
Abstract
Thalidomide was first developed as a sedative around 60 years ago, but exhibited teratogenicity, leading to serious defects such as limb deformities. Nevertheless, thalidomide is now recognized as a therapeutic drug for the treatment of Hansen’s disease and myeloma. Immunomodulatory drugs (IMiDs), a new class of anti-cancer drug derived from thalidomide, have also been developed and exert potent anti-cancer effects. Although the molecular mechanism of thalidomide and IMiDs remained unclear for a long time, cereblon, a substrate receptor of the CRL4 E3 ubiquitin ligase was identified as a primary direct target by a new affinity technique. A growing body of evidence suggests that the effect of IMiDs on myeloma and other cancer cells is mediated by CRBN. Each IMiD binds to CRBN and alters the substrate specificity of the CRBN E3 ubiquitin ligase complex, resulting in breakdown of intrinsic downstream proteins such as Ikaros and Aiolos. Here we give an overview of the current understanding of mechanism of action of IMiDs via CRBN and prospects for the development of new drugs that degrade protein of interest.
Similar content being viewed by others


History of immunomodulatory drugs (IMiDs)
Immunomodulatory drugs (IMiDs) are a new class of anti-cancer drugs for which the parent molecule is thalidomide. Thalidomide (Fig. 1) was developed as a sedative in 1950s by the German pharmaceutical company Grunenthal. Experiments using rodents initially suggested it to be safe for use in humans, and the drug was sold over 40 countries, including Japan. However, as is widely known, thalidomide was found to have serious teratogenic effects. Use during pregnancy is associated with developmental defects of the limbs and ears. In the 1960s, thalidomide treatment resulted in thousands of children born with phocomelia or amelia. It is estimated that more than 10,000 children worldwide have been affected by thalidomide teratogenicity [1–7].

Structure of IMiDs. Thalidomide is composed of a glutarimide ring and a pthalimide ring. Lenalidomide and pomalidomide are very similar to thalidomide. Lenalidomide is 4-amino analog of thalidomide lacking a carbonyl group of pthalidmide. Poamlidomide is 4-amino analog of thalidomide. Although CC-122 possesses a glutarimide ring, the structure is quite different from other IMiDs
Due to these serious side effects, thalidomide was withdrawn from the market in the early 1960s. However, research on thalidomide continued behind the scenes, and it was found to exert favorable effects against erythema nodosum leprosum (a form of Hansen’s disease), as well as to have anti-inflammatory and anti-angiogenic effects [8–10]. In 1999 it was also found to exert highly favorable effects against multiple myeloma [11], a type of blood cancer, and in 2006 the use of thalidomide was approved by the Food and Drug Administration (FDA) in the United States for the treatment of multiple myeloma. This drug was subsequently approved in Japan, and has been sold in this country since 2008. However, due to its teratogenicity, the use of thalidomide is strictly controlled by the System for Thalidomide Education and Prescribing Safety (S.T.E.P.S) in the US and the Thalidomide Education and Risk Management System (TERMS) in Japan [12, 13].
Numerous thalidomide derivatives have been developed and are collectively referred to as IMiDs. Of these, lenalidomide and pomalidomide (Fig. 1) show potent anti-cancer activity [3, 14]. The use of these IMiDs has been approved by the FDA for the treatment of multiple myeloma. Lenalidomide (but not pomalidomide) is shown to be specifically effective for the treatment of myelodysplastic syndrome del(5q) (5q− syndrome).
Cereblon, a direct target of thalidomide
The mechanisms underlying the therapeutic actions and adverse effects of thalidomide remained a mystery for many years, as the target of thalidomide, i.e., the proteins to which it directly binds to in vivo to exert its pharmacological effects, were unknown.
In previous work, our group developed a technique using magnetic nanoparticles (known as Handa beads) by which proteins [including target protein(s) of thalidomide] that bind specifically to a chemical compound can be affinity purified in a single step. The diameter of these beads is ~200 nm, which allows to them have numerous linkers for ligand immobilization (Fig. 2). Using this technique, we have been identifying the targets of many drugs and natural compounds [15], and during this work we became interested in identifying the target of thalidomide.

Handa beads. Electron microscopic image of Handa beads (left). Structure of thalidomide-immobilized Handa beads (right)
We first covalently bound a thalidomide derivative onto the surface of Handa beads (Fig. 2), and performed affinity purification using these beads and cell lysates of various tissues to identify proteins specifically binding to thalidomide. As described above, thalidomide is known to exert a variety of effects, which suggest that it binds to multiple protein targets. However, unexpectedly, we identified only two proteins that specifically bind to thalidomide among the various lysates tested. Mass spectrometry analysis identified these proteins to be damage-specific DNA binding protein 1 (DDB1) and cereblon (CRBN). Binding analyses using affinity beads and DDB1 and CRBN recombinant proteins demonstrated that CRBN binds directly to thalidomide, whereas DDB1 forms a complex with CRBN, and hence binds indirectly to thalidomide.
Cereblon, the direct target of thalidomide, is a 442-amino acid protein that is evolutionary conserved from plants to humans. However, the cellular function of CRBN was largely unknown at the time we identified it as a candidate binding partner of thalidomide. An important clue to understanding the function of CRBN was DDB1. DDB1 has been shown to be an adaptor subunit of the E3 ubiquitin ligase complex formed together with Cul4, Roc1, and a substrate receptor [16–18]. This type of E3 ubiquitin ligase complex is known as Cullin-RING ligase 4 (CRL4). CRBN competed with DDB2, another substrate receptor, for binding to DDB1. By demonstrating its similarly to other substrate receptors, including DDB2, we found that CRBN ubiquitinates itself (autoubiquitination). We suggest that CRBN itself functions as a substrate receptor. Furthermore, we found that thalidomide inhibits the autoubiquitination of CRBN. We concluded that CRBN is a direct target of thalidomide.
Next we examined whether CRBN was involved in teratogenic effects of thalidomide. We chose zebrafish (Danio rerio) as a primary animal model. Zebrafish exhibit rapid growth and have transparent bodies, so thalidomide defects are easier to observe and analyze. Thalidomide-treated embryos showed developmental defects in their pectoral fins and otic vesicles (ear), similar to the teratogenic effects seen in humans. To demonstrate that CRBN is the bona fide target of thalidomide in its teratogenic activity, we constructed a thalidomide-binding-deficient mutant (Y384A and W386A) of CRBN by genetic engineering techniques. We expressed this mutant in embryos in zebrafish and chicks (Gallus gallus), and treated the resulting embryos with thalidomide. When wild-type CRBN was expressed, both animals showed developmental defects, however, when the CRBN mutant was expressed, these developmental defects were largely suppressed. We concluded that CRBN is a bona fide target for thalidomide teratogenicity [19].
Therapeutic effects of IMiDs via CRBN
CRBN, which was identified as a primary direct target for thalidomide, hence became the focus of attention in the field of thalidomide and IMiDs. The next important question is whether or not CRBN is involved in the therapeutic effects of IMiDs. IMiDs have been shown to upregulate p21, leading to G1 arrest, and downregulate IRF4 (interferon regulatory factor 4), which forms a positive feedback loop with c-myc for myeloma survival [20–22]. The mechanism of these expression changes by IMiDs were largely unknown.
Stewart and colleagues first reported that RNAi knockdown of CRBN in multiple myeloma cells results in nearly complete cancellation of the anti-myeloma effects of IMiDs [23]. Furthermore, some patients who had become resistant to lenalidomide were found to have decreased expression of CRBN. We began a research project with Celgene, the company that developed and now sells IMiDs, and demonstrated that lenalidomide and pomalidomide bind more strongly than thalidomide to CRBN [24]. Furthermore, it was shown that the expression change of c-myc, IRF4, and p21 by IMiDs are mediated by CRBN. These results suggest that CRBN is essentially required for the anti-cancer effects of IMiDs.
The next issue was the substrate of CRBN, which was associated with the anti-cancer effects of IMiDs. Ebert’s gourp and Kaelin Jr’s group independently demonstrated that when lenalidomide binds to CRBN, a new function of CRBN is induced, namely, to ubiquitinate and degrade Ikaros (IKZF1) and Aiolos (IKZF3), which are transcription factors that are known to be responsible for the maturation of B cells [25, 26]. Concurrently, our group, in collaboration with Celgene, demonstrated that in T cells, the ubiquitination and degradation of Ikaros and Aiolos induced by IMiDs leads to the enhanced expression of IL-2 [27]. These results demonstrated at the molecular level that IMiDs are excellent anti-cancer drugs, exerting anti-cancer effects in B cell derived cancer cells as well as inducing the activity of T cells.
Subsequently it was found that even when thalidomide is bound to CRBN, the ubiquitination and degradation of Ikaros and Aiolos are weakly induced, resulting in a new issue. In other words, although when IMiDs bind to CRBN, their common substrates Ikaros and Aiolos are ubiquitinated and degraded, the question arose as to whether there could be specific substrates for each IMiD.
Regarding this point, research has begun to show that IMiDs exert favorable therapeutic effects against blood cancers other than multiple myeloma. In particular, it has been shown that lenalidomide exerts highly favorable effects against 5q− syndrome, a type of myelodysplastic syndrome [28, 29]. Ebert and colleagues who previously identified Ikaros and Aiolos reported that when lenalidomide binds to CRBN, casein kinase 1a (Ck1α) is specifically ubiquitinated and degraded [30].
In 5q− syndrome, there is a deletion in the long arm of one of the two chromosome 5, resulting in haploinsufficiency; the genetic locus coding for Ck1α is located right within this deleted region. In 5q− cells, the expression level of Ck1α is thus half of that of wild-type cells. When Ck1α was further degraded upon lenalidomide treatment, this resulted in the activation of p53 leading to the induction of apoptosis. The results are consistent with previous reports of the involvement of p53 in the Ck1α pathway [31, 32]. It was also found that the ubiquitination and degradation of Ck1α by CRBN is specific to lenalidomide, and that thalidomide and pomalidomide do not exhibit this activity. These results explain why lenalidomide is specifically effective against 5q− syndrome, and why each type of IMiD has its own specific substrate.
Structural analysis of CRBN and novel findings on its interspecies specificity
Following the discovery of CRBN, in addition to analyses on the association between CRBN and the therapeutic effects of IMiDs, extensive structural analyses were performed on complexes containing an IMiD and CRBN.
The relationship between the structure of CRBN and its activity is intriguing. We hence identified its binding region to DDB1 and thalidomide using genetic and biochemical techniques. DDB1 was found to bind to the intermediate region (187–260) of CRBN, whereas thalidomide was found to bind to the C-terminus region (339–442) [19]. The aforementioned thalidomide-binding deficient mutant of CRBN has alanine substitutions in tyrosine 384 and tryptophan 386. Furthermore, we demonstrated that the glutarimide moiety (Fig. 1) of thalidomide on its own is sufficient for its binding to CRBN. In addition, we were interested in the differences between the optical isomers of thalidomide, and found that the S enantiomer binds more strongly to CRBN than the R enantiomer.
In 2014 Thoma’s group and our group independently revealed the X-ray crystallographic structure of the complex containing CRBN, DDB1, and IMiD [33, 34]. Thoma and colleagues used chicken CRBN, whereas our group used human CRBN. Both groups concluded that IMiDs, via their glutarimide moiety, become inserted into the triple-tryptophan (tri-Trp) pocket composed of three tryptophan residues (W380, W386, and W400) located at the C-teminus of CRBN. We had previously identified the W386 region, and furthermore, this data confirmed our previous data showing that CRBN is able to bind to the glutarimide moiety of thalidomide on its own.
DDB1 was found to bind to the internal region of CRBN (amino acids 188–248), which was consistent with the region identified biochemically, as mentioned above. DDB1 contains three β-propeller structures, namely BPA, BPB, and BPC [35, 36]. CRBN binds to BPA and BPC. Known substrate receptors (such as DDB2 and SV5V) bind solely to BPC, and the region is thought to be the same as a binding region of DDB1 to CRBN. Therefore, CRBN, which also binds to BPA, is unique.
CRBN contains a CCCC-type zinc finger motif (C323, C326, C391, and C394) in the vicinity of its tri-Trp pocket, but its function and significance is yet unknown.
During our structural biological analyses, our group made an unexpected discovery. One long-standing mystery of thalidomide has been its lack of teratogenicity in rodents. Even more mysterious was that the therapeutic effects of IMiDs could hardly be detected in rodents. The homology between human and mouse CRBN is 94 %, and because they both bind to thalidomide, it was expected that solving this mystery would be difficult. However, with regard to the therapeutic effects of IMiDs, it was found that four amino acids in CRBN around its thalidomide-binding region differ between humans and rodents, and that this is responsible for the observed species specificity [34]. As mentioned above, Ck1α is degraded in humans by lenalidomide. However, it was demonstrated that because the equivalent of V388 of humans is I391 in mice, Ck1α is not degraded in mice [31]. Consistently, when mCRBN I391V was expressed in mouse cells, treatment with lenalidomide led to the degradation of Ck1α. Although there are many unresolved issues regarding the induction of substrates upon the binding of IMiDs to CRBN, it has become clear that a few amino acids in the vicinity of the tri-Trp pocket in CRBN, as well as IMiDs play crucial roles.
Concluding remarks
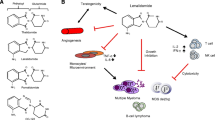
As described in the first half of this review, because IMiDs including thalidomide exert multiple actions, numerous target molecules were thought to exist. However, when we searched for target molecules using a novel affinity bead technology, only a single thalidomide-binding molecule was identified, namely, CRBN (Fig. 3). Subsequently, CRBN was found to function as a substrate receptor of an E3 ubiquitin ligase; the binding of IMiDs to CRBN enables it to bind to “neo-substrates”, leading to their ubiquitination. It was also found that upon the binding of IMiDs to CRBN, original substrates (e.g. MEIS2) of CRBN are spared from ubiquitination and degradation, and accumulate [32]. Regarding neo-substrates, Ikaros and Aiolos are ubiquitinated and degraded by thalidomide, lenalidomide, and pomalidomide, whereas Ck1α is only ubiquitinated and degraded by lenalidomide, suggesting that there may be unidentified substrates of IMiDs. Although IMiDs have a wide range of functions, rather than using different targets for each of its functions, these agents appear to bind a single target (CRBN), and by modulating the substrate specificity of CRBN in numerous ways, IMiDs are thought to exert diverse effects (Fig. 4).

Comparison between the classic model and the current model of the target of IMiDs. In the old model, IMiDs have multiple direct targets resulting in pleiotropic effects. In the current model, IMiDs basically binds CRBN, a primary direct target, and induce pleiotropic effects by breakdown of various neo-substrates by CRBN E3 ubiquitin ligase

Schematic representation of IMiDs-mediated CRBN modulation. In the absence of any IMiDs, CRBN degrades original substrates and have a certain role of physiological reactions (e.g. glutamine metabolism). Internal ligands of CRBN are possibly involved in this process. On the other hand, in the presence of an IMiD, the substrate specificity of CRBN is changed and neo-substrates are recognized and undergo subsequent protein degradation. IMiDs (Thalidomide, Lenalidomide, Pomalidomide and CC-122) induces the breakdown of Ikaros and Aiolos. Instead, original substrates (e.g. MEIS) are accumulated. Lenalidomide specifically induces the degradation of Ck1α by binding to CRBN. CC-122 might induce breakdown of its intrinsic neo-substrate(s) but these have not been identified, yet
IMiDs are now known to be effective against not only multiple myeloma and 5q− syndrome, but also diffuse large B-cell lymphoma (DLBCL), a type of non-Hodgkin lymphoma, as well as mantle cell lymphoma, and these effects are known to also depend on CRBN [37, 38]. In addition, the development of CC-122 (Fig. 1), a next-generation IMiD, is also underway [39], and clinical trials in patients with blood cancers such as DLBCL, as well as in patients with solid tumors have been initiated. Research towards the isolation and identification of the substrate(s) of CC-122 is now actively being pursued (Fig. 4). Many novel CRBN-binding compounds are also currently being investigated. One of these, CC-885, has strong anti-cancer activity and preclinical data suggest that CC-885 induces breakdown of GSPT1 (G1 to S1 phase transition 1) resulting in anti-cancer effects. This compound is now being studied for use in the treatment of acute myeloid leukemia (AML) [40].
In addition, the tri-Trp pocket of CRBN, which was revealed by X-ray crystallography, actually looks as if it binds an intracellular natural ligand (internal ligand). Recently, it was reported that uridine, a component of RNA, binds to the tri-Trp pocket of CRBN [41]. Identifying internal ligands that bind to the tri-Trp pocket of CRBN under physiological conditions will deepen our understanding of the physiological function of CRBN, and lead to further elucidation of various biological phenomena.
More recently, it was reported that glutamine induces acetylation-dependent breakdown of glutamine synthetase via CRBN. When cells are exposed to high concentrations of glutamine, glutamine synthetase is acetylated at lysines 11 and 14, resulting in degradation. That study indicated that CRBN controls glutamine metabolism and acetylation is needed for degradation of some substrates of CRBN [42].
Since our discovery of CRBN in 2010, the body of knowledge relating to this protein and associated phenomena have increased; however, a few questions remain unsolved. Firstly, the substrate of CRBN responsible for exerting the teratogenic effects of thalidomide is unknown, as is the role of CRBN in the initially identified effects of thalidomide as a sedative. Several groups are also working towards elucidation of these unresolved issues.
Understanding CRBN will ultimately lead to the establishment of methods to regulate substrate specificity of CRBN. Very recently, Bradner and colleagues designed a new chemical compound of thalidomide fused with the BRD4 (bromodomain-containing protein 4) inhibitor JQ1 [43]. BRD4 is a pathogenic target of acute myeloid leukemia [44]. Surprisingly, this chemical compound was found to enhance the degradation of BRD4 via its binding to CRBN. In addition, Crews and colleagues have established a technique called PROTAC (PROteolysis Targeting Chimera), and by developing a chemical compound of IMiDs fused with BCR-ABL, a well-known critical target of chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL), they are well on their way to designing a degradation system for BCR-ABL [45]. Although there are many uncovered issues, such as off-target protein degradation and the interference of internal ligands to therapeutic effects, the development of drugs via controlling the degradation of various proteins of interest using CRBN is currently a topic of utmost interest.
References
McBride WG. Thalidomide embryopathy. Teratology. 1977;16:79–82.
Lenz W. A short history of thalidomide embryopathy. Teratology. 1988;38:203–15.
Bartlett JB, Dredge K, Dalgleish AG. The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer. 2004;4:314–22.
Franks ME, Macpherson GR, Figg WD. Thalidomide. Lancet. 2004;363:1802–11.
Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol. 2007;39:1489–99.
Vargesson N. Thalidomide-induced limb defects: resolving a 50-year-old puzzle. BioEssays. 2009;31:1327–36.
Ito T, Ando H, Handa H. Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci. 2012;68:1569–79.
Sheskin J. Thalidomide in the treatment of lepra reactions. Clin Pharmacol Ther. 1965;6:303–6.
Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173:699–703.
D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA. 1994;91:4082–5.
Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341:1565–71.
Zeldis JB, Williams BA, Thomas SD, Elsayed ME. S.T.E.P.S.: a comprehensive program for controlling and monitoring access to thalidomide. Clin Ther. 1999;21:319–30.
Ooba N, Sato T, Watanabe H, Kubota K. Resolving a double standard for risk management of thalidomide: an evaluation of two different risk management programmes in Japan. Drug Saf. 2010;33:35–45.
Muller GW, Chen R, Huang SY, et al. Amino-substituted thalidomide analogs: potent inhibitors of TNF-alpha production. Bioorg Med Chem Lett. 1999;9:1625–30.
Sakamoto S, Hatakeyama M, Ito T, Handa H. Tools and methodologies capable of isolating and identifying a target molecule for a bioactive compound. Bioorg Med Chem. 2012;20:1990–2001.
Groisman R, Polanowska J, Kuraoka I, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–67.
Lee J, Zhou P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol Cell. 2007;26:775–80.
Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–61.
Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–50.
Escoubet-Lozach L, Lin IL, Jensen-Pergakes K, et al. Pomalidomide and lenalidomide induce p21 WAF-1 expression in both lymphoma and multiple myeloma through a LSD1-mediated epigenetic mechanism. Cancer Res. 2009;69:7347–56.
Shaffer AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454:226–31.
Lopez-Girona A, Heintel D, Zhang LH et al. Lenalidomide downregulates the cell survival factor, interferon regulatory factor-4, providing a potential mechanistic link for predicting response. Br J Haematol. 2011;154:325–36.
Zhu YX, Braggio E, Shi CX, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood. 2011;118:4771–9.
Lopez-Girona A, Mendy D, Ito T, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–35.
Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2013;343:301–5.
Lu G, Middleton RE, Sun H, et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science. 2013;343:305–9.
Gandhi AK, Kang J, Havens CG, et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN). Br J Haematol. 2014;164:811–21.
List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–65.
List A, Kurtin S, Roe DJ, et al. Efficacy of lenalidomide in myelodysplastic syndromes. N Engl J Med. 2005;352:549–57.
Kronke J, Fink EC, Hollenbach PW, et al. Lenalidomide induces ubiquitination and degradation of Ck1αlpha in del(5q) MDS. Nature. 2015;523:183–8.
Jaras M, Miller PG, Chu LP, et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J Exp Med. 2014;211:605–12.
Jadersten M, Saft L, Smith A, et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol. 2011;29:1971–9.
Fischer ES, Bohm K, Lydeard JR, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512:49–53.
Chamberlain PP, Lopez-Girona A, Miller K, et al. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21:803–9.
Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–17.
Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–3.
Yang Y, Shaffer AL, 3rd, Emre NC et al. Exploiting synthetic lethality for the therapy of ABC diffuse large B cell lymphoma. Cancer Cell. 2012;21:723–37.
Zhang LH, Kosek J, Wang M, Heise C, Schafer PH, Chopra R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br J Haematol. 2013;160:487–502.
Hagner PR, Man HW, Fontanillo C, et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood. 2015;126:779–89.
Matyskiela ME, Lu G, Ito T, et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature. 2016;. doi:10.1038/nature18611.
Hartmann MD, Boichenko I, Coles M, Zanini F, Lupas AN. Hernandez Alvarez B. Thalidomide mimics uridine binding to an aromatic cage in cereblon. J Struct Biol. 2014;188:225–32.
Nguyen TV, Lee JE, Sweredoski MJ, et al. Glutamine triggers acetylation-dependent degradation of glutamine synthetase via the thalidomide receptor cereblon. Mol Cell. 2016;61:809–20.
Winter GE, Buckley DL, Paulk J, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–81.
Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–8.
Lai AC, Toure M, Hellerschmied D, et al. Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. Angew Chem Int Ed Engl. 2016;55:807–10.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Ito, T., Handa, H. Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs. Int J Hematol 104, 293–299 (2016). https://doi.org/10.1007/s12185-016-2073-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2073-4