
Abstract
With the world’s fossil fuels being finite in nature, an increasing interest focuses on the application of alternative renewable resources such as biomass. Biomass-derived platform chemicals with abundant functional groups have the potential to replace bulk chemicals for the production of value-added chemicals, fuels, and materials. The upgrading of these platform chemicals relies on the development of efficient catalytic systems. Hydrotalcite, with its wide compositional variety, tuneable anion-exchange capacity, and controlled acidity/basicity sites demonstrates great potential in the catalytic upgrading of biomass and the derived platform chemicals. The past decade has witnessed the emergence of research achievements on the development of efficient and robust hydrotalcite-derived metal catalysts and their applications in the upgrading of biomass or the derived platform chemicals. In this review, we aim to summarize the recent advances on the catalytic upgrading of biomass-derived platform chemicals (e.g., furfural, 5-hydroxymethylfurfural, levulinic acid, and glycerol) via hydrotalcite-derived metal catalysts. We also observed that the crucial role of using hydrotalcite-derived catalysts relies on their strong metal–support interactions. As a result, a section focusing on the discussion of the metal–support interactions of hydrotalcite-derived catalysts was provided.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The current society has been extensively shaped by the widely available fossil fuels, polymer materials, and commodity chemicals. Given that fossil fuel reserves are finite in nature, increasing concern has focused on the exploration of alternative renewable resources such as biomass [1, 2]. Other negative climate effects of fossil fuel burning that have been substantiated include the periodic occurrence of oil crises, emission of massive greenhouse gas, and the increasingly serious environmental problems caused by petroleum-derived polymers [3].
The full utilization of biomass involves the development of new biorefinery technologies, such as the oxidative gasification process (above 800 °C), for the conversion of biomass into CO2, CO, H2, and CH4. The further purified syngas can be converted into energy density fuels via Fischer–Tropsch technology [4]. Pyrolysis in the absence of oxygen is another highly attractive thermal approach (typically between 450 °C and 650 °C). This process is typically coupled with a second hydrodeoxygenation process due to the substantial oxygen content in raw products [5, 6].
Although the production of important bulk chemicals is desired, these strategies normally suffer from high energy input and low final product yield. If higher value products are preferred, milder depolymerization and selective defunctionalization strategies will be desired more [7]. The obtained building blocks normally retain a part of the structural complexity inherent to the biomass raw material. The US Department of Energy summarized a list containing 12 potential biobased platform chemicals that can be produced from biomass biologically or chemically in high yield [8]. These building-block chemicals have the potential to be subsequently valorized into a variety of valuable biobased chemicals and materials. Naturally, the greatest challenge to achieving the whole process is the development of suitable catalytic methods. Hydrotalcite, which has previously revealed widespread application, shows tremendous potential.
Hydrotalcite is one of the most important layered anionic clays; it has a special layered double hydroxide structure. Its general structure can be denoted as [M2+1−xM3+x(OH)2]x+An−x/n·mH2O, where M2+ and M3+ are bivalent and trivalent metal ions, and An− is the interlayer anion (Fig. 1) [9]. The anionic species (e.g., CO32−, NO3−, F−, and Cl−) and water are located in the interlayer and form hydrogen bonds in the brucite layers [10].

Schematic representation of general hydrotalcite structure
The special structure of hydrotalcite results in several unique properties, including (1) good adsorption capacity, (2) excellent cation-exchange capability, (3) tunable surface basicity, (4) high compositional variety; (5) uniform dispersion of M2+ and M3+ cations [9, 10]. Numerous preparation methods, such as (1) induced hydrolysis [11], (2) sol–gel [12], (3) rehydration/reconstruction [13], (4) hydrothermal methods, or (5) coprecipitation at low supersaturation while keeping a constant pH of 7–10, have been developed to control its properties [10].
Hydrotalcite itself is a good solid base catalyst and is widely used as a catalyst support. In addition, the calcination of amorphous porous metal oxides (PMOs) can be obtained after. Its application for catalytic conversions, such as catalytic oxidation and hydrogenation reactions [14, 15], organic transformations [16], and catalytic conversion of lignocellulose [17], has been extensively explored and reviewed. In numerous catalytic systems, the high catalytic activities of hydrotalcite-derived catalysts lie in strong metal–support interactions. However, as far as we know, the metal–support interactions of hydrotalcite-derived catalyst have never been summarized. In this review, we start with the summary of hydrotalcite-derived catalysts during the transformation of biomass platform chemicals and then shift to the discussion of metal–support interactions.
Catalytic Upgrading of Biomass Platform Chemicals
As a natural, renewable, available resource, biomass is expected to be used in the production of energy materials and chemicals. In the production of chemicals, biomass can only be used in various commercial chemicals and special materials as the starting material [18]. Biobased platform chemicals that are readily available from biomass must be upgraded and converted to obtain high-value-added biobased chemicals. Biobased platform chemicals, including furfural, hydroxymethylfurfural (HMF), levulinic acid (LA), and glycerol, are easily prepared from biomass feedstock [19]. The formation of furfural from xylose can occur via acid-catalyzed conversion [20]; glycerol is produced on a large scale as a by-product of biodiesel [21]. Moreover, given the abundant functional groups, numerous studies focused on the upgrading and transformation of these platform chemicals. From catalyst design to catalytic mechanism studies, these reactions have been extensively studied. Hence, these platform chemicals were selected, and their applications through hydrotalcite-based catalyst upgrading and conversion were summarized in this review.
Catalytic Upgrading of Furfural
Furfural, which is composed of an aldehyde group and a furan ring, is an important chemical building block derived from lignocellulose. Currently, large quantities of xylose can be obtained by hydrolysis of lignocellulose, and xylose is then converted to xylulose via Lewis acid catalysts. Furfural is finally obtained after the dehydration of xylulose under the catalysis of Brønsted acid [22]. As a potential energy chemical selected by the US Department of Energy [23], furfural has attracted great attention as a penitential feedstock for biofuels. Thus far, various upgrading strategies, including hydrogenation, rearrangement, C–C coupling, and their combinations, have been reported [24]. As shown in Fig. 2, hydrotalcite-derived catalysts, given their tunable properties and high activity, have extensively been involved in the above-mentioned reactions. In this section, all related catalytic systems will be summarized to determine the special role of hydrotalcite in the upgrading of furfural.

Summary of catalytic systems for the transformation of furfural to value-added chemicals via hydrotalcite catalysts
Furfural is restricted in fuel applications (diesel fuels and jet fuels) because of its preferential chain length. Aldol condensation reaction shows excellent potential in increasing the molecular weight of furfural and achieving target fuel precursors with desirable chain length. Wang et al. [25] used hydrotalcite for the catalytic condensation of furfural with acetone and successfully achieved high yields of 2-furfurylideneacetone (FAC). Meanwhile, mixed oxides obtained from calcined hydrotalcite exhibited a high activity due to the formation of basicity MgO during calcination at 500 °C. Salvador et al. [26] prepared a hydrotalcite-derived metal oxide catalyst (Mg3AlOx) and evaluated its performance under the three different aldolization reactions. Regarding the reaction of aldol condensation of furfural with acetone, they observed that not only C8 products (FAC) but also C13 products were formed through the recondensation of C8 and furfural. CO2 and NH3-temperature-programmed desorption (TPD) analysis revealed the presence of basic sites; they proposed that the basic sites favor condensation, which is consistent with the work by Wang et al. [25].
Svobodová et al. [27] performed similar work and used furfural and cyclohexanone in the aldol condensation reaction to produce high energy–density aviation fuel. The reaction between them can be used to produce long chain ketones containing 11 or 16 carbon atoms. In this study, the main objective was to evaluate the properties of hydrotalcite catalyst, and a good catalytic activity (100% conversion and yield of (2E)-2-[2-furyl-methylene]-cyclohexanone up to 68%) of rehydrated mixed oxides was finally observed after investigating textural properties and the total amount of basic and acidic sites.
Tong et al. [28] reported the oxidative condensation reaction of furfural based on hydrotalcite-derived Au catalysts. After the oxidative condensation of furfural with ethanol under oxygen atmosphere, furan-2-acrolein was obtained, the promoting effects of the base were further investigated, and the combination of Au/HT and K2CO3 produced 63% furan-2-acrolein at 130 °C.
Furfural can also produce alcohols through hydrogenation and further ring opening reactions of furan ring. Kaneda et al. [29] reported that Pt nanoparticles supported on hydrotalcite (Pt/HT) can promote the direct transformation of furfural to 1,2-pentanediol. Given the synergistic effect of Pt and HT, the Pt/HT catalyst exhibited excellent catalytic performance (Fig. 3). First, the furan ring was adsorbed on PT nanoparticles, and the formyl group generated alcohol species under the action of the base site on HT. Subsequently, the furan ring was broken under the action of hydrogen to obtain pentanone. 1,2-Pentanediol (with a yield of 73% at 150 °C) was obtained in the subsequent hydrogenation process.

Reproduced with permission from Ref. [29]. Copyright 2014, American Chemical Society
Proposed reaction scheme for the transformation of furfural to 1,2-pentanediol via Pt/HT catalyst.
Xiao et al. [30] proposed that furfural can be hydrogenated to obtain cyclopentanol under the catalysis of a hydrotalcite-derived Cu–Mg–Al metal oxide catalyst. This kind of catalyst was prepared by calcination methods, and the Cu:Mg:Al mole ratio of 2.5:12.5:5.0 showed the best catalytic activity. Under 140 °C and 40 bar of hydrogen, 98.5% furfural was converted, and cyclopentanol selectivity reached 94.8%. In 2015, they continued to explore the Cu/Zn/Al catalysts for the conversion of furfural to cyclohexanol [31]. Scanning electron microscopy (SEM) images (Fig. 4) indicated that the layer structure of hydrotalcite was clear and uniform, and the catalyst can restore the structure similar to the precursor after calcination and reduction. In the activity test, 100% furfural conversion with a yield of 84% for cyclopentanol was achieved over Cu/Zn/Al (6:9:5)-600 catalyst because of its high Cu dispersion and small particle size.

Reproduced with permission from Ref. [31]. Copyright 2015, Springer Nature
SEM images of a Cu/Zn/Al (6:9:5) precursor, b Cu/Zn/Al (6:9:5)-600 used for four times, c Cu/Zn/Al (6:9:5)-400 °C, d Cu/Zn/Al (6:9:5)-500 °C, e Cu/Zn/Al (6:9:5)-600 °C, and f Cu/Zn/Al (6:9:5)-700 °C.
As an important platform molecule, the industrial production of furfuryl alcohol attracts wide attention, and the catalytic hydrogenation of furfural is considered a promising solution. Raja et al. [32] developed a highly efficient Ni-based hydrotalcite catalyst and used it for the hydrogenation of furfural to furfuryl alcohol. In this work, the interactions between supported Ni particles and the carrier, metal dispersion, and surface alkalinity were well studied. The chemical state and nature of Ni and support species were measured by the Raman experiment. The increase in Ni loadings caused the peaks to shift to a lower frequency and become narrow. When the molar ratio of Ni was 3, the peak was similar to that of pure NiO. The researchers concluded that the strong interactions between Ni particles and the support were excited, which decreased with the increase in nickel content. The good dispersion of Ni on the support was also confirmed by transmission electron microscopy (TEM) analysis. The effect of the ratio of Mg to Ni on the alkalinity of the catalyst was tested by CO2-TPD. The results indicated that the alkalinity of the catalyst should be attributed to the increase in Mg content. MAN-2 (Ni:Mg = 2) with a moderate Ni surface area and the largest basic sites showed the best performance (furfural conversion of 98% and furfural alcohol selectivity of 95%).
In the above furfural conversion experiment, the use of hydrogen is inevitable, but numerous problems, such as safety, flammability, and storage, exist with the use of high-pressure molecular hydrogen. Several groups proposed the alternative route by using transfer hydrogenation systems (CTH) to solve these challenges. Zhang and Chen [33] proposed that the CTH route with formic acid and various alcohols as hydrogen donors is suitable for the transfer hydrogenation of furfural. Using methanol as the hydrogen donor and solvent and using copper-based hydrotalcite catalyst for CTH of furfural, a yield of 94% was achieved for furfuryl alcohol. The reaction mechanism was also well discussed. As shown in Fig. 5, methanol reformed on the CuO species to provide molecular hydrogen and hydrogenation active sites Cu(0). The reformation of methanol includes two parts: (a) methanol-generated dimethyl ether and water under the action of acidic sites; (b) the generation of H2 that can be ascribed to the presence of water and Cu2+. The Cu2+ species in the catalyst was reduced to Cu(0) sites by means of H2. Given the formation of molecular hydrogen, the C = O bond was then attacked by H atoms from the surface of the catalyst. If the resulting furfural alcohol was further hydrogenated, the H atom attacked the dehydration reaction to obtain a new target compound 2-methylfuran.

Reproduced with permission from Ref. [33]. Copyright 2017, American Chemical Society
Proposed reaction mechanism for CTH of furfural to furfural alcohol over Cu-based hydrotalcite catalyst using methanol as a hydrogen donor.
Romana et al. [34] also studied the CTH reaction of furfural with 2-propanol as the hydrogen donor and Co3O4–Al2O3 hydrotalcite-derived catalyst. First, they prepared three different catalysts containing Ni, Co, and Zn. After the investigation, Co3O4–Al2O3 exhibited the highest catalytic activity (97% selectivity and 74% yield) due to its high basic sites.
In general, hydrotalcite-based catalysts have numerous applications in the catalytic conversion of furfural. In the above studies, most of the studies used mixed metal oxide catalysts by the calcination of hydrotalcite. The calcined hydrotalcite has a higher surface area and enhanced surface basicity. The content of basic sites promotes the condensation reaction and hydrogenation reaction. In addition, the synergy between the basic site and active metal center is important for the high activity in the conversion of furfural.
Catalytic Upgrading of HMF
HMF is one of the key platform chemicals given its convenient preparation and unique structure with different functional groups [35, 36]. HMF can be produced via acid (e.g., H2SO4 [37], H3BO3 [38], and CO2 [39])-catalyzed dehydration of fructose. However, considering its high cost, more studies shifted to the direct conversion of glucose [40] in recent years. As one of the most widely used renewable platform chemicals, HMF is crucial in connecting the utilization of biomass resources and industrial chemicals, such as the production of biobased fine chemicals, polymers, and fuels.
Various new chemicals (e.g., 2,5-dimethylfuran (DMF), 2,5-furandimethanol (FDM), 2,5-furandicarboxylic acid (FDCA), etc.) have been obtained by the functionalization/defunctionalization of hydroxyl or/and aldehyde groups with different catalytic systems [41]. Hydrodeoxygenation or acidolysis of the furan ring can produce alkanes, alcohols, furancarboxylic acids, LA, or γ-valerolactone (GVL) [42, 43]. During all these transformations, catalysts are vitally important in improving yield and selectivity. Owing to their good metal dispersibility, porous structure, and inherent surface basicity, hydrotalcite-derived catalysts are also extensively used in the conversion of HMF, especially for the preparation of DMF and FDCA. As a result, this section will focus on the catalytic systems in upgrading DMF and FDCA via hydrotalcite-derived catalysts.
Catalytic Oxidation of HMF to FDCA
FDCA is a valuable fine chemical that is widely used as an antibacterial agent, pharmaceutical intermediate, and monomer to synthesize polyesters [44]. The oxidation transformation of HMF to FDCA as an atom economic pathway is the most promising route for future industrialization. In this reaction system, the hydroxyl and aldehyde groups on HMF are oxidized to form 2,5-diformylfuran (DFF) and 5-hydro-xymethyl-2-furancarboxylic acid (HMFCA) intermediates, respectively. These intermediates undergo further oxidation to generate FDCA.
As summarized in Table 1, numerous catalytic systems using hydrotalcite as support or co-catalyst have been reported. Most of the support hydrotalcite catalysts are prepared by the impregnation method. Thus, metal particles can be distributed uniformly, and more active sites are provided. In addition, the inherent alkalinity of the hydrotalcite catalyst ensures that the use of homogeneous alkali is avoided in the formation of FDCA.
In 2011, Riisager et al. [45, 46] reported the oxidation of HMF using ruthenium hydroxide catalysts supported on different supports, e.g., hydrotalcite, MgO, and spinel (MgAl2O4). After the oxygenation at 140 °C, the Ru(OH)x/HT catalyst exhibited a promising activity, and a 95% yield of FDCA was achieved. Although the HT catalyst showed high efficiency to generate FDCA, the inductively coupled plasma (ICP) analysis revealed that magnesium ions appeared in the reaction product, which implied the leaching of Mg2+ during the reaction. However, differences were observed in the amount of dissolution of different magnesium-containing catalyst supports. Unlike HT and MgO, spinel (MgAL2O3) contained a small amount of dissolved Mg (< 0.02%), which was easier to recycle while ensuring the relatively high yield of FDCA (60%).
Given the production of acidic by-products, the recyclability of the catalyst is adversely affected. On this basis, two years later, Xie et al. [47] conducted further research on the Ru catalyst and used the combination of hydrotalcite and Ru/C. This catalytic system showed excellent efficiency, and the conversion rate of 100% and selectivity of 78.2% were achieved under 20 bar O2 at 110 °C. In this bi-catalyst system, hydrotalcite as a solid base catalyst is important for the selective production of products. In an alkali-free environment, the intermediates DFF (48.6%) and FFCA (38.1%) are the main products. Hydrotalcite is favored for the oxidation of the formyl group, increasing the FFCA selectivity to 82.8% from 38.1% (no base), decreasing DFF selectivity to 1.4% or 0.9% from 48.6% (no base) at high conversion (90%–100%), and with the final FDCA selectivity reaching 78.2% at 150 °C. However, in the presence of a strong base, such as NaOH, the selectivity to DFF, FDA, and FFCA is extremely low. Therefore, the addition of HT improves the selectivity and yield of the product due to its inherent and suitable alkalinity.
In the addition of Ru, Au revealed excellent catalytic activity in various oxidation systems. The oxidation of HMF to FDCA has also been extensively studied with Au-based catalytic systems. Ebitani et al. [48] reported a new and environment-friendly gold nanoparticle catalyst supported on hydrotalcite (Au/HT) for the generation of FDCA. The complete HMF conversion was achieved at 95 °C without the addition of a homogeneous base under O2 atmosphere, and the product conversion rate, selectivity, and yield were all higher than 99%. Experiments have proven that the formation of metal active sites and alkaline contained in the hydrotalcite carrier plays a vital role in the reaction. Researchers used Au/C, Au/HT, and hydrotalcite as catalysts to conduct the same experiment to explore the influence of metal Au and hydrotalcite support on the reaction selectivity. When using hydrotalcite without loading Au as a catalyst, the conversion rate and selectivity were 0, which proved that hydrotalcite without metal sites has no activity. When using Au/C as the catalyst, the HMF conversion rate was 28%, and the FDCA selectivity was 4%, indicating that the catalyst without hydrotalcite was far inferior to Au/HT. The time course of product formation was also investigated in this study. At the beginning of the reaction, HMFCA was quickly generated and then converted to FDCA after 1 h. DFF was not detected in the reaction system, which meant that the aldehyde group of HMF can be oxidized much easier than the hydroxyl group.
For the further improvement of the catalyst’s stability, in 2019, Fang et al. [49, 50] reported that a new support can be easily prepared by physically mixing active carbon (AC) and hydrotalcite. Combining the immobilization of Au nanoparticles stabilized by polyvinylpyrrolidone, a robust Au/HT-AC catalyst was prepared. The produced catalyst exhibited a significant improvement in stability and still achieved 99% conversion and 98% selectivity after six cycles. As shown in Fig. 6, unlike the previously reported Au/HT-based catalysts, the Au/AC catalyst enabled the activation of aldehyde and hydroxyl groups simultaneously. However, for the Au/HT-AC catalyst, the catalytic route was still the same as the Au/HT catalytic route. Continuing this work, this group also investigated the size effects. The catalyst with 1% loading showed the best results, whereas the Au nanoparticles were narrowly distributed with a mean particle size of 2.5 nm (Fig. 7).

Reproduced with permission from Ref. [45]. Copyright 2020, Wiley VCH
Two different reaction pathways with Au catalysts on different supports for aerobic oxidation of HMF to synthesis FDCA.

Reproduced with permission from Ref. [49]. Copyright 2020, Wiley VCH
High-resolution TEM images of Au/2HT-1AC catalysts containing different amounts of Au. a 1.0 wt%, b 2.0 wt%, and c 4.9 wt% Au. d Conversion, selectivity, and the initial conversion rate over the Au/2HT-1AC catalysts containing different amounts of Au.
In addition to Ru and Au, bimetal catalytic systems were reported. Ebitani and Choudhary [51] prepared the hydrotalcite-supported bimetallic catalyst-Pd20Pt80-PVP/HT and achieved unexpectedly high conversion rate (99%), selectivity (99%), and yield (99%) under relatively mild conditions (98 °C and O2 flow). Motivated by this study, Xia et al. [52] introduced Pd into a gold-based catalyst and obtained another bimetallic catalyst that can be used to synthesize FDCA from HMF with improved activity. After the successful doping of Pd, the particle sizes significantly decreased from 10.2 nm to 3.4 nm for Au/HT and Pd-Au/HT (2:1), respectively. The X-ray diffraction (XRD) patterns showed no evident diffraction peaks corresponding to Pd nanoparticles, indicating the good dispersion of Pd nanoparticles on hydrotalcite support.
Catalytic Upgrading to HMF to DMF
DMF is a promising renewable liquid fuel with excellent physical and chemical properties. Compared with ethanol and butanol, DMF as a fuel exhibits a higher energy density. Furthermore, DMF is easier to mix with gasoline than ethanol, making it more conducive to transportation and storage. These characteristics make DMF an ideal substitute for liquid fuels such as bioethanol [53].
Although developing efficient strategies to produce DMF from HMF is attractive, the selective transformation of HMF remains a challenge because it requires a series of chemical transformations. On this basis, researchers are searching for an effective and achievable reaction method that can selectively generate DMF. As summarized in Table 2, hydrotalcite-derived catalysts, especially the PMOs generated by the calcination of hydrotalcite, exhibited excellent results. In this section, we will compare all these systems and determine the role of hydrotalcite in this transformation.
In 2012, Barta et al. [54] presented an efficient strategy for the hydrolysis of HMF to DMF by a copper-doped porous metal oxide catalyst (Cu-PMO) in supercritical methanol. This catalyst with a large surface area and porous structure was prepared by the calcination of Cu-doped hydrotalcite. At 260 °C, the DMF yield reached 48% after 3 h with several undesirable by-products (e.g., 2,5-dimethyltetrahydrofuran and 2-hexanol). The low selectivity can be attributed to the reactive intermediates generated during the concomitant methanol-reforming process.
To further improve the selectivity of DMF, the group modified the catalyst composition by adding small amounts of Ru to the hydrotalcite precursor [55]. The best product selectivity was achieved when using Cu20Ru2-PMO at 220 °C, 50 bar H2, and with ethanol as the solvent. Experimental results demonstrated that Ru played an important role in increasing the reduction efficiency of the main reaction intermediates, such as 2,5-furandimethanol and 5-methyl-2-furanmethanol, and finally generated DMF and 2,5-dimethyltetrahydrofuran (DMTHF) in high yield (81%). Inspired by the above findings, Zhang et al. [33] developed a catalytic system by using methanol as a hydrogen donor and the solvent. Under optimized conditions (240 °C and 10 bar N2), a high yield of 96.7 mol% was achieved using Cu3Al–A catalyst.
In addition to Cu, the Ni-based systems were explored. Zhu et al. [56] suggested that Ni–Al2O3 catalysts prepared by the calcination of NiAl hydrotalcite pressures can selectively and efficiently convert HMF to DMF. The superior activity of this catalyst can be demonstrated by the good dispersion of Ni active sites and strong interaction between Ni and Al2O3 over the resulting catalysts. Interestingly, by fine-tuning the surface-active sites and optimization of the reaction conditions, different products, including DMF (91.5%), 2,5-dihydroxymethyltetrahydrofuran (DHMTHF) (96.2%), and DMTHF (97.4%), can be achieved with high yield.
Fang et al. [57] reported a similar Ni-based catalyst using NiZnAl hydrotalcite as the precursor. The Ni–Zn alloy catalyst was highly selective, and a yield of 93.6% for DMF was obtained at 180 °C, which was significantly improved compared with the monometallic Ni catalyst (63.5%). The remarkable effect of this catalyst was due to its unique structure containing the intermetallic β1-NiZn in the shell and bulk α-NiZn alloy in the core obtained by the reduction of NiZnAl hydrotalcite.
In general, the mixed oxide obtained by the calcination of hydrotalcite showed excellent performance as a support in the process of HMF conversion to DMF. The simple calcination treatment favored the adjustment of the active metal and acid centers of the catalyst to achieve selective hydrogenation and deoxygenation, respectively.
Catalytic Conversion of LA to GVL
LA is an important oxocarboxylic acid that can be produced by the hydrolysis of HMF [58, 59]. In the past decades, the upgrading of LA into high-value-added fuel chemicals has attracted great attention. LA can be upgraded to various value-added chemicals, including LA esters and LA-derived ketals, 2-methyltetrahydrofuran, GVL, and fuel additives [59, 60].
Intense research efforts have been carried out to convert LA into GVL. GVL is highly attractive because of its good fuel properties, such as excellent lubricity, high energy density, high boiling point, and low solubility in water [61]. In addition, GVL has a number of applications, such as a food and fuel additive [62], green solvent [63], and precursor in the production of fine chemicals. GVL can be produced by the catalytic conversion of LA or its esters [64]. The successful synthesis of GVL can be operated in homogeneous and heterogeneous catalytic systems [65, 66]. Although homogeneous catalysts presented superior activity (99% yield of GVL) toward the selective transformation of LA into GVL [66], catalyst separation and reuse at the end of reaction limited their upgrading for industrial application. In this case, heterogeneous catalysts, which are promising for recycling and separation, are easy to exploit. Hydrotalcite-derived catalysts have been intensively investigated for this reaction (Table 3) due to their advantages, which include high activity, high dispersion of active phases, and large surface area [67].
Yan et al. [61] prepared three different Cu-based catalysts, including Cu–Cr, Cu–Al, and Cu–Fe, by the coprecipitation method using hydrotalcite as the precursor. The best performance (90.7% yield) was achieved with the Cu–Cr catalyst. The results indicated that Cu sites activated H2 and governed the catalytic reaction, and the difference in catalysis was probably caused by electronegativity (1.61 for Al3+, 1.66 for Cr3+, and 1.83 for Fe3+). The high electronegativity enhanced the hydrogenation capability for C=O bond, leading to a decrease in the yield of GVL.
Chen et al. [67] introduced magnetic Fe and Ni into hydrotalcite-like catalyst and obtained a group of magnetic Ni/Cu/Al/Fe catalysts. Activity tests showed that increasing the nickel content can also increase the reaction rate. However, with the continuous increase in the nickel content to 30 wt%, the reaction activity was significantly reduced. On the one hand, Ni2+ in the precursor was difficult to reduce to Ni0 in the subsequent activation process, and on the other hand, the actual content of active Cu0 was relatively low. After optimization, the best molar ratio was Ni:Cu:Mg:Fe:Al = 4.59:1:1.58:1.96:0.7.
Ma et al. [68] synthesized a similar Ni/Cu/Al/Fe catalyst and discussed its reaction pathway. Based on their study, two possible reaction pathways were proposed (Fig. 8): pathway (1), which involves the hydrogenation of LA into the intermediate HA and then esterification with methanol to form methyl hydroxypentanoic (MHV), followed by further acid catalyzation into GVL; pathway (2), in which the esterification reaction was first carried out to obtain methyl levulinate (ML), followed by the hydrogenation into GVL. These different pathways proceeded at different reaction conditions; meanwhile, a low reaction temperature favored pathway (1), and a high temperature was beneficial for pathway (2).

Reproduced with permission from Ref. [68]. Copyright 2017, Elsevier
Two possible reaction pathways from LA to GVL via the hydrotalcite-derived Ni/Cu/Al/Fe catalyst.
For the Cu/Ni hydrotalcite-derived catalyst, the total conversion of LA to GVL at 140 °C was achieved by Gupta and Kantam [69]. They revealed that copper is an active metal for hydrogenation because it adsorbed the hydrogen dissociatively, and the presence of Ni further increased the concentration of dissociative hydrogen due to the formation of Cu–Ni alloy. Thus, increasing the copper content significantly improved the catalytic activity, whereas the addition of Ni increased the selectivity toward GVL.
One advantage of hydrotalcite-derived catalysts is their tunable surface acidic/basic activity. Aytam et al. [70] investigated the effect of surface acid sites of hydrotalcite-derived Cu–Ni catalysts with the catalytic conversion of LA as the model reaction. Pyridine-adsorbed DRIFTS spectra showed that all catalysts exhibited abundant Lewis acid sites (LAS) at 1450 cm−1 and a small number of Brønsted acid sites (BAS) at 1540 cm−1, and LAS/BAS depended on the molar ratio of Ni/Cu/Al. The catalytic activity test proved that the higher the ratio of LAS/BAS, the better the catalytic activity. When the molar ratio of Ni:Cu,:AL was 4:2:3, the LAS/BAS ratio was 16.2, and a high rate of GVL production was observed, achieving 99% selectivity to GVL and a yield of 99.8%. The LAS facilitated the formation of AL and its subsequent conversion to GVL at the Ni site on the surface.
A Co/Al2O3 catalyst derived by hydrotalcite was reported by Li et al. [71]. The generated core–shell structure was proved by TEM images, and the strong interactions between Co and Al species increased the stability of Co particles, making it robust against leaching and sintering. Similar work was also carried out by Aytam et al. [72]. With a Ni–Ti hydrotalcite-derived catalyst, the quantitative yield of GVL was achieved at 280 °C. In addition to Mg–Al hydrotalcite, several studies reported the use of Mg–La hydrotalcite. Jaya et al. [73] prepared Ru nanoparticles supported on Mg–LaO hydrotalcite, which achieved a 92% conversion rate for LA and a 99% selectivity for GVL under extremely mild conditions (80 °C and 5 bar H2).
Although the above catalytic systems have achieved excellent LA conversion rates, most of them used external hydrogen sources. Numerous studies have adopted the catalytic transfer hydrogenation process to achieve the same results under mild conditions. Varkolu Mohan et al. [74] employed formic acid as a hydrogen donor for the hydrogenation of LA. Three kinds of catalysts (Ni/Al2O3, Ni/MgO, and Ni/HT) were evaluated under the same conditions. After investigation, only Ni/Al2O3 catalyst achieved a high yield of GVL (94%). The by-product water produced in the reaction had a poisonous effect on the catalyst containing the MgO component, resulting in the severe deactivation of Ni/MgO and Ni/HT. Chary et al. [75] used the hydrotalcite catalyst to convert LA under the same catalytic system and achieved 100% conversion of LA. They observed that Mg–Al pure mixed oxide was formed after the calcination of hydrotalcite. Among the different screened Mg/Al catalysts, the catalyst with a Mg/Al ratio of 3 had the highest conversion rate (Fig. 9). Alumina and MgO formed a homogeneous mixed oxide from the mutually dispersed hydrotalcite structure, which has a larger surface area than a single oxide structure, thereby increasing the availability of active base sites.

Reproduced with permission from Ref. [75]. Copyright 2018, Wiley VCH
Correlation between the conversion of GVL to the ratio of Mg:Al in hydrotalcite.
Sanchai Kuboon et al. [76] selected methyl levulinate (ML) instead of LA as raw material and used different alcohols as H donors. By analyzing the role of different alcohols under the system, they proposed good synergies between secondary alcohols and the CuNiO catalysts. In their study, the high yield of GVL resulted in the complementary cooperation between the secondary alcohol and the catalyst. Combining H2-TP, in situ XRD, alcohol-assisted simultaneous reaction, and X-ray photoelectron spectroscopy (XPS) analysis methods, the surface of metal oxide was reduced by hydrogen species derived from alcohol dehydrogenation to generate metal active sites. As shown in Fig. 10, ML reacted with the hydrogen species on the catalyst surface and generated a methyl γ-hydroxypentanoate intermediate, which underwent a cyclization reaction to form GVL. 2-PrOH dehydrogenation supplemented the active sites for the next GVL production cycle.

Reproduced with permission from Ref. [76]. Copyright 2019, Elsevier
Proposed reaction mechanism for ML-to-GVL conversion on the surface of CuNiO catalyst using isopropanol as a hydrogen donor.
Hydrotalcite-derived catalysts have shown excellent application potential in transforming LA or LA-derived esters to GVL. In the above-mentioned research reports, although the loaded metal particles were different, all achieved the high-efficiency conversion of LA. The hydrotalcite-derived catalyst has the properties of high specific surface area and capability to disperse and support metal particles, which enabled the synthesized catalyst to effectively contact the substrate, and provide sufficient reactive sites, which greatly accelerated the reaction efficiency.
Catalytic Upgrading of Glycerol
Glycerol, which includes two primary hydroxyl groups and one secondary hydroxyl group, is derived from biodiesel production as an undesired by-product. Since 2010, the global annual production of crude glycerin has been increasing, and the global annual output in 2020 has exceeded 8 million tons [77]. During the production of biodiesel, given that glycerol has limited applications as raw material and the market price of glycerol has dropped rapidly, the inevitable formation of glycerol implies the importance of further upgrading of glycerol into valuable products in the biodiesel industry [78, 79].
As an important chemical building block, the upgrading of glycerol to valuable chemicals has become a hot research topic. Given its different functional groups, a large number of high-value compounds can be synthesized through oxidation, dehydration, hydrogenolysis, gasification, and carboxylation reactions [21, 79, 80].
Through different reactions, glycerol carbonate (GC), glyceric acid, H2, dihydroxyacetone, epichlorohydrin, and hydroxyacetone(acetol) can be obtained through glycerol conversion. As summarized in Fig. 11, hydrotalcite-derived catalysts exhibited wide applications in the conversion of glycerol, and in this section, a detailed discussion about these catalytic systems will be carried out.

Summary of hydrotalcite-derived catalysts for the conversion of glycerol to different products
From Glycerol to GC
GC is an attractive derivative of glycerol, and it is prepared by transesterification reaction under mild conditions with a base catalyst [81]. Ebitani et al. [82] reported the synthesis of GC by transesterification of glycerol and dimethyl carbonate over hydrotalcite catalyst. They observed that the yield of GC increased simultaneously along with the increase in Mg/Al ratio, and the highest yield (75%) was achieved when the ratio of Mg/Al = 5. The XRD patterns indicated the formation of hydrotalcite structure at Mg/Al = 5, and the excess Mg ions precipitated as hydromagnesite. Hou et al. [83] conducted the same experiment using a calcined Mg–Al hydrotalcite catalyst. Their results indicated that for the calcined catalyst, the best Mg/Al ratio was 2, the glycerol conversion rate reached 66.9%, and the GC selectivity reached 97%.
Both the above-mentioned hydrotalcite-based catalysts achieved ideal catalytic activities. The hydromagnesite phase is formed in the uncalcined hydrotalcite and forms a composite material with hydrotalcite, and glycerol is easily adsorbed on hydromagnesite, thereby reacting with the alkali sites on hydrotalcite. For the calcined hydrotalcite, given the loss of water, carbonate, and hydroxide during calcination, a dispersed catalyst with medium alkali density, large pores, and high crystallinity is obtained.
Intensive research is focusing on GC synthesis under solvent-free conditions to explore the economic feasibility of the reported process. Sangeetha et al. [84] achieved the GC synthesis over a Cu-based mixed oxide catalyst derived from hydrotalcite under solvent-free conditions. The textural properties of Cu-based mixed oxides showed a significant change in surface area, total basic sites, and strong basic sites with various Cu molar ratios compared with the hydrotalcite precursor. The CO2-TPD analysis confirmed the formation of strong base sites in the sample, which increased with the increase in Cu content. After kinetic studies and detailed characterization of catalysts, a reasonable mechanism was drawn by this group (Fig. 12). First, glycerol oxide anions are produced on the basic sites by extracting acid protons of the primary hydroxyl groups. Next, the reaction of glyceroxide anion and dimethylcarbonate generated methyl glyceryl carbonate intermediate and methanol. Finally, the intermediate underwent a cyclization reaction to generate GC and methanol.

Reproduced with permission from Ref. [84]. Copyright 2018, Elsevier
Proposed surface catalytic mechanism from glycerol to GC via Cu-based mixed oxides.
Debecker et al. [78] used the hydrotalcite promoted by NaAlO2 as a catalyst to obtain an excellent conversion of glycerol (92% conversion and almost 100% selectivity). The strong basic sites of the catalyst significantly increased after the addition of NaAlO2. This finding also proved that the basic sites are the active center of transesterification reaction, which is indispensable for the synthesis of GC [85].
From Glycerol to Glyceric Acid
The catalytic oxidation of glycerol is also one of the promising pathways in glycerol transformation. Glyceric acid can be obtained by selectively oxidizing the terminal hydroxyl groups of glycerol. Ebitani et al. [86] achieved the highly selective preparation of glyceric acid by using hydrotalcite-supported platinum catalyst under mild conditions (60 °C). Wei et al. [87] synthesized the rehydrated Pt/hydrotalcite catalyst by impregnation–reduction and in situ rehydration method. XRD and TEM analyses showed that Pt particles were uniformly distributed in re-Pt/HT, and the layered structure of hydrotalcite was restored. The re-Pt/HT exhibited an excellent catalytic activity (conversion: 87.6%, glyceric acid yield: 58.6%).
The utilization of bimetallic catalysts for catalytic oxidation of glycerol is also a major topic of the current research. Xu et al. [88] used Au–Pt bimetallic catalyst for the selective oxidation of glycerol and studied the effect of different supports (hydrotalcite, MgO, and γ-Al2O3). They pointed out that the selectivity of the product is related to the nature of the supports, among which MgO exhibited the highest basic strength but no acid strength. Hydrotalcite has a similar acid strength and higher alkali strength than γ-Al2O3. On this basis, the researchers proposed the relationship between the nature of supports and reaction rates. As shown in Fig. 13, during the oxidation process, reaction (1) occurred irrespective of the nature of supports, and reactions (2)–(6) were in direct proportion to the strength of basic sites. The amount of glyceric acid was affected mainly by the rates of reactions (2) and (3). Basic strength effectively improved the rates of reactions (2) and (3) and was attributed to the excessive oxidation of glycerol. The high catalytic activity of hydrotalcite led to the doping of basic and acid sites contained in the supports, which reduced the reaction rate of reaction (3). Therefore, the bimetallic catalyst with hydrotalcite as the supports exhibited the highest glyceric acid.

Reproduced with permission from Ref. [88]. Copyright 2015, Elsevier
Relationship between the nature of supports and reaction rates for the catalytic conversion of glycerol to generate glyceric acid via Au–Pt bimetallic catalyst.
Jin et al. [89] reported PtCo/hydrotalcite bimetallic catalysts with adjustable selectivity to produce glyceric acid (low temperature and high O2 pressure) or lactic acid (high temperature and low O2 pressure) from glycol. With a small amount of Co as a dopant, electrons can be transferred to the Co sites at high temperatures, resulting in a decreased binding force of Pt and O. This finding means that glycerol was easily desorbed from the surface of the catalyst to undergo a dehydration reaction. Under low temperature, doping Co can enhance the Pt–O bond strength, causing the strong adsorption of glycerol on the catalyst surface to generate glyceric acid. Thus, the selective production of glycerol oxidation products can be achieved through PtCo/hydrotalcite bimetallic catalyst by adjusting the reaction mechanism.
Renewable Hydrogen Production Via Aqueous-Phase Reformation of Glycerol
Hydrogen is one of the most potential sustainable fuels due to its high energy density, pollution-free condition, and renewability. The aqueous-phase reformation of glycerol as a clean and efficient hydrogen production technology has also become an attractive method for glycerol conversion [90].
In 2013, Weckhuysen et al. [91] reported the successful application of Pt–Cu bimetallic catalyst in the catalytic aqueous reformation of glycerol. The Mg–Al mixed oxide obtained by the calcination of hydrotalcite was selected as the support to prepare a bimetallic Pt–Cu catalyst. Compared with the uncalcined catalyst, the oxide support formed after calcination exhibited a higher hydrogen selectivity. The improvement of selectivity was related to the increase in alkalinity and surface area of the supports after calcination. Moreover, the addition of Cu can reduce the formation of CH4 by-product, further improving the hydrogen selectivity.
Rajaram et al. [92] developed a K-promoted Pt–hydrotalcite catalyst that can be used for the aqueous-phase reformation of glycerol. Inspired by previous studies showing that a basic support promotes high hydrogen yield and low alkane yield [93], they proposed the use of K-modified Pt/HT catalyst to further increase the basic strength.
Table 4 shows that the number of basic sites increased dramatically after the addition of K and further increased the selectivity to hydrogen. However, the increase in K content will decrease the surface area due to the change in hydrotalcite morphology, forming a needle-like structure. The catalyst with 28% K (Pt-KHT-28) showed the greatest hydrogen selectivity, and no formation of agglomerates occurred.
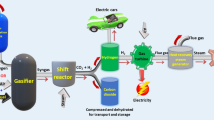
Yu et al. [94] reported a novel process by combining the steam reformation of glycerol with CH4 reformation. As shown in Fig. 14, in this process, the steam reforming reaction of glycerin produced hydrogen and CO2. Subsequently, CO2 was immediately captured by CaO in the catalyst and converted to CaCO3. After the first step, the reacted catalyst entered the CH4-reforming cycle, and CaCO3 was decarbonated to CaO, which led to catalyst regeneration. In this process, the bi-functional catalyst Ni–CaO–Ca12Al14O33, which was prepared by the calcination of Ni–Ca–Al hydrotalcite precursor, played a key role in preventing CaO sintering. Given its high activity and stability, a hydrogen selectivity of 98.5 was achieved, along with a high CH4 conversion rate (95.3%) and H2/CO molar ratio (< 2).

Reproduced with permission from Ref. [94]. Copyright 2019, Elsevier
Schematic of the catalytic system by the combination of steam reformation of glycerol and reformation of CH4 to co-produce hydrogen and syngas.
Upgrading of Glycerol to Other Value-Added Chemicals
Xu et al. [88] confirmed that the basic-promoted Au/HT catalyst can effectively produce glyceric acid in a high yield (41%) and has low selectivity to dihydroxyacetone. Continuing this research, they modified the catalyst to obtain dihydroxyacetone through further oxidation of glycerol; in addition to glyceric acid [95], Au/CuMgAl-HT catalyst was prepared. The addition of Cu metal improved the selectivity of dihydroxyacetone (72%) at a 53% conversion of glycerol under 60 °C for 6 h.
Mondelli et al. [96] developed a sustainable process for producing epichlorohydrin from glycerol and achieved the recycling of catalyst and reaction materials. Different from the traditional method of synthesizing epoxides from glycerol, the homogeneous base was replaced with a hydrotalcite-derived catalyst in the process of dehydrochlorination of the dichloropropanol intermediate. This modification not only avoided the formation of salt by-products, made the product easier to separate but also avoided serious corrosion problems and prolonged the service life of the equipment. Elemental analysis showed that the catalyst contained a high chlorine content, which was equivalent to the release amount formed by epichlorohydrin. This result proved that the catalyst was fully involved in the conversion of dichloropropanol, and OH was replaced by Cl during the rehydration process. Subsequently, the calcination of the catalyst can restore the structural characteristics and basic properties of the mixed oxide and release HCl to complete the closed chlorine circuit.
The catalytic dehydration of glycerol to hydroxyacetone (acetol) also received great interest. A certain ratio of acidic and basic Lewis centers is conducive to the formation of hydroxyacetone [97]. Domine et al. [98] synthesized Cu–MgAl mixed oxide with Cu-doped MgAl hydrotalcite, and the catalyst resulted in the excellent selectivity of acetol. The copper-free catalyst lacked any reactivity, indicating that Cu is essential for high reactivity. Therefore, they studied the role of Cu in the reaction and observed that Cu promoted the formation of intermediates containing C=O groups by using 1,2-propanediol as a probe molecule. The production of acetol involved the adsorption of glycerol on the weak acid site of the carrier, Cu-promoted formation of carbonyl intermediates, and the synergistic effect between basic and acid sites, resulting in the final dehydration reaction.
In summary, hydrotalcite-based catalysts have extensive applications in glycerol conversion not only because of their simple synthesis and non-toxicity but also because of their tunable physical and chemical properties. In addition, a variety of catalytic activities can be possessed by supporting various metals.
Metal–Support Interactions of Hydrotalcite-Derived Metal Catalysts
In Sect. 2, hydrotalcite-related catalytic systems for the upgrading of biomass-derived platform chemicals have been summarized. This kind of catalyst demonstrated excellent activity due to its tunable properties. For example, the surface acidic and basic properties can be modified by the adjustment of Mg/Al ratio and/or change of the calcination temperature. Its high surface area and electronegative surface make hydrotalcite a good candidate for the intensive stabilization of metal particles and the generation of a robust catalyst. For most of the reactions, the supported metal and hydrotalcite work efficiently as a whole, which means that metal–support interactions are important. Thus, in this section, we aimed to further investigate the details of metal–support interactions during the reaction process.
Metal–Support Interactions for Catalytic Hydrogenation/Hydrogenolysis Reactions
In 2015, Zhu et al. [56] presented a study about the rational designation of Ni-based hydrotalcite catalyst (NiAl-HT) and its application in the hydrogenation of HMF. The homogeneous distributions of Ni and Al in the hydrotalcite precursor resulted in the good dispersion of Ni species in the final catalysts and strong interactions between Ni and Al2O3. As shown in Fig. 15, the interactions between Ni and Al2O3 can be proven by various characterization technologies. XRD patterns (Fig. 15a) showed the high dispersion of NiO in the hydrotalcite structure at atomic levels. After calcination and hydrogen pretreatment, the Ni particle size increased slightly (Fig. 15b), which implied the finite sintering of Ni nanoparticles during treatment at high temperature due to the strong interaction between Al2O3 and Ni. Raman spectra (Fig. 15c) showed the band (550 cm−1) related to the surface Ni oxide component, which means that Ni2+ incorporated well into the subsurface of Al2O3. Moreover, after the calcination at high temperatures, the band broadened and moved significantly to around 600 cm−1, revealing the enhanced interactions between the metal and support. FTIR spectra (Fig. 15d) also revealed that after the increase in the calcination temperature, the vibrations of AlO4 enhanced and became broader, which can be attributed to the modification of structure from AlO6 to AlO4. This structural change was also related to the strong interactions between Ni2+ and Al3+ species. TPR experiments were also carried out to further explore structural evolutions. H2-TPR (Fig. 15e) demonstrated that the peak shifted toward a higher temperature after the increase in the calcination temperature and CO-TPR (Fig. 15f) and revealed the decrease in the desorption temperature of CO with the increase in the calcination temperature. Both results indicated the existence of strong metal–support interactions. In summary, all the above results demonstrated the successful designation of NiAl catalysts. The highly dispersed Ni particles result in strong interactions between metal and supports, which are ideal for HMF hydrogenation.

Reproduced with permission from Ref. [56]. Copyright 2015, The Royal Society of Chemistry
Characterization results of Ni-based hydrotalcite catalysts. a XRD results of NiAl-HT and calcined catalysts (NiAl-CT); b XRD results of reduced NiAl-CT catalysts; c Raman spectra, d Fourier transform infrared (FT-IR) spectra, e H2-TPR, and f CO-TPD results of NiAl-HT and NiAl-CT.
Another system for the catalytic conversion of furfural to furfuryl alcohol by using Pd–Cu hydrotalcite catalyst also proved the strong interactions between active metal species and the support [99]. After a series of characterizations by TPR, CO chemisorption, XRD, and XPS, the authors concluded that metal–support interaction affected the reducibility of Cu, which resulted in its stabilization as an oxidized form (i.e., Cu+) and created special defect sites at the Cu–MgO interface. The defect sites located at the metal–support interfacial region favored the coordination of the carbonyl group and thus accelerated its hydrogenation to alcohol. A similar study was also carried out by Thirumalaiswamy et al. [32] over Ni-based hydrotalcite catalyst. After systematic characterizations, they concluded that the enhanced interactions between Ni and support can be attributed to the incorporation of Mg because it improved the Ni dispersion and further led to a good catalytic performance.
In the work on the efficient hydrogenation of LA to GVL with magnetic Co/Al2O3 catalyst reported by Li et al. [71], the formation of core–shell structure and the strong interaction between Co and Al species stabilized the Co particles against sintering and leaching during the reaction. This easily separated catalyst also exhibited excellent recyclability (Fig. 16a, b). The ICP-atomic emission spectroscopy analysis also demonstrated limited leaching (8 ppm of Co after reaction), which indicated that it was sufficiently robust to avoid severe deactivation under harsh reaction conditions. XRD analysis (Fig. 16c) also revealed that the used catalyst maintained the Co and CoAl2O4 phases, as shown by the fresh catalyst.

Reproduced with permission from Ref. [71]. Copyright 2015, Elsevier
General scheme showing the separation process of magnetic Co/Al2O3 catalyst prepared by the calcination of hydrotalcite and used for the hydrogenation of LA to GVL. a General procedure for the separation of catalyst; b recycling test of 4Co/Al2O3 catalyst at 50% LA conversion. c XRD patterns of fresh and reused catalyst.
As discussed in Sect. 2.2, a NiZnAl catalyst derived from hydrotalcite demonstrated an improved DMF yield (93.6%) compared with the catalyst (63.5%) without the addition of Zn. Its superior performance can be attributed to the geometric properties and modulated electron density of the formed NiZn alloy. As shown in Fig. 17a, improving the C=O/C–O hydrogenolysis capability, but at a limited C=C saturation rate, is crucial to obtaining the high selectivity of DMF. Inspired by the work reported by Nørskov et al. [100], who first predicted the unique role of NiZn alloy and its significant role in the immediate removal of acetylene and prevention of its further C=C saturation, Fang et al. [57] proposed a fabrication strategy to synthesize a NiZnAl catalyst derived from hydrotalcite (Fig. 17b). The structural evolution shown in Fig. 18c during the generation of NiZn alloy catalyst included two steps: (1) the formation of NiO–ZnO–Al2O3 oxide by the calcination of NiZnAl hydrotalcite and resulting strong metal–support interaction; (2) formation of NiZn alloy through Zn inclusion to the Ni lattice via hydrogen treatment. Various characterizations technologies, including TPR, XRD, XPS, and CO-IR, demonstrated the structural evolution process from hydrotalcite to NiO doped with Zn after calcination and the formation of NiZn alloy after the reduction with an intermetallic surface (β1-NiZn) and an alloy bulk (α-NiZn). Finally, the altered geometric and electronic properties were proven by H2-TPD and CO-IR (Fig. 17d).

Reproduced with permission from Ref. [57]. Copyright 2017, American Chemical Society
Catalytic hydrolysis of HMF to DMF over NiZn alloy catalysts and NiZnAl hydrotalcite-derived catalysts. a A general scheme showing the different products during the selective conversion of HMF; b proposed modulating process of NiZn alloy; c structural evolutions during the treatment of the NiZnAl catalyst; d catalytic behavior of NiAl and NiZnAl catalysts during C−O to C-C conversion.

Reproduced with permission from Ref. [87]. Copyright 2019, Elsevier
Pt/HT-catalyzed aerobic oxidation of glycerol to glyceric acid. a Normalized quasi-in situ XANES spectra; b in situ FT-IR spectra, and c H2-TPR measurement of re-MgxAl1-LDH-Pt samples; d Proposed reaction pathway of glycerol oxidation over re-MgxAl1-LDH-Pt catalyst.
Metal–Support Interactions for Catalytic Oxidation Reactions
The strong metal–support interactions were also reported during oxidation reactions of biomass-derived platform chemicals. For example, in the catalytic oxidation of HMF to FDCA, several research groups proved the significant metal–support interaction between Au and support in either Au/HT-AC [49, 50] and Pd-Au/HT [52] catalysts. In these systems, the physicochemical properties of supports, particle size, dispersion of active metals, and metal–support interactions all played important roles in the ultimate catalytic behaviors.
Another classical system is the catalytic oxidation of glycerol to glyceric acid. In the work reported by Wei et al. [87], a series of Pt-HT catalysts was prepared through wet impregnation and in situ rehydration procedure. To investigate the metal–support interaction and structure–activity relationship in this system, the researchers carried out a combination study including in situ FT-IR spectroscopy, X-ray absorption fine structure spectroscopy, H2-TPR, and catalytic evaluations to confirm the significant role of electron-deficient Ptd+ species in the enhancement of catalytic activities via enhancing the C–H bond cleavage (Fig. 18a–c).
Figure 18d shows the proposed catalytic mechanism based on the characterizations results and catalytic evaluations. Four processes were involved in the first step of glycerol oxidation. (1) The terminal –OH group of glycerol was absorbed on the basic site of hydrotalcite and afforded an α-Cδ+–Hδ− structure. In addition, the O2 molecule underwent dissociation adsorption and generated active O species, which then adsorbed on the metallic Pt0 site. (2) The proton H in the terminal –OH group reacted with the basic site on hydrotalcite and released water, simultaneously forming alkoxide species. (3) Glycerol aldehyde was formed by the cleavage of Ptδ+–Hδ−and α-Cδ+–Hδ− species. (4) Finally, the positively charged Ptδ+ site significantly accelerated the rate-determining step of cleaving α-Cδ+–Hδ− bond and enhanced the glycerol oxidation activity.
The above information showed that the strong interaction between the supported metal and the support is essential for the high catalytic effect of hydrotalcite. Among catalysts, the active metals present a uniform distribution on hydrotalcite, ensuring the wide contact between the reaction substrate and active sites. In addition, an alloy of the active metal and carrier is formed, which changes the valence state of the active metal, thereby affecting its redox properties. The strong interaction between the supported metal and support effectively avoids the leaching and sintering of the active metal, which provides the catalyst with better recyclability. However, thus far, most of the studies on metal–support interactions are based on hydrogenation/hydrogenolysis and oxidation reactions, and their effects on other reactions (e.g., C–C coupling, amination) should be determined.
Conclusion
This review summarized the catalytic systems using hydrotalcite-derived catalysts for upgrading various important biomass-derived platform chemicals. Compared with the bulk chemicals obtained from fossil fuels, biomass-derived chemicals normally retain a higher oxygen content, which means that large amounts of catalytic systems need to be designed for the hydro-processing and/or oxidation reactions. Owing to its tuneable properties and strong interactions with metal particles, hydrotalcite shows great potential in this field.
In addition to biomass-derived platform chemicals, the direct transformation of raw biomass is practical and challenging. We speculate that the established catalytic systems via platform chemicals can also be used in the upgrading and conversion of raw biomass. Biomass raw materials, such as lignocellulose, are more complex than platform chemicals. Using hydrotalcite-based catalysts to directly convert lignocellulose will simplify the reaction process and has great economic benefits. Moreover, we believe that it can be widely used not only in hydrogenation/hydrogenolysis and oxidation reactions but also in the application of hydrotalcite-based catalysts in amination and C–C coupling reactions in the future.
Although great achievements have been obtained, numerous challenges remain regarding the design of an efficient and robust catalyst. For example, (1) metal nanoparticles can be transformed to nanocluster or single atoms to allow the maximum utilization of active sites. (2) Hydrotalcite-based materials have shown great potential in electrocatalysis, supercapacitors, and secondary batteries; however, their application for electrocatalytic conversion of biomass-derived platform chemicals is still scarce. (3) Although the strong metal–support interactions of hydrotalcite-derived catalysts have been proven in numerous systems, mechanism studies of the interactions between active metal sites and support, especially under working conditions, are still limited, requiring additional assistance from in situ characterization research and theoretical calculation. With all these improvements, we believe that more excellent studies will be published and encourage more industrial applications of hydrotalcite-derived catalysts for biomass conversion.
References
Huber GW, Iborra S, Corma A (2006) Synthesis of transportation fuels from biomass: chemistry, catalysts, and engineering. Chem Rev 106(9):4044–4098
Tuck CO, Pérez E, Horváth IT et al (2012) Valorization of biomass: deriving more value from waste. Science 337(6095):695–699
Krane J (2017) Climate change and fossil fuel: an examination of risks for the energy industry and producer states. MRS Energy Sustain 4(2):1–12
Unruh D, Pabst K, Schaub G (2010) Fischer−Tropsch synfuels from biomass: maximizing carbon efficiency and hydrocarbon yield. Energy Fuels 24(4):2634–2641
Kim S, Kwon EE, Kim YT et al (2019) Recent advances in hydrodeoxygenation of biomass-derived oxygenates over heterogeneous catalysts. Green Chem 21:3715–3743
Lin Z, Chen R, Qu Z et al (2018) Hydrodeoxygenation of biomass-derived oxygenates over metal carbides: from model surfaces to powder catalysts. Green Chem 20:2679–2696
Lin Q, Li YH, Tang ZR et al (2020) Valorization of biomass-derived platform molecules via photoredox sustainable catalysis. Trans Tianjin Univ 26(5):325–340
Werpy T, Petersen G (2004) Top value added chemicals from biomass: volume I—results of screening for potential candidates from sugars and synthesis gas. National Renewable Energy Lab., Golden, CO (US)
Nishimura S, Takagaki A, Ebitani K (2013) Characterization, synthesis and catalysis of hydrotalcite-related materials for highly efficient materials transformations. Green Chem 15(8):2026
Jabłońska M, Palkovits R (2016) Nitrogen oxide removal over hydrotalcite-derived mixed metal oxides. Catal Sci Technol 6(1):49–72
Gutmann N, Müller B (1996) Insertion of the dinuclear dihydroxo-bridged Cr(III) aquo complex into the layered double hydroxides of hydrotalcite-type. J Solid State Chem 122(1):214–220
Lopez T, Bosch P, Ramos E et al (1996) Synthesis and characterization of sol–gel hydrotalcites. Struct Text Langmuir 12(1):189–192
Kooli F, Rives V, Ulibarri MA (1995) Preparation and study of decavanadate-pillared hydrotalcite-like anionic clays containing transition metal cations in the layers. 1. samples containing nickel-aluminum prepared by anionic exchange and reconstruction. Inorg Chem 34(21):5114–5121
Feng JT, He YF, Liu YN et al (2015) Supported catalysts based on layered double hydroxides for catalytic oxidation and hydrogenation: general functionality and promising application prospects. Chem Soc Rev 44(15):5291–5319
Li YX, Li JX, Yang X et al (2019) Preparation of CeO2-modified Mg(Al)O-supported Pt-Cu alloy catalysts derived from hydrotalcite-like precursors and their catalytic behavior for direct dehydrogenation of propane. Trans Tianjin Univ 25(2):169–184
Baskaran T, Christopher J, Sakthivel A (2015) Progress on layered hydrotalcite (HT) materials as potential support and catalytic materials. RSC Adv 5(120):98853–98875
Hernández WY, Lauwaert J, van der Voort P et al (2017) Recent advances on the utilization of layered double hydroxides (LDHs) and related heterogeneous catalysts in a lignocellulosic-feedstock biorefinery scheme. Green Chem 19(22):5269–5302
Mika LT, Cséfalvay E, Németh Á (2018) Catalytic conversion of carbohydrates to initial platform chemicals: chemistry and sustainability. Chem Rev 118(2):505–613
Parihar A, Bhattacharya S (2020) Cellulose fast pyrolysis for platform chemicals: assessment of potential targets and suitable reactor technology. Biofuels, Bioprod Bioref 14(2):446–468
Cai CM, Zhang TY, Kumar R et al (2014) Integrated furfural production as a renewable fuel and chemical platform from lignocellulosic biomass. J Chem Technol Biotechnol 89(1):2–10
Tan HW, Abdul Aziz AR, Aroua MK (2013) Glycerol production and its applications as a raw material: a review. Renew Sustain Energy Rev 27:118–127
Machado G, Leon S, Santos F et al (2016) Literature review on furfural production from lignocellulosic biomass. Nat Resour 7(3):115–129
Bozell JJ, Petersen GR (2010) Technology development for the production of biobased products from biorefinery carbohydrates—the US Department of Energy’s “Top 10” revisited. Green Chem 12(4):539
Lange JP, van der Heide E, van Buijtenen J et al (2012) Furfural: a promising platform for lignocellulosic biofuels. Chemsuschem 5(1):150–166
Liu HH, Xu WJ, Liu XH et al (2010) Aldol condensation of furfural and acetone on layered double hydroxides. Kinet Catal 51(1):75–80
Ordóñez S, Díaz E, León M et al (2011) Hydrotalcite-derived mixed oxides as catalysts for different C-C bond formation reactions from bioorganic materials. Catal Today 167(1):71–76
Tišler Z, Vondrová P, Hrachovcová K et al (2019) Aldol condensation of cyclohexanone and furfural in fixed-bed reactor. Catalysts 9(12):1068
Tong XL, Liu ZH, Hu JL et al (2016) Au-catalyzed oxidative condensation of renewable furfural and ethanol to produce furan-2-acrolein in the presence of molecular oxygen. Appl Catal A Gen 510:196–203
Mizugaki T, Yamakawa T, Nagatsu Y et al (2014) Direct transformation of furfural to 1, 2-pentanediol using a hydrotalcite-supported platinum nanoparticle catalyst. ACS Sustain Chem Eng 2(10):2243–2247
Zhou MH, Zeng Z, Zhu HY et al (2014) Aqueous-phase catalytic hydrogenation of furfural to cyclopentanol over Cu-Mg-Al hydrotalcites derived catalysts: model reaction for upgrading of bio-oil. J Energy Chem 23(1):91–96
Wang Y, Zhou MH, Wang TZ et al (2015) Conversion of furfural to cyclopentanol on Cu/Zn/Al catalysts derived from hydrotalcite-like materials. Catal Lett 145(8):1557–1565
Manikandan M, Venugopal AK, Prabu K et al (2016) Role of surface synergistic effect on the performance of Ni-based hydrotalcite catalyst for highly efficient hydrogenation of furfural. J Mol Catal A: Chem 417:153–162
Zhang J, Chen JZ (2017) Selective transfer hydrogenation of biomass-based furfural and 5-hydroxymethylfurfural over hydrotalcite-derived copper catalysts using methanol as a hydrogen donor. ACS Sustain Chem Eng 5(7):5982–5993
Oleg L, David K, Romana V et al (2018) Using Mg–Al mixed oxide and reconstructed hydrotalcite as basic catalysts for aldol condensation of furfural and cyclohexanone. Chem Catal Chem 10(6):1464–1475
Axelsson L, Franzén M, Ostwald M et al (2012) Jatropha cultivation in southern India: assessing farmers’ experiences. Biofuels Bioprod Bioref 6(3):246–256
Menegazzo F, Ghedini E, Signoretto M (2018) 5-hydroxymethylfurfural (HMF) production from real biomasses. Molecules 23(9):2201
Körner P, Jung D, Kruse A (2018) The effect of different Brønsted acids on the hydrothermal conversion of fructose to HMF. Green Chem 20(10):2231–2241
Sthlberg T, Rodriguez-Rodriguez S, Fristrup P et al (2011) Metal-free dehydration of glucose to 5-(hydroxymethyl)furfural in ionic liquids with boric acid as a promoter. Chem Eur J 17(5):1456–1464
Labauze H, Camy S, Floquet P et al (2019) Kinetic study of 5-hydroxymethylfurfural synthesis from fructose in high pressure CO2–water two-phase system. Ind Eng Chem Res 58(1):92–100
Cuervo OHP, Romanelli GP, Cubillos JA et al (2020) Selective catalytic dehydration of xylose to furfural and fructose and glucose to 5-hydroximethylfurfural (HMF) using preyssler heteropolyacid. ChemistrySelect 5(14):4186–4193
van Putten RJ, van der Waal JC, de Jong E et al (2013) Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem Rev 113(3):1499–1597
Nakagawa Y, Yabushita M, Tomishige K (2021) Reductive conversion of biomass-derived furancarboxylic acids with retention of carboxylic acid moiety. Trans Tianjin Univ 27(3):165–179
Hou QD, Qi XH, Zhen MN et al (2021) Biorefinery roadmap based on catalytic production and upgrading 5-hydroxymethylfurfural. Green Chem 23(1):119–231
Sousa AF, Vilela C, Fonseca AC et al (2015) Biobased polyesters and other polymers from 2, 5-furandicarboxylic acid: a tribute to furan excellency. Polym Chem 6(33):5961–5983
Gorbanev YY, Kegnæs S, Riisager A (2011) Selective aerobic oxidation of 5-hydroxymethylfurfural in water over solid ruthenium hydroxide catalysts with magnesium-based supports. Catal Lett 141(12):1752–1760
Gorbanev YY, Kegnæs S, Riisager A (2011) Effect of support in heterogeneous ruthenium catalysts used for the selective aerobic oxidation of HMF in water. Top Catal 54(16–18):1318–1324
Xie JH, Nie JF, Liu HC (2014) Aqueous-phase selective aerobic oxidation of 5-hydroxymethylfurfural on Ru/C in the presence of base. Chin J Catal 35(6):937–944
Gupta NK, Nishimura S, Takagaki A et al (2011) Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2, 5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem 13(4):824
Sun WX, Gao TQ, Zhu GH et al (2020) Influence of support properties and particle size on the gold-catalyzed base-free aerobic oxidation of 5-hydroxymethylfurfural. ChemistrySelect 5(4):1416–1423
Gao TQ, Gao T, Fang WH et al (2017) Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2, 5-furandicarboxylic acid in water by hydrotalcite-activated carbon composite supported gold catalyst. Mol Catal 439:171–179
Choudhary H, Ebitani K (2016) Hydrotalcite-supported PdPt-catalyzed aerobic oxidation of 5-hydroxymethylfurfural to 2, 5-furandicarboxylic acid in water. Chem Lett 45(6):613–615
Xia HA, An JH, Hong M et al (2019) Aerobic oxidation of 5-hydroxymethylfurfural to 2, 5-difurancarboxylic acid over Pd-Au nanoparticles supported on Mg–Al hydrotalcite. Catal Today 319:113–120
Wang HY, Zhu CH, Li D et al (2019) Recent advances in catalytic conversion of biomass to 5-hydroxymethylfurfural and 2, 5-dimethylfuran. Renew Sustain Energy Rev 103:227–247
Hansen TS, Barta K, Anastas PT et al (2012) One-pot reduction of 5-hydroxymethylfurfural via hydrogen transfer from supercritical methanol. Green Chem 14(9):2457
Kumalaputri AJ, Bottari G, Erne PM et al (2014) Tunable and selective conversion of 5-HMF to 2, 5-furandimethanol and 2, 5-dimethylfuran over copper-doped porous metal oxides. Chemsuschem 7(8):2266–2275
Kong X, Zheng RX, Zhu YF et al (2015) Rational design of Ni-based catalysts derived from hydrotalcite for selective hydrogenation of 5-hydroxymethylfurfural. Green Chem 17(4):2504–2514
Kong X, Zhu YF, Zheng HY et al (2017) Inclusion of Zn into metallic Ni enables selective and effective synthesis of 2, 5-dimethylfuran from bioderived 5-hydroxymethylfurfural. ACS Sustainable Chem Eng 5(12):11280–11289
Jiang ZW, Hu D, Zhao ZY et al (2021) Mini-review on the synthesis of furfural and levulinic acid from lignocellulosic biomass. Processes 9(7):1234
Kohli K, Prajapati R, Sharma B (2019) Bio-based chemicals from renewable biomass for integrated biorefineries. Energies 12(2):233
Sheldon RA (2014) Green and sustainable manufacture of chemicals from biomass: state of the art. Green Chem 16(3):950–963
Yan K, Liao JY, Wu X et al (2013) A noble-metal free Cu-catalyst derived from hydrotalcite for highly efficient hydrogenation of biomass-derived furfural and levulinic acid. RSC Adv 3(12):3853
Horváth IT, Mehdi H, Fábos V et al (2008) Γ-Valerolactone—a sustainable liquid for energy and carbon-based chemicals. Green Chem 10(2):238–242
Bond JQ, Alonso DM, Wang D et al (2012) Integrated catalytic conversion of g-valerolactone to liquid alkenes for transportation fuels 1110:1110–1115
Termvidchakorn C, Faungnawakij K, Kuboon S et al (2019) A novel catalyst of Ni hybridized with single-walled carbon nanohorns for converting methyl levulinate to γ-valerolactone. Appl Surf Sci 474:161–168
Omoruyi U, Page S, Hallett J et al (2016) Homogeneous catalyzed reactions of levulinic acid: to γ-valerolactone and beyond. Chemsuschem 9(16):2037–2047
Fábos V, Mika LT, Horváth IT (2014) Selective conversion of levulinic and formic acids to γ-valerolactone with the shvo catalyst. Organometallics 33(1):181–187
Zhang J, Chen JZ, Guo YY et al (2015) Effective upgrade of levulinic acid into γ-valerolactone over an inexpensive and magnetic catalyst derived from hydrotalcite precursor. ACS Sustain Chem Eng 3(8):1708–1714
Zhang RH, Ma YB, You F et al (2017) Exploring to direct the reaction pathway for hydrogenation of levulinic acid into γ-valerolactone for future clean-energy vehicles over a magnetic Cu-Ni catalyst. Int J Hydrog Energy 42(40):25185–25194
Gupta SSR, Kantam ML (2018) Selective hydrogenation of levulinic acid into γ-valerolactone over Cu/Ni hydrotalcite-derived catalyst. Catal Today 309:189–194
Gundeboina R, Velisoju VK, Gutta N et al (2019) Influence of surface Lewis acid sites for the selective hydrogenation of levulinic acid to γ-valerolactone over Ni-Cu-Al mixed oxide catalyst. React Kinetics Mech Catal 127(2):601–616
Long XD, Sun P, Li ZL et al (2015) Magnetic Co/Al2O3 catalyst derived from hydrotalcite for hydrogenation of levulinic acid to γ-valerolactone. Chin J Catal 36(9):1512–1518
Gundeboina R, Gadasandula S, Velisoju VK et al (2019) Ni-Al-Ti hydrotalcite based catalyst for the selective hydrogenation of biomass-derived levulinic acid to γ-valerolactone. ChemistrySelect 4(1):202–210
Swarna Jaya V, Sudhakar M, Naveen Kumar S et al (2015) Selective hydrogenation of levulinic acid to γ-valerolactone over a Ru/Mg–LaO catalyst. RSC Adv 5(12):9044–9049
Varkolu M, Velpula V, Burri DR et al (2016) Gas phase hydrogenation of levulinic acid to γ-valerolactone over supported Ni catalysts with formic acid as hydrogen source. New J Chem 40(4):3261–3267
Hussain S, Velisoju VK, Rajan NP et al (2018) Synthesis of γ-valerolactone from levulinic acid and formic acid over Mg–Al hydrotalcite like compound. ChemistrySelect 3(22):6186–6194
Tanwongwan W, Eiad-Ua A, Kraithong W et al (2019) Simultaneous activation of copper mixed metal oxide catalysts in alcohols for gamma-valerolactone production from methyl levulinate. Appl Catal A: Gen 579:91–98
Crnomarkovic M, Panchaksharam Y, Spekreijse J et al (2018) Road to bio business cases: case studies on potentially attractive opportunities for bio-based chemicals in Europe Consortium 73–81
Ramesh S, Devred F, van den Biggelaar L et al (2018) Hydrotalcites promoted by NaAlO2 as strongly basic catalysts with record activity in glycerol carbonate synthesis. ChemCatChem 10(6):1398–1405
Zhou CHC, Beltramini JN, Fan YX et al (2008) Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem Soc Rev 37(3):527–549
Christoph R, Schmidt B, Steinberner U et al (2006) Glycerol. Ullmann’s Encycl Ind Chem 754–756
Ochoa-Gómez JR, Gómez-Jiménez-aberasturi O, Ramírez-López C et al (2012) A brief review on industrial alternatives for the manufacturing of glycerol carbonate, a green chemical. Org Process Res Dev 16(3):389–399
Takagaki A, Iwatani K, Nishimura S et al (2010) Synthesis of glycerol carbonate from glycerol and dialkyl carbonates using hydrotalcite as a reusable heterogeneous base catalyst. Green Chem 12(4):578
Zheng LP, Xia SX, Hou ZT et al (2014) Transesterification of glycerol with dimethyl carbonate over Mg-Al hydrotalcites. Chin J Catal 35(3):310–318
Marimuthu M, Marimuthu P, S K AK, et al (2018) Tuning the basicity of Cu-based mixed oxide catalysts towards the efficient conversion of glycerol to glycerol carbonate. Mol Catal 460:53–62
Bai RX, Wang Y, Wang S et al (2013) Synthesis of glycerol carbonate from glycerol and dimethyl carbonate catalyzed by NaOH/γ-Al2O3. Fuel Process Technol 106:209–214
Tsuji A, Rao KT, Nishimura S et al (2011) Selective oxidation of glycerol by using a hydrotalcite-supported platinum catalyst under atmospheric oxygen pressure in water. Chemsuschem 4(4):542–548
Zhang JB, Li XL, Xu M et al (2019) Glycerol aerobic oxidation to glyceric acid over Pt/hydrotalcite catalysts at room temperature. Sci Bull 64(23):1764–1772
Xu CL, Du YQ, Li C et al (2015) Insight into effect of acid/base nature of supports on selectivity of glycerol oxidation over supported Au–Pt bimetallic catalysts. Appl Catal B Environ 164:334–343
Liu MY, Yan WJ, Wu JW et al (2020) Electronically coupled PtCo/MgAl hydrotalcite catalysts display tunable selectivity toward glyceric acid and lactic acid for glycerol conversion. Catal Lett 150(9):2590–2598
Ramesh S, Venkatesha NJ (2017) Template free synthesis of Ni-perovskite: an efficient catalyst for hydrogen production by steam reforming of bioglycerol. ACS Sustain Chem Eng 5(2):1339–1346
Boga DA, Oord R, Beale AM et al (2013) Highly selective bimetallic Pt–Cu/Mg(Al)O catalysts for the aqueous-phase reforming of glycerol. ChemCatChem 5(2):529–537
Pendem C, Sarkar B, Siddiqui N et al (2018) K-promoted Pt-hydrotalcite catalyst for production of H2 by aqueous phase reforming of glycerol. ACS Sustain Chem Eng 6(2):2122–2131
Wen GD, Xu YP, Ma HJ et al (2008) Production of hydrogen by aqueous-phase reforming of glycerol. Int J Hydrog Energy 33(22):6657–6666
Dang CX, Wu SJ, Cao YH et al (2019) Co-production of high quality hydrogen and synthesis gas via sorption-enhanced steam reforming of glycerol coupled with methane reforming of carbonates. Chem Eng J 360:47–53
Yin YR, Tang T, Xu CL (2017) Au/CuMgAl-hydrotalcite catalysts promoted by Cu+ and basic sites for selective oxidation of glycerol to dihydroxyacetone. Gold Bull 50(4):319–326
Lari GM, Pastore G, Mondelli C et al (2018) Towards sustainable manufacture of epichlorohydrin from glycerol using hydrotalcite-derived basic oxides. Green Chem 20(1):148–159
Carvalho DC, Pinheiro LG, Campos A et al (2014) Characterization and catalytic performances of copper and cobalt-exchanged hydroxyapatite in glycerol conversion for 1-hydroxyacetone production. Appl Catal A Gen 471:39–49
Mazarío J, Concepción P, Ventura M, Domine ME (2020) Continuous catalytic process for the selective dehydration of glycerol over Cu-based mixed oxide. J Catal 385:160–175
Fulajtárova K, Soták T, Hronec M et al (2015) Aqueous phase hydrogenation of furfural to furfuryl alcohol over Pd-Cu catalysts. Appl Catal A Gen 502:78–85
Studt F, Abild-Pedersen F, Bligaard T et al (2008) Identification of non-precious metal alloy catalysts for selective hydrogenation of acetylene. Science 320(5881):1320–1322
Acknowledgements
This work is supported by Beijing Forestry University Outstanding Young Talent Cultivation Project (2019JQ03005) and the Young Tip-top Talent Project of Science and Technology Innovation by State Forestry and Grassland Administration of China (2019132609).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that there are no conflicts of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, ZH., Sun, Z. & Yuan, TQ. Recent Advances in the Catalytic Upgrading of Biomass Platform Chemicals Via Hydrotalcite-Derived Metal Catalysts. Trans. Tianjin Univ. 28, 89–111 (2022). https://doi.org/10.1007/s12209-021-00307-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-021-00307-6