcome, whether through large increases in methanogenesis, increased storm activity, or solubility effects of increased temperatures.
Here we report the results of a 3-year study investigating seasonal and spatial variability of methane dynamics in a eutrophied boreal estuary with a legacy of eutrophication. The legacy effect is the result of decades of heightened autochthonous and allochthonous carbon loading in response to eutrophication in the catchment of the estuary, as well as the Baltic Sea in general. This has decreased the depth of the sulfate-methane transition zone (SMTZ), leading to increased CH4 fluxes from the sediment. Data presented here contain both water column and sediment porewater concentrations and calculated flux estimates from sediments to water column and from there to the atmosphere.
Materials and Methods
Study Area
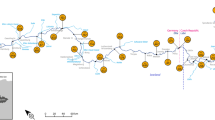
This study was conducted in an estuary and adjacent archipelago area located in Southern Finland, east of the Hanko Peninsula (Fig. 1.). The estuary goes by several names in the literature (Jilbert et al. 2018) but will be referred to as Pojo Bay here. It has been a location for numerous scientific studies for more than a century (Stipa 1999). It is a microtidal, fjord-like estuary that receives fresh water primarily from the river Mustionjoki (also known as Karjaanjoki) and opens in to the Gulf of Finland. The catchment area of Mustionjoki is 2046 km2, consisting of 46% forest, 19% agriculture, 11% lakes, and 10% urban area (Asmala et al. 2012). There is a shallow sill (< 5 m water depth) near the city of Ekenäs separating the inner estuary from the outer archipelago and restricts currents, which creates a strong salinity gradient in the basin (< 1 in the inner bay to 7–8 in the outer archipelago). The inner bay is typically strongly stratified and the stagnant deep water is renewed only during late autumn or winter, when wind-driven inflows push more saline water over the sill into the inner bay (Stipa 1999). The estuary freezes intermittently during winter, with the inner bay typically freezing over completely and featuring several decimeters of ice. The inner bay has long suffered from periods of anoxia and sediment cores from the estuary feature distinct accumulation of solid-phase sulfur since the early 1970s, indicative of increased input of organic matter in response to eutrophication in the area (Jilbert et al. 2018). However, with improvements in communal wastewater treatment, limitations imposed on agricultural fertilizer usage and decreases in upstream industrial activity, modern nutrient and organic matter loading via the river is limited compared to similar estuaries along the Finnish coast (Meeuwig et al. 2000; Asmala et al. 2013). Annual mean loadings of carbon and nutrients determined during 2010–2011 are as follows: TOC = 4037 t year−1, TN = 378 t year−1, and TP = 12 t year−1 (Asmala et al., 2013). Two large lakes in the catchment also retain nutrients effectively (Koskiaho et al. 2015).

Map of the study area and the stations along the transect. Stations J and L were used in the seasonal study. The inset in the top left corner shows the location of the weather stations used for wind data, and the dashed arrow the predominant wind direction between the stations. Average wind direction and speed in June 2015 is shown next to the station symbols. The inset in the lower right corner shows the position of the estuary in the Baltic Sea. The estuarine depth contour is shown in the bottom of the figure
Sampling Strategy
Spatial methane dynamics in the estuary were studied along a 33-km transect consisting of 11 sites, which span from the river mouth of Mustionjoki in the inner estuary, out to open sea. The sites were selected for being representative of the various biogeochemical conditions along the estuary, both in the water column and in the sediments. The transect sites are labeled from A to K alphabetically and ordered by their distance offshore. The transect can be broadly split into three distinct parts: inner bay (A–E), sill (F–G), and archipelago (H–K) (Fig. 1). Sites D and J are both long-time monitoring stations also known in the literature as Sällvik deep and Storfjärden, respectively.
Seasonality of methane dynamics was studied at two sites: J and L, of which J is situated within the main channel and is deeper, relatively open and more dynamic, whereas L is a more sheltered site surrounded by islands and features stronger seasonal stratification and is periodically hypoxic or anoxic below the pycnocline.
Sites along the transect were sampled three times for water column parameters (years 2015, 2016, and 2017) and twice for sediments (2014 and 2016). Three upstream sites in the Mustionjoki River (max 2 km from river mouth) were also sampled once in September 2015. The seasonal sites (J and L) were sampled four times during 2016 and once during 2017 (April, June, August, October, and March).
Water Sampling and CH4 Analysis
Water samples were collected using a 5-L LIMNOS™ water sampler. Each site along the transect was sampled at 5-m depth intervals, with the first sample immediately below the surface (covering approximately 0–50 cm depth due to the length of the sampler). The two seasonal sites J and L were sampled at 2-m intervals. Complimentary temperature and salinity profiles were measured with a handheld CTD device at every station (Fig. S2). Samples were collected from small boats in the summer and from a hovercraft during winter sampling (March 2017).
Dissolved CH4 samples were collected and prepared for analysis using a headspace equilibration method. Briefly, 30 mL of water was retrieved directly from the water sampler through a rubber tube, into a 60-mL plastic syringe and stored in a cooler. Within 6 h, a headspace of 30 mL of 5.0 purity N2 was added and the samples were left to warm up for 30 min at room temperature. The samples were shaken vigorously for 3 min prior to transferring the headspace/gas in to a dry syringe through a three-way stopcock. From the dry syringe, the samples were injected into 12 mL glass tubes with butyl rubber septa (pre-evacuated LabCo Exetainer™ model 839W). The concentration of methane in the headspace was measured with a gas chromatograph equipped with a FID sensor (Agilent Technologies 7890B) against a three-point calibration of known gas concentrations (0.46, 5, and 47 ppm). Standards were measured before and after each sample series, and a 5-ppm standard was measured after every 10th sample, to account for between- and within-series drift, respectively. The original dissolved gas concentration of methane was then calculated using Henry’s law and Bunsen solubility (Wiesenburg and Guinasso 1979). Samples were always analyzed within 2 weeks of sampling. For a more detailed description of the method, we refer the reader to Myllykangas et al. (2017). The method has limited sensitivity at CH4 concentrations close to atmospheric equilibrium (1–3 nM). However, the lowest concentrations measured in this study were an order of magnitude higher than these values; hence, we consider the method reliable in this setting.
Sediment Sampling and Porewater Geochemical Analysis
Sediment cores were retrieved with a GEMAX™ twin gravity corer with 3 cm diameter holes pre-drilled at 1.5 cm intervals on the side of the core tube. The holes were closed with wide water-resistant electrical tape prior to sampling. After recovery, the holes were cut open and a cutoff syringe was quickly inserted into the core and filled with 10 cm3 of sediment. The sediment in the syringe was then transferred to a 65-mL glass bottle containing supersaturated NaCl solution (Egger et al. 2015). The bottles were instantly capped with a butyl rubber septum and a screw cap and stored upside down while awaiting analysis. The transfers were always performed immediately after the core was brought on deck and as rapidly as possible to minimize degassing. Within 24 h, a headspace of 10 mL of 5.0 purity N2 was introduced into the samples (with an equivalent amount of NaCl-slurry flowing out) and the samples were shaken to ensure all methane had evolved from the dissolved phase to the headspace.
Two 1-mL subsamples were taken from the headspace of each sample with a gas-tight 1-mL glass syringe and transferred to an evacuated 12-mL glass tube with a butyl rubber septum (LabCo Exetainer™ model 839W) and pressurized with 20 mL of 5.0 N2, creating a dilution of 1:21. The mole fraction of methane in headspace of the samples was analyzed with a FID-equipped gas chromatograph (Agilent Technologies 7890B) against a standard series of known gas concentrations (5, 1000, and 10,000 ppm). As with water column samples, standard series were analyzed before and after each sample series and a 5-ppm standard was inserted after every 10 samples. The original porewater methane concentration was calculated assuming quantitative evolution of methane into the headspace. The volume of porewater in the 10-mL wet sediment sample was calculated directly from porosity values measured in parallel cores or estimated using assumed porosity profiles based on data from neighboring sites.
Vertical sulfate (SO42−) and hydrogen sulfide (H2S) profiles were generated from a parallel GEMAX™ core after Rhizon™ sampling. Two vertical series of holes at 2 cm intervals were drilled into the core tube, one for each sulfur species. The holes were taped prior to sampling, and after recovery, Rhizons™ were inserted into the holes and porewater was collected into 10 mL polyethylene syringes. The syringes for the H2S series were pre-loaded with 1 mL 10% zinc acetate prior to sampling in order to trap sulfide in the form of zinc sulfide. The samples in the SO42− series were acidified post-sampling with 1 M HNO3.
The acidified SO42− samples were analyzed using inductively coupled plasma-optical emission spectrometry (ICP-OES). The post-sampling acidification removes H2S from the samples (Jilbert and Slomp 2013); hence, the measured S pool was considered to represent only SO42−. The H2S series was analyzed spectrophotometrically at 670 nm wavelength according to a modified version of the methods of Cline (1969) and Reese et al. (2011). For a more detailed description of the method, see Jilbert et al. (2018).
Sediment Flux
Flux of methane from the sediment to water column was calculated using Fick’s first law:
Where J is the calculated flux of methane (mol m−2 s−1), ϕ is the porosity of the surface sediment (determined from water content assuming sediment density of 2.65 g cm−3), ∂C is the concentration difference between the surface sample from sediment (mol m−3) and the deepest water sample (concentration assumed uniform between deepest sample and sediment surface), ∂x is the distance between the two different measurements (m), and Ds is the bulk sediment diffusion coefficient for methane, which was estimated according to (Berner 1980):
where D0 is the molecular diffusion coefficient for methane in seawater at 4 °C (0.87 × 10−10m2s−1) (Iversen and Jørgensen 1993) and θ is tortuosity, estimated as per (Boudreau 1997) as:
In addition to surface flux calculations described above, methane dynamics were studied also deeper in the sediment at seasonally studied sites J and L. A simple 1-D diagenetic model of CH4 production and consumption was generated by using the software PROFILE (Berg et al. 1998). The software utilizes a simplified version of mass conservation equation of Boudreau (1997):
where R is the net production rate of CH4. The software uses F tests and least squares fitting routines to determine optimum numbers of zones of production and consumption based on the concentration gradients of the porewater profiles. The model domain is defined by the whole interval sampled for porewater methane concentrations. In situ porosity values were used at each depth interval.
Atmospheric Flux
Flux of methane to the atmosphere was calculated with a two-layer model (Liss and Slater 1974):
where F is the diffusive flux (mol m−2 s−1), Caq (mol m−3) is the surface water concentration of CH4 and Ceq the atmospheric equilibrium concentration calculated based on Henry’s law, and k is the gas transfer velocity (m s−1). Ceq was calculated using the atmospheric CH4 concentrations of 1.91, 1.92, and 1.96 ppm for the years 2015, 2016, and 2017, respectively (data from the Utö monitoring station of the Finnish Meteorological Institute, approximately 100 km west of the estuary). The gas transfer velocity can be quantified in a number of ways, but it is commonly parameterized as a function of wind speed (e.g., Cole and Caraco 1998; Liss and Merlivat 1986; Wanninkhof 1992). Here we opted to use the exponential wind relationship formulated especially for estuaries by Raymond and Cole (2001), 1.91e0.35u, where u is the mean wind speed at 10 m height above sea level. Schmidt number is the ratio between the kinematic viscosity of water and the molecular diffusion coefficient (Jähne et al. 1987) and k values are commonly normalized to 600, which is the Schmidt number of CO2 in freshwater at 20 °C.
Hence, the final atmospheric flux was calculated as:
where ScCH4 is the Schmidt number for methane, calculated individually for all samples using in situ salinity and temperature values according to Wanninkhof (2014). Wind speed for each site was estimated by interpolating monthly wind speed averages between two weather stations of the Finnish Meteorological Institute situated roughly SW–NW along the transect (W1: Kiikala, Salo; W2: Tulliniemi, Hanko; Fig. 1). SW (the main fetch axis of the estuary) was the predominant wind direction and wind speeds were consistently higher at station W2 (Fig. S1, Online Resource). Due to wind speed having a strong effect on atmospheric exchange, atmospheric flux estimates in this study are presented as ranges of ± 1 standard deviation from the value estimated using the monthly mean wind speed.
Results
Transect
Water Column
Salinity in the inner bay was < 5 on all sampling occasions, with salinities offshore ranging between 6 and 8. Both summer transects displayed clear thermal stratification, although the thermocline was markedly deeper in the inner bay than in the outer archipelago. At the two most offshore sites, the depth of the thermocline increased again (Fig. S2, Online Resource).
The highest dissolved CH4 concentrations in the water column were consistently found near the river mouth at the surface of station A, with the highest value of 665 nM found in June of 2016 (Fig. 2). These values were similar to those measured in the inflowing river water in September 2015 (450 ± 7 nM, not shown), indicating a clear signal of river-derived CH4 in the surface waters of the inner estuary. Surface concentrations decreased offshore consistently throughout all the sampling years, with a midwater minimum at 15 m depth in the estuary. CH4 concentrations were elevated in the near-bottom samples at most sites on most sampling occasions, suggesting efflux from the sediments. However, these values were consistently lower than those close to the river mouth. For example, in June 2015, the deepest estuary site D had a near-bottom value of 156 nM, while in June 2016, the deepest open sea site K had a near-bottom value of 153 nM. In the archipelago areas, CH4 concentrations were comparatively high throughout the water column, while concentrations at the offshore site K were consistently among the lowest measured. During winter, ice cover had a strong influence on the CH4 distribution in the estuary; concentrations at the river mouth were lower than during summer, the CH4-rich surface plume expanded further into the inner estuary with concentrations of 130–245 nM in the surface, and also the midwater minimum was expanded vertically from summer. Overall, the surface water concentrations were the highest in winter throughout the whole estuary.

Transect water column dissolved CH4 concentrations (nmol L−1) along the transect sites during three sampling campaigns. Note the irregular depths and that station distances are not to scale. The colors represent the relative change in the concentration throughout the whole data set, with the highest concentration presented in bright red and the lowest concentration in deep green. The shaded sites in March represent ice cover. For references to color, we refer the reader to the online version of the paper
During both summers, the highest atmospheric CH4 fluxes were found at site A near the river mouth, from which the fluxes steadily decreased offshore (Fig. 3). The highest mean flux was calculated in June of 2016 (− 1.56 mmol m−2 day−1, min − 0.7, max − 3.46), while the lowest fluxes were consistently found from the furthest offshore site K during all years. During winter, sites under ice were considered to have an atmospheric flux of 0, but potential fluxes are still shown in Fig. 3 because they are indicative of fluxes at the ice margins or gaps in the ice. At completely ice-free sites in March 2017 (H, J, and K), the mean atmospheric fluxes were higher than at the same sites during summer, and due to stronger winds, the maximum flux at site J (−1.12 mmol m−2 day−1) was among the highest in the whole study.

Calculated atmospheric flux of methane along the transect during June 2015, June 2016, and March 2017. Dots indicate values estimated from monthly mean wind speed using Eq. 6. Gray-shaded areas indicate range of fluxes estimated from ± 1 standard deviation from monthly mean wind speed. Due to the nonlinearity of Eq. 6, the range is asymmetric about the mean. Also presented in the middle panel is the calculated sediment flux along the transect during June 2016. The hatched area in March 2017 represents ice cover, where true flux is assumed to be 0. Hence, the values presented are potential ice-free fluxes only
Sediment
Sediment porewater CH4 concentrations measured in June 2016 are typically in the millimolar range, indicating significant methanogenesis in shallow sediments throughout the transect (Fig. 4). The highest CH4 concentration, 6.19 mM, was found at site K from 12 cm depth, although the offshore trends in absolute CH4 concentrations and the depth of the SMTZ are complex. Sediments near the sill (sites E–G) were typically devoid of methane, most likely due to the fact that these locations are characterized by transport rather than accumulation bottoms and, hence, lower concentrations of degrading organic matter. Sediments in bathymetric depressions (e.g., sites D and K), conversely, show elevated values. However, the shape of SO42− profiles suggests that SO42− reduction occurs at all sites.

Sediment porewater concentration profiles of methane from June 2016 (black circles) and sulfate from September 2014 (white circles) along the whole transect. The red circles represent bottom water sulfate concentration, and the horizontal gray line the depth of SMTZ at the sites (defines as equivalent concentrations of CH4 and SO42−
The depth of the SMTZ (here defined as the depth of equivalent CH4 and SO42− concentration in the porewater profiles) is determined both by the upwards flux of methane and the bottom water SO42− concentration. The latter show a clear increasing trend with distance offshore (Fig. 4). The depth of the SMTZ, in turn, is the primary control on the flux of methane across the sediment–water interface. Fluxes are typically highest in the bathymetric depressions (Fig. 3), consistent with high methane concentrations in the sediments at these locations. However, despite higher absolute porewater CH4 concentrations at site D, site C shows a higher flux across the sediment–water interface, due to its lower bottom water SO42− concentration and hence shallower SMTZ.
The highest fluxes anywhere on the transect were calculated at the offshore site K (− 9.66 mmol m−2 day−1), a bathymetric depression where porewater CH4 concentrations in the upper centimeter of the sediments are > 1 mM (Figs. 3 and 4). The sediment and atmospheric fluxes showed no correlation along the transect sites (Pearson’s product-moment correlation p > 0.5).
Seasonal
Water Column
Water column CH4 concentration patterns, as well as atmospheric and sediment fluxes, varied significantly during the seasonal cycle at both study sites J and L. Bottom water CH4 concentrations at site L were generally higher than at site J. The highest bottom water value of 611 nM was found in June from the deepest sampling point at site L (Fig. 5), consistent with the stronger stratification at this site and consequent accumulation of sediment-derived CH4 in the bottom water. However, surface water concentrations at both sites were similar, and both sites showed their highest respective surface water concentrations in March. In terms of total CH4 inventories per square meter, the seasonal evolution of the two sites varied. Site J featured the largest inventory in April with 4.49 mmol L−1 m−2 from where the inventory decreased throughout the year before increasing again in next March. At site L two maxima were observed, in June and October.

Water column CH4 concentrations at seasonally sampled sites J and L. The red dashed line represents in situ atmospheric equilibrium concentration. Note the different scale in the x axis of the inset. The number in the top right corner of each panel is the total CH4 inventory in mmol L−1 m−2
Atmospheric fluxes were generally higher at site J (Table 1), although the highest atmospheric flux in the dataset was calculated at L in October (− 0.39 mmol m−2 day−1). No flux is reported for site L in March because the site was under ice. Site J was ice-free and in March had the second highest seasonal flux value in the study of − 0.34 mmol m−2 day−1.
Sediments
Surface sediment porewater CH4 concentrations were generally higher at site L, with the highest concentration of 0.96 mM found in June. Accordingly, the SMTZ at this site was consistently shallower in the sediment column at site L (Fig. 6). The SMTZ, as defined by the depth of equivalent concentrations of CH4 and SO42−, is also indicated by the maximum in porewater H2S, which was also generally observed close to this depth. The accumulation of CH4 above this suggests intensive methanogenesis despite active sulfate-mediated AOM, evidenced by the presence of H2S.

Porewater concentrations of methane (black circles), sulfate (white circles), and hydrogen sulfide (triangles) at the seasonally sampled sites J and L. The concentrations of methane are from 2016, while concentrations of SO42− and H2S are from 2015. The red line represents modeled production and consumption of methane (secondary x axis) and the dashed gray line represents approximate depth of the SMTZ, as given by the equivalent concentration of CH4 and SO42−
The overall shape of the CH4 profiles at the two sites also differs considerably. Site J shows a concave profile in the uppermost 15 cm, while site L shows a convex profile in this interval. Also, concentrations at site L begin decreasing after 20 cm, whereas they generally keep increasing with depth at site J, with the highest concentration 5.33 mM found from 40 cm depth in June. The profiles from site J in April and October show some evidence for decreasing concentrations in the deepest samples.
The PROFILE outputs, too, show a clear zone of net methane consumption in the surface sediments at site J, while the corresponding interval at site L shows net production. Conversely, site L shows net production below ~ 15 cm, with evidence for net consumption in the deepest interval during April and October, while site L always shows a clear zone of net consumption below ~ 25 cm.
The sediment CH4 flux was considerably higher at site L throughout all sampling months (Table 1). At site J, the highest sediment fluxes (− 0.61 to − 0.66 mmol m−2 day−1) are found in April and October with lower fluxes (− 0.25 mmol m−2 day−1) during the summer, while site L displays an opposite trend, with the highest sediment fluxes (− 5.69 to − 5.73 mmol m−2 day−1) found during summer and lower in spring and autumn (− 4.11 to − 4.32 mmol m−2 day−1).
Discussion
Pojo Bay and Its Archipelago as Net Sources of Methane to the Atmosphere
The Pojo Bay estuary surface waters were consistently supersaturated with CH4 throughout the whole study across all sampling years and sites, making it a net source of CH4. In terms of site-specific values on our study transect, highest fluxes were measured in the estuary. However, due to the spatial extent of the archipelago regions along the coast of the Gulf of Finland, the contribution of these areas is expected to dominate the net methane flux to the atmosphere in this coastal setting.
Many of the patterns in CH4 fluxes to the atmosphere seen along the transect are well established in previous literature. The seaward decrease of surface water CH4 concentrations is a commonly observed phenomenon in estuaries (Bartlett et al. 1987; de Angelis and Scranton 1993; Middelburg et al. 1996; Upstill-Goddard et al. 2000), and overall CH4 concentrations in estuaries are lower than in fresh waters (Wik et al. 2016b), which was also true at Pojo Bay. The average surface water saturation in the whole Pojo Bay estuary was 4148% (range 561–21,234%), which is broadly comparable to other European estuaries (Bange 2006; Upstill-Goddard and Barnes 2016). Surface waters of the open Baltic are typically only slightly supersaturated and rarely exceed 500% (Bange et al. 1994; Gülzow et al. 2013). This shows that while coastal areas and estuaries are smaller in area, they are still likely an important part in total CH4 emissions from the Baltic Sea.
Major Sources of Methane to the Water Column in Pojo Bay and Its Archipelago
Mustionjoki River
Rivers effectively accumulate methane from their drainage and are very active in methanogenesis (Stanley et al. 2016) and are therefore strong sources of atmospheric methane themselves (Upstill-Goddard et al. 2000). In this study, the inflowing river was a perpetual source of CH4 in to the inner bay of the estuary. Although the input of CH4 from the river to the estuary during the sampling period in June 2016 was limited due to low discharge at this time (~ 7 m3 s−1), the average input is likely to be higher. Concentrations up to 2.5 times higher in the river mouth have been measured at this location (e.g., 1660 nM, unpublished data). Also, based on long-term monitoring data from The Finnish Environment Institute, periods of discharge up to six times higher (~ 50 m3 s−1) were observed in 2016 alone.
Methanogenesis in the Sediment Column
The strong gradients in CH4 concentrations in the sediment porewaters indicate a diffusive flux of CH4 from sediments to the water column throughout the transect (Fig. 4). Only in the area of organic-poor sediments close to the sill (sites F, G) are low porewater CH4 concentrations observed at all depths in the sediments. Coastal sediments in this region are characterized by high concentrations of organic carbon (3–6%) and sedimentation rates of 0.5–0.9 cm/year (Jilbert et al. 2018), leading to short oxygen exposure times for sedimenting organic matter and high rates of anaerobic remineralization processes, including methanogenesis (Middelburg and Levin 2009; Sobek et al. 2009). The flux of CH4 from sediments to the water column leads to elevated bottom water CH4 concentrations, especially in deeper, more stratified locations (e.g., sites C, D, L, Figs. 2 and 5).
Potential Role of Ebullition
The flux from sediments to water column may be enhanced by ebullition. If porewater CH4 concentrations exceed local hydrostatic pressure, the formation of gas bubbles may occur (Wever et al. 1998). Although we did not observe supersaturation of CH4 in the porewaters within the sampled depth interval in our sediment cores, bubble formation may initiate well below saturation (Chanton et al. 1989), implying that ebullition is possible in this setting. While we did not measure ebullition directly, we made several visual observations of bubbles in the water column during sampling at various sites along the transect. Also, at site L, water column gas replicates occasionally had extremely high CH4 content (> 20 μM), suggestive of gas bubbles becoming trapped within the sampling syringe.
The simultaneous presence of CH4 and H2S and SO42− in the shallow sediments at site L (Fig. 6) indicates a strong overlap of the diagenetic zones in the sediments, as observed previously in the northern Baltic Sea (Sawicka and Brüchert 2017; Jilbert et al. 2018). This shows that methanogenesis is occurring in the upper sediments simultaneously with sulfate reduction, potentially due to the use of noncompetitive substrates by the microbial communities (Maltby et al. 2018). These high CH4 concentrations close to the sediment–water interface increase the possibility that ebullition could occur, due to subannual temperature changes or sediment destabilization. Furthermore, ebullition could also partially explain the stochastic nature of the water column CH4 concentrations observed in the archipelago in this study. Not only does ebullition commonly display high spatiotemporal variability (e.g., Scandella et al. 2016), it also has been shown to enhance diffusive sedimentary fluxes by making the sediment more porous (Flury et al. 2015).
Regulation of Sediment Flux by Oxidation Processes
AOM in the SMTZ
The accumulation of H2S in conjunction with a downward decline of SO42− and upward decline of CH4 at around 10 cm depth at sites J and L is suggestive of SO42−-AOM. The depth of the SMTZ was considerably deeper at site J compared to site L and varied in depth between sites along the transect (Fig. 4, Table 1).
At all sites, the SMTZ is expected to act as a strong filter to CH4 fluxes from sediments to the water column (Knittel and Boetius 2009). However, the significant differences in the depth of the SMTZ between sites indicate that the efficiency of this filter function is variable with space in the coastal environment. Sites such as D, K, and L are characterized by high sedimentations rates and organic matter contents, leading to relatively compressed redox zonation and a shallow SMTZ (Figs. 4 and 6). We did not observe a consistent effect of bottom water sulfate concentration on the depth of the SMTZ as given by the depth of equivalent concentrations of CH4 and SO42− (Fig. 4), implying that the flux of organic matter to the sediments is the main factor controlling the SMTZ depth. Our detailed comparison of sites J and L confirms that locations with a shallower SMTZ (site L) also show higher fluxes of CH4 to the water column (Table 1), implying a less efficient filter function of SO42−-AOM.
MOX at the Sediment Surface
The uppermost 10–15 cm of the sediments at site J showed consistent net consumption of porewater CH4 (Fig. 6.). We interpret this as evidence of enhanced MOX in the zone above the SMTZ due to the presence of benthic fauna. Benthic animals have an important role in many sedimentary solute fluxes (Middelburg and Levin 2009). Our results show strong evidence of bioirrigation, i.e., the introduction of oxic bottom water via animal burrows down to 10 cm or more in the sediment, which greatly reduces sediment flux of methane. Indeed, site J was recently shown to have an active community of benthic fauna that affects solute fluxes seasonally (Kauppi et al. 2018), whereas in comparison at site L, there is no benthic fauna, likely due to recurrent seasonal hypoxia or anoxia (e.g., Gammal et al. 2017 report 0.0 mg/L O2 at this site for August 2010). We suggest that this is reflected in the porewater profiles, with CH4 found much closer to the sediment surface at the hypoxic site L due to the absence of bioirrigation. Similarly, Abril and Iversen (2002) observed that sediment cores that contained large amounts of burrowing animals exhibited porewater CH4 minima close to the sediment surface, not observed in parallel cores without animals.
While we do not have detailed CH4 porewater profiles from all transect stations, we expect non-hypoxic bottom areas in both the estuary and archipelago to exhibit some degree of bioirrigation; hence, this may be an important process regulating the sediment CH4 flux in this system.
In contrast to our findings, Bonaglia et al. (2017) observed that macrofauna enhanced CH4 fluxes from the sediment by a factor of 8 compared to fauna-free sediment. However, they based their flux estimates on changes in concentration over time in an oxic incubation chamber, which makes direct comparison with our results difficult. For example, it is possible that the addition of benthic organisms to previously uncolonized sediments stimulated transient release of CH4, including through ebullition, whereas in our study locations, the porewater signals of bioirrigation-induced MOX appear to be largely stable over time (Fig. 6).
Quantitative Budget of Methane Fluxes in Pojo Bay and Its Archipelago
A highly simplified budget of CH4 flows in the estuary and archipelago areas was estimated assuming a set of constraints as outlined in Table S1 (Online Resource). The goal of this exercise is to establish the relative orders of magnitude and ranges of CH4 flows in different parts of the system. Due to the required extrapolation, it is not possible to refine these estimates further with the available data. Nevertheless, the exercise yields some key findings. For example, the results suggest that the surface waters of the estuary at the time of sampling contained a reservoir of 12,853 mol CH4 (Fig. 7). The estimated efflux to the atmosphere from the estuary was 5589 mol/day, indicating a turnover time of the surface-water inventory on a timescale of days, which is considerably less than the residence time of the bay (1.5 years, Meeuwig et al. 2000). The estimated input of CH4 from the river at the time of sampling was 374 mol/day, while the calculated diffusive flux from the isolated deep-water layer was only 0.5 mol/day. This indicates that the combined inputs to the surface waters at the time of sampling were significantly less than the amount required to balance the efflux to the atmosphere (Fig. 7). Hence, we conclude that the estuarine system was far from steady state with respect to CH4 at the time of sampling. Furthermore, the system likely displays continuous high-amplitude variation in reservoir size, as well as riverine input and atmospheric fluxes, on very short timescales as a consequence of variable weather conditions.

Quantitative budget of the main sinks and sources of CH4 in the estuary and archipelago compartments of the study system. The arrows in the figure indicate the direction of fluxes. Derivation of all values is presented in Table S1 (Online Resource) and the areal definition of the archipelago is presented in Fig. S3 (Online Resource). The gray-shaded area behind the sediment flux value conceptually represents the oxidative filter, i.e., real sediment flux is likely much less than presented here as most of the upwards-diffusing CH4 will be oxidized before it enters the water column
The estimated range for atmospheric fluxes shown in Fig. 7 is based on the measured surface water CH4 concentrations and the climatological range of wind stress in the month of June. Hence, the values are strongly dependent on the conditions during our sampling campaigns. The true range of values is also expected to be influenced by variability in the size of the surface water CH4 reservoir over time. This in turn is likely to be controlled by variable inputs from the river. The budget shows that the river is the principal source of CH4 to surface waters in the estuary, being orders of magnitude higher than the diffusive flux from the isolated deep-water layer. Theoretically, periods of high river discharge may lead to accumulation of the surface-water inventory, followed by rapid pulse-like expulsion to the atmosphere during high-wind stress storm events (Gelesh et al. 2016).
The flux of CH4 from the sediments to the deep waters is estimated to be 5676 mol/day, a value that appears high compared to the deep-water reservoir of 4209 mol. However, the coarse resolution of our sediment sampling method is insufficient to resolve the fine-scale details of the porewater CH4 gradient close to the sediment–water interface. It is likely that the vast majority of the upwards-diffusing CH4 is oxidized by AOM and MOX in the uppermost sediment layers and, hence, that the true flux to the bottom waters is considerably lower than calculated here.
Despite extensive oxidation at the sediment–water interface, a fraction of sediment-derived CH4 is clearly observed as elevated values in the deep waters of the estuary (Fig. 2). This drives an upwards diffusive flux toward the pycnocline and, hence, an additional, if small, contribution to surface water CH4 concentrations in the estuary (Fig. 7). Processes in the water column, including dilution and oxidation, appear to attenuate the sediment-derived CH4 signal in the intermediate-depth layers of the estuarine water column, leading to the development of a midwater minimum (CH4 < 30 nmol L−1). This is likely maintained by the strong stratification, which also prevents downward advection of CH4 introduced by the river water.
The budget shows that only a small fraction of the CH4 introduced to surface waters of the estuary is transported across the sill to the archipelago area (Fig. 7). This indicates that the estuary behaves as a largely self-contained system with respect to CH4 cycling. Conversely, the archipelago area displays its own internal CH4 cycle, in which inputs are dominated by diffusion from sediments. Although the same oxidation filters are active in the archipelago as in the estuary, the flux from sediments is sufficient to raise deep-water CH4 concentrations in some areas up to 600 nmol L−1 (Fig. 5). The heterogeneous nature of the archipelago environment makes spatial extrapolation of fluxes even more difficult than in the estuary. However, even if a small fraction of the archipelago area (as defined in Fig. S3, Online Resource) behaves similarly to our study locations, the estimated atmospheric fluxes are orders of magnitude higher than the flux of CH4 introduced to the archipelago via the sill (Fig. 7), indicating the importance of the sediment CH4 source in this environment.
Overall, we can conclude from the budget that sedimentary methanogenesis obviously contributes to the total inventory of CH4 in the estuary, especially during times of low discharge. However, in terms of atmospheric methane emissions originating from the estuary, current allochthonous CH4 loading (i.e., input from the river) is likely far more important than the autochthonous loading caused by the legacy of eutrophication and sediment methanogenesis. This finding is similar to Abril and Iversen (2002) who calculated that annual CH4 input by the river in Randers Fjord was equivalent to two thirds of total atmospheric emissions in that system, implying a lesser role for autochthonous estuarine CH4 production. In the archipelago, the situation is reversed and methanogenesis fueled by the legacy of eutrophication is the main source of atmospheric methane.
Controls on Intra-annual Variability in Water Column Methane Concentrations
Seasonal Solubility and Oxidation Effects
Apart from the river-influenced surface layer, CH4 concentrations in the estuary were overall lower in winter than in summer (Fig. 2). This runs contrary to the expectation of higher solubility of CH4 at lower temperature, indicating that some additional processes control CH4 saturation in the estuary in winter. Rates of water column MOX are sensitive to both temperature and salinity. MOX has an inverse relationship with salinity and is more efficient at low-salinity environments (de Angelis and Scranton 1993; Abril and Iversen 2002). The low salinity in the estuary could therefore favor MOX and cancel out the MOX inhibition caused by the cold temperatures (Steinle et al. 2017). Water column AOM, which may be active in low-oxygen regions of the study transect, has also been shown to have relatively high temperature optimum of 25–37 °C (Zehnder and Brock 1980). Furthermore, both AOM and MOX are most effective at the interfaces of strong gradients such as the pycnocline (Borges and Abril 2011), which are not present in a mixed water column. Therefore, the cold, more saline, and well-mixed conditions in the archipelago might be inhibiting both MOX and AOM, thus explaining the elevated concentrations of CH4 in the archipelago during winter (Fig. 2).
Seasonal Changes in Stratification and Mixing
In the archipelago areas, CH4 accumulated in the deeper waters is transported to the surface through physical mixing processes during winter, leading to a more uniform profile of CH4 concentrations with higher surface concentrations (Figs. 2 and 5). Breakdown of stratification in late autumn could potentially lead to intermittent, “pulse-like,” atmospheric emissions with large amounts of CH4 escaping into the atmosphere over a relatively short period of time (Gelesh et al. 2016). Our results from site L suggest that stronger stratification coupled with low-oxygen bottom water in summer leads to decreased atmospheric flux of methane, whereas a large flux is detected during autumn, when mixing events are more likely (Fig. 5, Table 1). Silvennoinen et al. (2008) also observed the highest supersaturations of CH4 during winter in Liminganlahti Bay in Northern Finland and also measured the highest atmospheric fluxes during winter in the unfrozen parts of the estuary.
In the Pojo Bay estuary, winter is characterized by extensive ice cover (Fig. 2). The inflowing river remained a strong source of methane during our winter sampling campaign, though the spatial signal of river-derived CH4 differed from summer. The signal extended further offshore but was more diffuse in terms of the absolute concentrations. This was likely due to the river water creating a more compressed, laterally expanded freshwater lens under the ice. In previous studies, the influence of the river and especially spring melt water has been found to extend all the way to site J (Niemi 1975; Heiskanen and Tallberg 1999).
Subseasonal Exchange of Water Masses in the Archipelago
There was a remarkable drop in the water column CH4 inventory from June to August at site L (Fig. 5), which can be explained by a large intrusion of saline water into the relatively secluded basin where this site is located, which displaced the CH4-rich deep-water layer (Fig. S2, Online Resource). The same phenomenon can be seen at site J also, but to a lesser extent, and there it seems that only the surface water becomes more saline. A combination of both oxidation and displacement is likely behind the large decrease in CH4 inventory (Schmale et al. 2016; Myllykangas et al. 2017).
Seasonal Migration of SMTZ
There were slight seasonal changes in the SMTZ depth and rates of methane production and consumption at the studied locations (Fig. 6). At site J, the SMTZ (defined as the depth of equivalent CH4 and SO42− concentration) was slightly deeper in spring and autumn and slightly shallower during summer (Fig. 6). Contrary to expectations, the calculated CH4 fluxes were slightly suppressed during summer months (Table 1). At site L, the SMTZ was deepest in spring and became shallower with the passage of the year. Yet, there was no coincident trend in methane flux (Table 1). We note that the seasonal pattern differs slightly if the SMTZ is defined by the maximum concentration of H2S (e.g., SMTZ at site L is deeper in October than August, Fig. 6). However, the small changes in methane-related processes in the sediments appear to be insufficient to drive major seasonal changes in the flux of CH4 to the water column in these locations. In the study of Schmaljohann (1996) from the Kiel harbor, clear seasonality was observed in the depth of the SMTZ over a range of 10–20 cm depth in sediment, potentially related to organic matter loading to the sediments. However, this author also observed a complex seasonal pattern of deep-water CH4 concentrations, which could not be readily attributed to the mobility of the SMTZ and sediment fluxes. In other studies by Dale et al. (2008) and Mogollón et al. (2011), significant seasonal changes in AOM rates in sediments occurred due to temperature variations. Still, in these study locations, the SMTZ was located at > 2 m sediment depth, making a comparison with our setting difficult. In conclusion, the relationship between seasonal changes in temperature and organic matter loading, and sediment methane fluxes, are difficult to constrain in our study.
Long-Term Evolution of Methane Emissions in Eutrophied Estuaries
Pojo Bay Estuary and Archipelago
Like many other coastal regions of the Baltic Sea, the Pojo Bay estuary and the adjacent archipelago suffered from anthropogenic eutrophication during the twentieth century, leading to the expansion of seasonally hypoxic bottom waters in stratified areas (Conley et al. 2011; Jokinen et al. 2018). In the Pojo Bay estuary itself, there was a brief recovery period in the early 1980s, but oxygen conditions soon deteriorated again and the estuary suffered from extensive hypoxia in the 1990s (Malve et al. 2000). During that time, enhanced primary production in the estuary and archipelago led to high rates of carbon accumulation in the sediments (Heiskanen and Tallberg 1999). In a recent study, Raateoja and Kauppila (2019) studied eutrophication development of three estuaries in Northern Baltic, including Pojo Bay, from the 1970s onwards. In accordance with other studies of SW Finland archipelago areas (Conley et al. 2011; Jokinen et al. 2018), they did not find any evidence of oligotrophication in the recent past, but note that the two large lakes in the catchment of Mustionjoki may buffer changes in coastal nutrient levels, by delaying land-to-sea nutrient transfer. Bryhn et al. (2017) also argue that direct nutrient loading reduction to coastal catchments rarely controls the recovery of these systems from eutrophication, due to import of nutrients from the open Baltic Sea. This phenomenon is expected to be particularly important in open archipelago areas such as the outer stations of our study transect. They found that in 95% of their 656 coastal study sites along the Swedish coast, the influence of offshore nutrient contributions had a much greater impact on local nutrient concentrations than either catchment loading or atmospheric deposition. Therefore, the future recovery from eutrophication in our study region is likely closely coupled to that of the Baltic Sea as a whole.
Eutrophication and increased oxygen demand in the northern Baltic Sea have been shown to strongly influence diagenetic zonation in sediments, favoring a shallow SMTZ and enhanced rates of methanogenesis (Egger et al. 2015; Rooze et al. 2016). This situation is favored by the low salinity and, hence, low bottom water sulfate concentrations in this system (Capone and Kiene 1988). The high porewater CH4 concentrations and shallow SMTZ depths observed in the sediments along the entire transect (Figs. 2 and 6) are therefore likely a modern phenomenon that is observed in response to nutrient loading in this region. This configuration of the diagenetic zones in the sediments contributes to high diffusive efflux of CH4. In the archipelago areas, where sediments contribute the majority of the CH4 fluxes to the atmosphere (Fig. 7), the legacy of eutrophication is therefore a potential key driver of methane emissions today. The theory of a recent acceleration of sedimentary methanogenesis is supported by the observation that CH4 concentrations at some locations decline in the deeper sediments (e.g., below 20 cm at station L, Fig. 6). This implies lower rates of methanogenesis in layers deposited before peak eutrophication and carbon loading. However, we acknowledge that Fe-mediated AOM in the sub-SMTZ sediments (Egger et al. 2015) may also impact on these profiles.
Outlook and Implications
In a recent study, Borges et al. (2018) suggested a close coupling between eutrophication of the Belgian coastal zone and methane emissions from sediments. These authors compared methane concentrations 26 years apart and found a significant decrease in water column CH4 concentrations, which they attribute to oligotrophication of the coastal zone during that time period. Our results confirm this close coupling, by showing that the shift toward eutrophication in the northern Baltic Sea has likely enhanced methanogenesis rates in the coastal sediments of this system.
Most findings of this study reinforce the patterns found in previous studies (e.g. Silvennoinen et al. 2008). The future evolution of methane emissions from our study area will likely depend on a combination of factors: first, the trajectory of nutrient loading from the catchment and import from the open Baltic Sea, which is expected to dictate rates of primary production and carbon loading to sediments. Secondly, climate change effects may play a strong role. For example, Wik et al. (2016b) found that the increased duration of the ice-free period caused by the warming climate was the main factor contributing to increased CH4 emissions from Northern lakes. Many areas of the northern Baltic Sea currently experience significant ice cover during winter months. According to our results, the distribution of sea ice strongly influences the distribution of methane in the water column in winter and the location of its eventual emission to the atmosphere (Figs. 2 and 3). In particular, the offshore propagation of terrestrial methane away from riverine sources may change if the extent and duration of ice cover is reduced in the future (Schneider et al. 2014). Climate change is also expected to increase precipitation especially in the Northern Hemisphere (Putnam and Broecker 2017). This would likely affect the freshwater balance in Pojo Bay in other similar estuaries and cause changes in stratification, solubility of CH4 and, combined with increased discharge and ground runoff, increase land-to-sea CH4 transport via increased methanogenesis in adjacent wetland areas (Corbett et al. 2015).
The role of ebullition in methane transfer from coastal sediments to the atmosphere requires further investigation. This process may be important both in our study system and in other coastal regions and may be sensitive to future changes in carbon loading, as well as bottom water temperatures and sediment resuspension importance of ebullition in controlling fluxes of methane directly from the sediment column. Bubbles rapidly rising through the water column to the surface may bypass the oxidative filter functions of AOM and MOX entirely (Knittel and Boetius 2009). The importance of ebullition as a source of atmospheric emissions of CH4 has become more evident in recent studies. Schilder et al. (2016) found that in many lakes ebullitive fluxes to the atmosphere were almost 10 times higher than the diffusive flux, and it has been suggested that ebullition is behind up to 90% of all CH4 emissions from aquatic systems (Wik et al. 2016a). It was also recently shown by Davidson et al. (2018) that while increased temperature and nutrient availability had little direct effect on diffusive fluxes of CH4, their effects combined caused a considerable increase in ebullition.
Summary
-
The whole estuary and its connecting archipelago were consistently a source of CH4 to the atmosphere.
-
According to our results, the allochthonous river input was consistently the main factor behind CH4 supersaturation in the estuary, while sedimentary methanogenesis fueled by past eutrophication was the main source of CH4 in the archipelago. The strong flux of CH4 from the sediments was primarily caused by a shallow SMTZ and, where present, the action of benthic biota.
-
Seasonal variability had a strong influence on atmospheric fluxes of methane and their spatial distribution. In the estuary, the CH4-rich river plume reached much farther offshore, and in the archipelago areas, physical mixing brought methane from the deeper water layers to the surface. Patterns of atmospheric and sedimentary fluxes of methane in the archipelago appeared more complex and less directly affected by seasonality.
-
The estuary displayed large variability in the sinks and sources of CH4 implying that CH4 fluxes to the atmosphere are equally variable and dependent on both local hydrodynamics as well as the larger climate system.
-
The strong CH4 fluxes from the sediments are likely a recent (decadal timescale) phenomenon. Our porewater CH4 profiles show declining concentrations at greater depth in the sediment column, implying that the main zone of methanogenesis in this system is the shallow sediments.
-
Despite the clear evidence of strong methanogenic activity throughout the sediments both in the estuary and the adjacent archipelago, atmospheric fluxes were generally at least an order of magnitude lower than respective sedimentary fluxes. This indicates that the oxidative filters are still functioning efficiently, although phenomena such as ebullition bypass these filters and future changes in temperature and eutrophication development might further modify the balance of these processes.
References
Abril, G., and N. Iversen. 2002. Methane dynamics in a shallow non-tidal estuary (Randers Fjord, Denmark). Marine Ecology Progress Series 230: 171–181. https://doi.org/10.3354/meps230171.
Abril, G., and A.V. Borges. 2004. Carbon dioxide and methane emissions from estuaries. In Greenhouse gas emissions—fluxes and processes, 187–207. Berlin/Heidelberg: Springer. https://doi.org/10.1007/3-540-26643-7_7.
de Angelis, M.A., and M.I. Scranton. 1993. Fate of methane in the Hudson River and Estuary. Global Biogeochemical Cycles 7: 509–523. https://doi.org/10.1029/93GB01636.
Asmala, E., R. Autio, H. Kaartokallio, L. Pitkänen, C.A. Stedmon, and D.N. Thomas. 2013. Bioavailability of riverine dissolved organic matter in three Baltic Sea estuaries and the effect of catchment land use. Biogeosciences 10: 6969–6986. https://doi.org/10.5194/bg-10-6969-2013.
Asmala, E., C.A. Stedmon, and D.N. Thomas. 2012. Linking CDOM spectral absorption to dissolved organic carbon concentrations and loadings in boreal estuaries. Estuarine, Coastal and Shelf Science 111: 107–117. https://doi.org/10.1016/j.ecss.2012.06.015.
Bange, H.W. 2006. Nitrous oxide and methane in European coastal waters. Estuarine, Coastal and Shelf Science 70: 361–374. https://doi.org/10.1016/j.ecss.2006.05.042.
Bange, H.W., U.H. Bartell, S. Rapsomanikis, and M.O. Andreae. 1994. Methane in the Baltic and North Seas and a reassessment of the marine emissions of methane. Global Biogeochemical Cycles 8: 465–480.
Bartlett, K.B., D.S. Bartlett, R.C. Harriss, and D.I. Sebacher. 1987. Methane emissions along a salt marsh salinity gradient. Biogeochemistry 4: 183–202. https://doi.org/10.1007/BF02187365.
Berg, P., N. Risgaard-Petersen, and S. Rysgaard. 1998. Interpretation of measured concentration profiles in sediment pore water. Limnology and Oceanography 43: 1500–1510. https://doi.org/10.4319/lo.1998.43.7.1500.
Berner, R.A. 1980. Early diagenesis: A theoretical approach. Princeton University Press, 1980.
Bianchi, T.S. 2007. Biogeochemistry of estuaries. New York: Oxford University Press, Inc..
Blasing, T.J. 2016. Recent greenhouse gas concentrations. https://doi.org/10.3334/CDIAC/atg.032.
Bonaglia, S., V. Brüchert, N. Callac, A. Vicenzi, E. Chi Fru, and F.J.A. Nascimento. 2017. Methane fluxes from coastal sediments are enhanced by macrofauna. Scientific Reports 7 (1): 1–10. https://doi.org/10.1038/s41598-017-13263-w.
Borges, A.V., and G. Abril. 2011. Carbon dioxide and methane dynamics in estuaries. Treatise on Estuarine and Coastal Science 5: 119–161. https://doi.org/10.1016/B978-0-12-374711-2.00504-0.
Borges, A.V., G. Speeckaert, W. Champenois, M.I. Scranton, and N. Gypens. 2018. Productivity and temperature as drivers of seasonal and spatial variations of dissolved methane in the Southern Bight of the North Sea. Ecosystems 21 (4): 583–599. https://doi.org/10.1007/s10021-017-0171-7.
Boudreau, B.P. 1997. Diagenetic models and their implementation. New York: Springer. https://doi.org/10.1007/978-3-642-60421-8.
Bryhn, A.C., P.H. Dimberg, L. Bergström, R.E. Fredriksson, J. Mattila, and U. Bergström. 2017. External nutrient loading from land, sea and atmosphere to all 656 Swedish coastal water bodies. Marine Pollution Bulletin 114: 664–670. https://doi.org/10.1016/j.marpolbul.2016.10.054.
Capone, D.G., and R.P. Kiene. 1988. Comparison of microbial dynamics in marine and freshwater sediments: Contrasts in anaerobic carbon catabolism. Limnology and Oceanography 33: 725–749. https://doi.org/10.4319/lo.1988.33.4part2.0725.
Chanton, J.P., C.S. Martens, and C.A. Kelley. 1989. Gas transport from methane-saturated, tidal freshwater and wetland sediments. Limnology and Oceanography 34: 807–819. https://doi.org/10.4319/lo.1989.34.5.0807.
Cicerone, R.J., and R.S. Oremland. 1988. Biogeochemical aspects of atmospheric methane. Global Biogeochemical Cycles 2: 299–327. https://doi.org/10.1029/GB002i004p00299.
Cline, J.D. 1969. Spectrophotometric determination of hydrogen sulfide in natural waters. Limnology and Oceanography 14: 454–458. https://doi.org/10.4319/lo.1969.14.3.0454.
Cole, J.J., and N.F. Caraco. 1998. Atmospheric exchange of carbon dioxide in a low-wind oligotrophic lake measured by the addition of SF 6. Limnology and Oceanography 43: 647–656. https://doi.org/10.4319/lo.1998.43.4.0647.
Conley, D.J., J. Carstensen, J. Aigars, P. Axe, E. Bonsdorff, T. Eremina, B.-M. Haahti, C. Humborg, P. Jonsson, J. Kotta, C. Lännegren, U. Larsson, A. Maximov, M.R. Medina, E. Lysiak-Pastuszak, N. Remeikaité-Nikiené, J. Walve, S. Wilhelms, and L. Zillén. 2011. Hypoxia is increasing in the coastal zone of the Baltic Sea. Environmental Science & Technology 45 (16): 6777–6783. https://doi.org/10.1021/es201212r.
Corbett, J.E., M.M. Tfaily, D.J. Burdige, P.H. Glaser, and J.P. Chanton. 2015. The relative importance of methanogenesis in the decomposition of organic matter in northern peatlands. Journal of Geophysical Research – Biogeosciences 120: 280–293. https://doi.org/10.1002/2014JG002797.
Dale, A.W., D.R. Aguilera, P. Regnier, H. Fossing, N.J. Knab, and B.B. Jørgensen. 2008. Seasonal dynamics of the depth and rate of anaerobic oxidation of methane in Aarhus Bay (Denmark) sediments. Journal of Marine Research 66: 127–155. https://doi.org/10.1357/002224008784815775.
Davidson, T.A., J. Audet, E. Jeppesen, F. Landkildehus, T.L. Lauridsen, M. Søndergaard, and J. Syväranta. 2018. Synergy between nutrients and warming enhances methane ebullition from experimental lakes. Nature Climate Change 8 (2): 156–160. https://doi.org/10.1038/s41558-017-0063-z.
Dean, J.F., J.J. Middelburg, T. Röckmann, R. Aerts, L.G. Blauw, M. Egger, M.S.M.M. Jetten, et al. 2018. Methane feedbacks to the global climate system in a warmer world. Reviews of Geophysics 56: 207–250. https://doi.org/10.1002/2017RG000559.
Diaz, R.J., and R. Rosenberg. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321: 926–929. https://doi.org/10.1126/science.1156401.
Egger, M., O. Rasigraf, C.J. Sapart, T. Jilbert, M.S.M. Jetten, T. Röckmann, C. van der Veen, et al. 2015. Iron-mediated anaerobic oxidation of methane in brackish coastal sediments. Environmental Science & Technology 49 (1): 277–283. https://doi.org/10.1021/es503663z.
Fenchel, T., C. Bernard, G. Esteban, B.J. Finlay, P.J. Hansen, and N. Iversen. 1995. Microbial diversity and activity in a Danish Fjord with anoxic deep water. Ophelia 43: 45–100. https://doi.org/10.1080/00785326.1995.10430576.
Flury, S., R.N. Glud, K. Premke, and D.F. McGinnis. 2015. Effect of sediment gas voids and ebullition on benthic solute exchange. Environmental Science and Technology 49: 10413–10420. https://doi.org/10.1021/acs.est.5b01967.
Gammal, J., J. Norkko, C.A. Pilditch, and A. Norkko. 2017. Coastal hypoxia and the importance of benthic macrofauna communities for ecosystem functioning. Estuaries and Coasts 40 (2): 457–468. https://doi.org/10.1007/s12237-016-0152-7.
Gelesh, L., K. Marshall, W. Boicourt, and L. Lapham. 2016. Methane concentrations increase in bottom waters during summertime anoxia in the highly eutrophic estuary, Chesapeake Bay, U.S.A. Limnology and Oceanography 61: S253–S266. https://doi.org/10.1002/lno.10272.
Gülzow, W., G. Rehder, J.S.V. Deimling, T. Seifert, Z. Tóth, and J. Schneider Von Deimling. 2013. One year of continuous measurements constraining methane emissions from the Baltic Sea to the atmosphere using a ship of opportunity. Biogeosciences 10: 81–99. https://doi.org/10.5194/bg-10-81-2013.
Heiskanen, A., and P. Tallberg. 1999. Sedimentation and particulate nutrient dynamics along a coastal gradient from a fjord-like bay to the open sea. Hydrobiologia 393: 127–140. https://doi.org/10.1023/A:1003539230715.
IPCC. 2014. Climate change 2013—The physical science basis. Edited by Intergovernmental Panel on Climate Change. Cambridge: Cambridge University Press. https://doi.org/10.1017/CBO9781107415324.
Iversen, N., and T.H. Blackburn. 1981. Seasonal rates of methane oxidation in anoxic marine sediments. Deep Sea Research Part B. Oceanographic Literature Review 28: 888. https://doi.org/10.1016/0198-0254(81)91600-9.
Iversen, Niels, and B. Jørgensen. 1993. Diffusion coefficients of sulfate and methane in marine sediments: Influence of porosity. Geochimica et Cosmochimica Acta 57: 571–578. https://doi.org/10.1016/0016-7037(93)90368-7.
Jähne, B., K.O. Münnich, R. Bösinger, A. Dutzi, W. Huber, and P. Libner. 1987. On the parameters influencing air-water gas exchange. Journal of Geophysical Research 92: 1937. https://doi.org/10.1029/JC092iC02p01937.
Jakobs, G., P. Holtermann, C. Berndmeyer, G. Rehder, M. Blumenberg, G. Jost, G. Nausch, and O. Schmale. 2014. Seasonal and spatial methane dynamics in the water column of the central Baltic Sea (Gotland Sea). Continental Shelf Research 91: 12–25. https://doi.org/10.1016/j.csr.2014.07.005.
Jilbert, T., E. Asmala, C. Schröder, R. Tiihonen, J.-P. Myllykangas, J.J. Virtasalo, A. Kotilainen, P. Peltola, P. Ekholm, and S. Hietanen. 2018. Impacts of flocculation on the distribution and diagenesis of iron in boreal estuarine sediments. Biogeosciences 15: 1243–1271. https://doi.org/10.5194/bg-15-1243-2018.
Jilbert, T., and C.P. Slomp. 2013. Iron and manganese shuttles control the formation of authigenic phosphorus minerals in the euxinic basins of the Baltic Sea. Geochimica et Cosmochimica Acta 107: 155–169. https://doi.org/10.1016/j.gca.2013.01.005.
Jokinen, S.A., J.J. Virtasalo, T. Jilbert, J. Kaiser, O. Dellwig, H.W. Arz, J. Hänninen, L. Arppe, M. Collander, and T. Saarinen. 2018. A 1500-year multiproxy record of coastal hypoxia from the northern Baltic Sea indicates unprecedented deoxygenation over the 20th century. Biogeosciences 15: 3975–4001. https://doi.org/10.5194/bg-15-3975-2018.
Kauppi, L., G. Bernard, R. Bastrop, A. Norkko, and J. Norkko. 2018. Increasing densities of an invasive polychaete enhance bioturbation with variable effects on solute fluxes. Scientific Reports 8: 1–12. https://doi.org/10.1038/s41598-018-25989-2.
Kirschke, S., P. Bousquet, P. Ciais, M. Saunois, J.G. Canadell, E.J. Dlugokencky, P. Bergamaschi, et al. 2013. Three decades of global methane sources and sinks. Nature Geoscience 6: 813–823. https://doi.org/10.1038/ngeo1955.
Knittel, K., and A. Boetius. 2009. Anaerobic oxidation of methane: Progress with an unknown process. Annual Review of Microbiology 63: 311–334. https://doi.org/10.1146/annurev.micro.61.080706.093130.
Koskiaho, J., S. Tattari, and E. Röman. 2015. Suspended solids and total phosphorus loads and their spatial differences in a lake-rich river basin as determined by automatic monitoring network. Environmental Monitoring and Assessment 187 (4): 187. https://doi.org/10.1007/s10661-015-4397-6.
Kummu, M., H. de Moel, G. Salvucci, D. Viviroli, P.J. Ward, and O. Varis. 2016. Over the hills and further away from coast: Global geospatial patterns of human and environment over the 20th–21st centuries. Environmental Research Letters 11: 034010. https://doi.org/10.1088/1748-9326/11/3/034010.
Liss, P.S., and L. Merlivat. 1986. Air-sea gas exchange rates: Introduction and synthesis. In The role of air-sea exchange in geochemical cycling, 113–127. Dordrecht: Springer. https://doi.org/10.1007/978-94-009-4738-2_5.
Liss, P.S., and P.G. Slater. 1974. Flux of gases across the air-sea interface. Nature 247: 181–184. https://doi.org/10.1038/247181a0.
Lotze, H.K. 2006. Depletion, degradation, and recovery potential of estuaries and coastal seas. Science 312: 1806–1809. https://doi.org/10.1126/science.1128035.
Maltby, J., L. Steinle, C.R. Löscher, H.W. Bange, M.A. Fischer, M. Schmidt, and T. Treude. 2018. Microbial methanogenesis in the sulfate-reducing zone of sediments in the Eckernförde Bay, SW Baltic Sea. Biogeosciences 15: 137–157. https://doi.org/10.5194/bg-15-137-2018.
Malve, O., M. Virtanen, L. Villa, M. Karonen, H. Aakerla, A.S. Heiskanen, K.M. Lappalainen, and R. Holmberg. 2000. Artificial oxygenation experiment in hypolimnion of Pojo Bay estuary in 1995 and 1996: Factors regulating estuary circulation and oxygen and salt balances. Helsinki .
Meeuwig, J.J., P. Kauppila, and H. Pitkänen. 2000. Predicting coastal eutrophication in the Baltic: A limnological approach. Canadian Journal of Fisheries and Aquatic Sciences 57: 844–855. https://doi.org/10.1139/f00-013.
Middelburg, J.J., G. Klaver, J. Nieuwenhuize, A. Wielemaker, W. De Haas, T. Vlug, and J.F.W.A. Van Der Nat. 1996. Organic matter mineralization in intertidal sediments along an estuarine gradient. Marine Ecology Progress Series 132: 157–168. https://doi.org/10.3354/meps132157.
Middelburg, J.J., and L.A. Levin. 2009. Coastal hypoxia and sediment biogeochemistry. Biogeosciences 6: 1273–1293. https://doi.org/10.5194/bg-6-1273-2009.
Mogollón, J.M., A.W. Dale, I. L’Heureux, and P. Regnier. 2011. Impact of seasonal temperature and pressure changes on methane gas production, dissolution, and transport in unfractured sediments. Journal of Geophysical Research 116: G03031. https://doi.org/10.1029/2010JG001592.
Myllykangas, J.-P., T. Jilbert, G. Jakobs, G. Rehder, J. Werner, and S. Hietanen. 2017. Effects of the 2014 major Baltic inflow on methane and nitrous oxide dynamics in the water column of the central Baltic Sea. Earth System Dynamics 8: 817–826. https://doi.org/10.5194/esd-8-817-2017.
Naqvi, S.W.A., H.W. Bange, L. Farías, P.M.S. Monteiro, M.I. Scranton, and J. Zhang. 2010. Marine hypoxia/anoxia as a source of CH4 and N2O. Biogeosciences 7: 2159–2190. https://doi.org/10.5194/bg-7-2159-2010.
Niemi, Å. 1975. Ecology of phytoplankton in the Tvärminne area, SW coast of Finland II. Primary production and environmental conditions in the archipelago and the sea zone. Annales Botanici Fennici 105. Finnish Zoological and Botanical Publishing Board
Paerl, H.W., L.M. Valdes, B.L. Peierls, J.E. Adolf, and L.J.W. Harding. 2006. Anthropogenic and climatic influences on the eutrophication of large estuarine ecosystems. Limnology and Oceanography 51: 448–462. https://doi.org/10.4319/lo.2006.51.1_part_2.0448.
Putnam, A.E., and W.S. Broecker. 2017. Human-induced changes in the distribution of rainfall. Science Advances 3: 1–15. https://doi.org/10.1126/sciadv.1600871.
Raateoja, M., and P. Kauppila. 2019. Interaction between the land and the sea: Sources and patterns of nutrients in the scattered coastal zone of a eutrophied sea. Environmental Monitoring and Assessment 191: 24. https://doi.org/10.1007/s10661-018-7143-z.
Raymond, P.A., and J.J. Cole. 2001. Gas exchange in rivers and estuaries: Choosing a gas transfer velocity. Estuaries 24: 312–317.
Reeburgh, W.S. 1969. Observations of gases in Chesapeake Bay sediments. Limnology and Oceanography 14: 368–375. https://doi.org/10.4319/lo.1969.14.3.0368.
Reeburgh, W.S. 2007. Oceanic methane biogeochemistry. Chemical Reviews 107: 486–513. https://doi.org/10.1021/cr050362v.
Reese, B.K., D.W. Finneran, H.J. Mills, M.-X. Zhu, and J.W. Morse. 2011. Examination and refinement of the determination of aqueous hydrogen sulfide by the methylene blue method. Aquatic Geochemistry 17 (4-5): 567–582. https://doi.org/10.1007/s10498-011-9128-1.
Rooze, J., M. Egger, I. Tsandev, and C.P. Slomp. 2016. Iron-dependent anaerobic oxidation of methane in coastal surface sediments: Potential controls and impact. Limnology and Oceanography 61: S267–S282. https://doi.org/10.1002/lno.10275.
Sawicka, J.E., and V. Brüchert. 2017. Annual variability and regulation of methane and sulfate fluxes in Baltic Sea estuarine sediments. Biogeosciences 14: 325–339. https://doi.org/10.5194/bg-14-325-2017.
Scandella, B.P., L. Pillsbury, T. Weber, C.D. Ruppel, H.F. Hemond, and R. Juanes. 2016. Ephemerality of discrete methane vents in lake sediments. Geophysical Research Letters 43: 4374–4381. https://doi.org/10.1002/2016GL068668.
Schilder, J., D. Bastviken, M. van Hardenbroek, and O. Heiri. 2016. Spatiotemporal patterns in methane flux and gas transfer velocity at low wind speeds: Implications for upscaling studies on small lakes. Journal of Geophysical Research – Biogeosciences 121: 1456–1467. https://doi.org/10.1002/2016JG003346.
Schmale, O., S. Krause, P. Holtermann, N.C. Power Guerra, and L. Umlauf. 2016. Dense bottom gravity currents and their impact on pelagic methanotrophy at oxic/anoxic transition zones. Geophysical Research Letters 43: 5225–5232. https://doi.org/10.1002/2016GL069032.
Schmaljohann, R. 1996. Methane dynamics in the sediment and water column of Kiel Harbour (Baltic Sea). Marine Ecology Progress Series 131: 263–273. https://doi.org/10.3354/meps131263.
Schneider, B., W. Gülzow, B. Sadkowiak, and G. Rehder. 2014. Detecting sinks and sources of CO2 and CH4 by ferrybox-based measurements in the Baltic Sea: Three case studies. Journal of Marine Systems 140: 13–25. https://doi.org/10.1016/j.jmarsys.2014.03.014.
Silvennoinen, H., A. Liikanen, J. Rintala, and P.J. Martikainen. 2008. Greenhouse gas fluxes from the eutrophic Temmesjoki River and its estuary in the Liminganlahti Bay (the Baltic Sea). Biogeochemistry 90 (2): 193–208. https://doi.org/10.1007/s10533-008-9244-1.
Sobek, S., E. Durisch-Kaiser, R. Zurbrügg, N. Wongfun, M. Wessels, N. Pasche, and B. Wehrli. 2009. Organic carbon burial efficiency in lake sediments controlled by oxygen exposure time and sediment source. Limnology and Oceanography 54: 2243–2254. https://doi.org/10.4319/lo.2009.54.6.2243.
Soja, A.J., N.M. Tchebakova, N.H.F. French, M.D. Flannigan, H.H. Shugart, B.J. Stocks, A.I. Sukhinin, E.I. Parfenova, F.S. Chapin, and P.W. Stackhouse. 2007. Climate-induced boreal forest change: Predictions versus current observations. Global and Planetary Change 56: 274–296. https://doi.org/10.1016/j.gloplacha.2006.07.028.
Stanley, E.H., N.J. Casson, S.T. Christel, J.T. Crawford, L.C. Loken, and S.K. Oliver. 2016. The ecology of methane in streams and rivers: Patterns, controls, and global significance. Ecological Monographs 86: 146–171. https://doi.org/10.1890/15-1027.
Steinle, L., J. Maltby, T. Treude, A. Kock, H.W. Bange, N. Engbersen, J. Zopfi, M.F. Lehmann, and H. Niemann. 2017. Effects of low oxygen concentrations on aerobic methane oxidation in seasonally hypoxic coastal waters. Biogeosciences 14: 1631–1645. https://doi.org/10.5194/bg-14-1631-2017.
Stipa, T. 1999. Water exchange and mixing in a semi-enclosed coastal basin (Pohja Bay). Boreal Environment Research 4: 307–317.
Syvitski, J.P.M., C.J. Vörösmarty, A.J. Kettner, and P. Green. 2005. Impact of humans on the flux of terrestrial sediment to the global coastal ocean. Science 308 (5720): 376–380. https://doi.org/10.1126/science.1109454.
Upstill-Goddard, R.C., and J. Barnes. 2016. Methane emissions from UK estuaries: re-evaluating the estuarine source of tropospheric methane from Europe. Marine Chemistry 180: 14–23. https://doi.org/10.1016/j.marchem.2016.01.010.
Upstill-Goddard, R.C., J. Barnes, T. Frost, S. Punshon, and N.J.P. Owens. 2000. Methane in the southern North Sea: Low-salinity inputs, estuarine removal, and atmospheric flux. Global Biogeochemical Cycles 14: 1205–1217. https://doi.org/10.1029/1999GB001236.
Wanninkhof, R.H. 1992. Relationship between wind speed and gas exchange. Journal of Geophysical Research 97: 7373–7382. https://doi.org/10.1029/92JC00188.
Wanninkhof, R.H. 2014. Relationship between wind speed and gas exchange over the ocean revisited. Limnology and Oceanography: Methods 12: 351–362. https://doi.org/10.4319/lom.2014.12.351.
Wever, T.F., F. Abegg, H.M. Fiedler, G. Fechner, and I.H. Stender. 1998. Shallow gas in the muddy sediments of Eckernforde Bay, Germany. Continental Shelf Research 18: 1715–1739. https://doi.org/10.1016/S0278-4343(98)00055-7.
Wiesenburg, D.A., and N.L. Guinasso. 1979. Equilibrium solubilities of methane, carbon monoxide, and hydrogen in water and sea water. Journal of Chemical & Engineering Data 24: 356–360. https://doi.org/10.1021/je60083a006.
Wik, M., B.F. Thornton, D. Bastviken, J. Uhlbäck, and P.M. Crill. 2016a. Biased sampling of methane release from northern lakes: A problem for extrapolation. Geophysical Research Letters 43: 1256–1262. https://doi.org/10.1002/2015GL066501.
Wik, M., R.K. Varner, K.W. Anthony, S. MacIntyre, and D. Bastviken. 2016b. Climate-sensitive northern lakes and ponds are critical components of methane release. Nature Geoscience 9: 99–105. https://doi.org/10.1038/ngeo2578.
Zehnder, A.J., and T.D. Brock. 1980. Anaerobic methane oxidation: Occurrence and ecology. Applied and Environmental Microbiology 39 (1): 194–204.
Acknowledgments
We thank the personnel and trainees of Tvärminne Zoological Station for providing invaluable support to the field campaigns of this study, as well as the staff of the Department of Food and Environmental Sciences at University of Helsinki for assistance with ICP-OES analyses. The comments of two anonymous reviewers greatly improved the manuscript.
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. This research was funded by the Academy of Finland projects 139267, 272964, and 267112; Walter and Andrée de Nottbeck Foundation project COOLGAS; and Onni Talas Foundation (personal grant to JPM).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by Margaret R. Mulholland
Electronic Supplementary Material
ESM 1
(PDF 332 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Myllykangas, JP., Hietanen, S. & Jilbert, T. Legacy Effects of Eutrophication on Modern Methane Dynamics in a Boreal Estuary. Estuaries and Coasts 43, 189–206 (2020). https://doi.org/10.1007/s12237-019-00677-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-019-00677-0