
Abstract
Histone deacetylases (HDACs) deacetylate the lysine residues of both histones and non-histone proteins. Histone acetylation results in a loose local chromatin structure that regulates gene-specific transcription. Non-histone proteins can also be acetylated, leading to dynamic changes in their activity and stability. For these reasons, HDAC inhibition has emerged as a potential approach for the treatment of MM. Specifically, combination treatment with HDAC inhibitors and proteasome inhibitors or immunomodulatory drugs shows remarkable anti-MM activity in both preclinical and clinical settings. However, the clinical studies using non-selective HDAC inhibitors also cause unfavorable side effects in patients, leading us to develop more isoform- and/or class–selective HDAC inhibitors to enhance tolerability without diminishing anti-MM activity, thereby improving patient outcome in MM.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a clonal plasma cell disorder derived from post-germinal center B cells [1, 2]. Progression occurs from monoclonal gammopathy of undetermined significance (MGUS) through smoldering MM to active MM and then plasma cell leukemia [3–5]. This progression is associated with multistep genetic abnormalities including chromosomal translocations, gene mutations, epigenetic dysregulations, and microenvironmental changes [1, 5]. These changes also lead to activation or dysregulation of MM-relevant intracellular signaling pathways including nuclear factor-κB (NF-κB) signaling, Janus kinase (JAK)-signal transducer and activator of transcription (STAT) signaling, and extracellular signal-regulated kinase (ERK) signaling [1, 5]. Indeed, proteasome inhibitors and immunomodulatory drugs (IMiDs) have been recognized to inhibit these multiple signaling pathways in the context of BM microenvironment and have significantly improved MM patient outcome; however, most patients eventually relapse even after the achievement of complete response [6, 7]. Therefore, further development of novel therapies is urgently needed, and epigenetic regulators represent promising novel therapeutic agents in various types of cancers including MM [8, 9].
Over the last decades, various types of epigenetic modifications in DNA and histone proteins have been reported. There are now at least 16 classes of histone modifications including acetylation, methylation, phosphorylation, ubiquitination, and sumoylation [8, 10]. Among these posttranslational modifications, acetylation marks on lysine residues of histone tails, neutralizes the positive charge of the lysine side chains, leading to chromatin decondensation and specific gene expression [10–12]. Lysine acetylation is generally mediated by the balance of activity between histone acetyltransferases (HATs) and histone deacetylases (HDACs) [13–15]. For example, activation of HATs and inhibition of HDACs can both hyperacetylate lysine residues in histones. Importantly, recent studies have revealed that HDACs target not only histones, but also non-histone proteins [16], indicating that HDAC inhibitors can hyperacetylate non-histones MM-relevant proteins including p53, Hsp90, and p65 NF-κB (RelA). However, the biologic impact of acetylation of these non-histone proteins has not yet been elucidated.
Histone and non-histone protein deacetylases have emerged as relevant therapeutic targets in cancer, including MM [8, 17]. Indeed, a large number of HDAC inhibitors have been developed and investigated in MM in both preclinical and clinical settings. Importantly, the US Food and Drug Administration (FDA) approved a non-selective HDAC inhibitor panobinostat (LBH589) to treat in relapsed/refractory MM in February 2015. In this review, we will discuss the underlying mechanisms of action of HDAC inhibitors in MM biology and future clinical implication.
HDACs and HDAC inhibitors in MM biology
HDACs
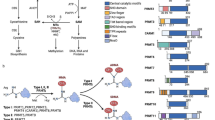
There are 18 HDAC isoforms in man which are divided into four groups class I, II, III and IV, based on homology to yeast HDACs, subcellular localization and non-cell based enzymatic activities (Fig. 1) [10, 13, 18]. Class I, II, and IV HDACs have zinc2+-dependent deacetylase domains, whereas class III HDACs has NAD+-dependent domains. Class I HDACs are HDAC1, 2, 3, and 8, which are homologous to the yeast RPD3 protein. Class II HDACs (HDAC4, 5, 6, 7, 9, and 10) share homologies with the yeast Hda1 protein. In addition, class II HDACs are divided into two subgroups; class IIa (HDAC4, 5, 7, and 9) and class IIb (HDAC6 and 10). Class IIa HDACs share an N-terminal domain distinct from other HDAC classes. Class III HDACs are sirtuins (SIRT1, 2, 3, 4, 5, 6, and 7), which are homologues of the yeast Sir2 protein [19]. Class III HDACs are different from other HDACs due to differences in their catalytic mechanism and their unrelated sequences. The only class IV HDAC is HDAC11, which shares sequence homology with the catalytic core regions of both class I and II enzymes, but does not have enough similarity otherwise to be placed in either class.

The classification of histone deacetylases (HDACs). Eighteen HDACs are divided into four classes in man. Class I, II, IV HDACs are Zn2+-dependent, whereas class III sirtuin enzymes are Zn2+-independent. The figure shows classical HDACs: class I (HDAC1, 2, 3, and 8), class IIa (HDAC4, 5, 7, 9), class IIb (HDAC6, 10), and class IV (HDAC11). Each HDAC has various structures and domains
In general, HDACs catalyze the removal of acetylation on lysine residues in target proteins (Fig. 2) [8, 10]. In the NH2-terminal tail of core histones, HDACs deacetylase acetylated lysine residues, resulting in a closed chromatin conformation associated with transcriptional repression. As described above, HDACs also deacetylate non-histone proteins including transcription factors such as tumor suppressor p53, STAT3 and NF-κB subunit RelA. Acetylation of non-histone proteins leads to changes in their function, protein–protein interaction, and protein stability [16, 17].

Lysine acetylation in both histone and non-histone proteins. Histone acetylation leads to a loose chromatin structure, resulting in gene expression. For example, treatment with HDAC inhibitors up-regulates p21 gene expression. Acetylation of non-histone proteins also modulates their function. For example, tumor suppressor p53 is degraded by proteasome via ubiquitination. Acetylation of p53 prevents its ubiquitination, resulting in accumulation of p53, followed by enhanced p53-DNA binding activity
Class II HDACs have unique characteristics in comparison with class I HDACs [20–22]. Class IIa HDACs shuttle between the cytoplasm and the nucleus, whereas class I HDACs are predominantly localized in the nucleus. The mechanism of this localization is due to phosphorylation of class IIa HDACs (Fig. 3). In the nucleus, class IIa HDACs form a complex with transcription factors, whereas in the cytoplasm they are phosphorylated in two or three conserved N-terminal serine residues and sequentially bind to 14-3-3 proteins. Class IIa HDACs mainly act as transcriptional suppressors of development and differentiation. In contrast, the localization of class IIb HDACs is the cytoplasm, unlike class I and IIa HDACs. Class IIb HDAC substrates are distinct from class I and IIa HDAC substrates. For example, HDAC6 co-localizes with the microtubule network, and possesses unique functions as a specific tubulin deacetylase [20].

Regulation of transcription by class IIa HDACs. Class IIa HDACs shuttle between the nucleus and the cytoplasm. In the nucleus, class IIa HDACs bind to transcription factors (TFs), resulting in transcriptional repression. Phosphorylation of class IIa by protein kinases such as Ca2+/calmodulin-dependent kinase family (CaMK), protein kinase D (PKD), and some members of microtubule affinity-regulating kinase (MAPK) family leads to its nuclear export. Phosphorylated-class IIa HDACs then bind to 14-3-3 proteins. Conversely, after dissociation with class IIa HDACs and 14-3-3 proteins, cellular phosphatases such as protein phosphatases PP1 and PP2A dephosphorylate class IIa HDACs, thereby promoting nuclear localization
Mechanisms of action of HDAC inhibitors in MM
HDAC inhibitors have been purified from natural sources or synthetically developed. HDAC inhibitors can be divided into six classes based on their chemical structure: short-chain fatty acid, hydroxamate, benzamide, cyclic tetrapeptide, electrophilic ketone, and miscellaneous (Table 1) [18, 23]. Although non-selective HDAC inhibitors may block a broad range of HDAC isoform activity, previous studies show that the majority of clinically relevant HDAC inhibitors target mainly HDAC 1, 2, 3, and 6, suggesting that the anti-tumor effect of non-selective HDAC inhibitors is due to class I and class IIb HDAC inhibition [24]. Among those non-selective HDAC inhibitors, panobinostat (LBH589) possesses potent HDAC inhibitory effect at nanomolar range concentrations [25]. Based on promising preclinical and clinical studies, panobinostat has already been approved to treat relapsed/refractory MM by the FDA in 2015.
HDAC inhibitors inhibit myeloma cell survival and proliferation by different mechanisms. Cancer cells including MM cells show dysregulation of cell cycle, leading to rapid cell proliferation. Treatment with non-selective HDAC inhibitors or class I HDAC inhibitors induces G0/G1 cell cycle arrest through upregulation of cell cycle regulators, p21WAF1 and/or p53 [26–30]. HDAC inhibitors can induce both apoptotic and non-apoptotic cell death. For example, vorinostat (suberanilohydroxamic acid; SAHA) does not trigger significant cleavage of caspase 8, 9, or 3 in apoptotic cells, suggesting that vorinostat triggers caspase-independent apoptosis [27]. In contrast, other studies demonstrate that HDAC inhibitor-induced apoptosis can be mediated via both the intrinsic and extrinsic pathways [26–30]. Regarding activation of the intrinsic pathway, HDAC inhibitors upregulate pro-apoptotic B cell lymphoma 2 (Bcl-2) family member proteins (Bim, Bid, Bak, Bax, Noxa, and Puma) [27, 31], whereas anti-apoptotic Bcl-2 family proteins such as Bcl-2, Bcl-xL [29], and Mcl-1 [28] are downregulated. Besides activation of caspases, vorinostat enhances expression of TNF-related apoptosis-inducing ligand (TRAIL) receptors, DR4 and DR5, enhancing their susceptibility to TRAIL-induced killing, which is associated with up-regulation of pro-apoptotic Bcl-2 family proteins [27, 31]. Vorinostat also suppresses autocrine insulin-like growth factor-1 (IGF-1) production, directly interrupting the IGF-1/IGF-1R signaling pathway critical for anti-apoptosis and survival of MM cells [32]. Interestingly, another hydroxamic acid-type HDAC inhibitor belinostat (PXD101) induces reactive oxygen species (ROS), which is blocked by the free radical scavenger N-acetyl-l-cysteine [30].
MM cells interact with other cellular components via soluble factors and direct cell–cell contact in the bone marrow (BM) microenvironment, which mediates MM cell survival, proliferation, and drug resistance. Therefore, inhibition of this interaction represents an important therapeutic strategy in MM. Importantly, vorinostat inhibits the secretion of IL-6 from BM stromal cells (BMSCs) without altering their viability, suggesting that HDAC inhibitors can overcome cell growth and anti-apoptosis in the context of BM milieu [26, 27]. HDAC inhibitors have also shown in vivo anti-MM activities in mouse xenograft models [26, 33]. Importantly, HDAC inhibitor-based combination treatments have also been studied. Specifically, HDAC inhibitors combined with conventional agents or bortezomib show remarkable anti-MM activities in preclinical settings [26, 29, 30, 32, 34].
Clinical studies of non-selective HDAC inhibitors in MM
Despite remarkable anti-MM activities as single agents in preclinical settings, panobinostat, vorinostat, or romidepsin has shown only modest clinical activity in relapsed/refractory MM [35–37]. Therefore, HDAC inhibitors have been clinically evaluated in combination with other agents, especially with proteasome inhibitors (Table 2). Among those HDAC inhibitors combined with bortezomib in clinical trials, vorinostat and panobinostat are the most extensively studied. Clinical efficacy of vorinostat has been studied in combination with bortezomib in phase I trials [38, 39]. In these studies, the maximum tolerated dose (MTD) of vorinostat was established as 400 mg with bortezomib 1.3 mg/m2. Subsequently, phase IIb and III “Vorinostat Clinical Trials in Hematologic and Solid Malignancies (VANTAGE)” trials were conducted combining vorinostat with bortezomib [40, 41]. In the phase II VANTAGE 095 trial (n = 143), 17 % overall response rate (ORR) (≥partial response: PR) and 31 % clinical benefit rate (CBR) (≥minimal response: MR) were observed. This combination therapy was generally well tolerated, with 19 % of patients discontinuing treatment due to adverse events (AEs) including thrombocytopenia and gastrointestinal toxicity. The phase III VANTAGE 088 trial has shown only a modest statistically significant difference in median progression-free survival (PFS) of 7.63 months in the bortezomib and vorinostat group vs 6.83 months in the bortezomib and placebo group. The most common grade 3/4 AEs were thrombocytopenia (45 % in the vorinostat group vs 24 % in the placebo group), neutropenia (28 vs 25 %), and anemia (17 vs 13 %).
The combination of panobinostat with bortezomib has been studied in relapsed or refractory MM patients in a phase Ib study [42]. In this clinical trial, the MTD was established at panobinostat 20 mg plus bortezomib 1.3 mg/m2. Frequent grade 3/4 AEs included thrombocytopenia, neutropenia, and asthenia. The next evaluation was panobinostat in combination with bortezomib and dexamethasone in phase II/III “Panobinostat Oral in Multiple Myeloma (PANORAMA)” trials [43, 44]. In the phase II PANORAMA 2 trial (n = 55), the ORR was 34.5 % and the CBR 52.7 %. Common grade 3/4 AEs included thrombocytopenia, fatigue, and diarrhea. In the randomized, double-blind phase III PANORAMA 1 trial (n = 768), panobinostat in combination with bortezomib and dexamethasone improved median PFS (11.99 months in panobinostat, bortezomib, and dexamethasone group vs 8.08 months in placebo, bortezomib, and dexamethasone group). Common grade 3/4 AEs included thrombocytopenia (67 % in the panobinostat group vs 31 % in the placebo group), lymphopenia (53 vs 40 %), diarrhea (26 vs 8 %), asthenia or fatigue (24 vs 12 %), and peripheral neuropathy (18 vs 15 %). The results of the phase III PANORAMA trial resulted in the FDA approval of panobinostat in combination with bortezomib and dexamethasone. Although the clinical relevance of the difference in PFS between the two groups is not clear, these were critical differences between PANORAMA trials and VANTAGE trials. First, VANTAGE trials didn’t include dexamethasone in their regimen. Second, panobinostat has more potent HDAC inhibition than vorinostat.
Clinical studies of combination treatments of panobinostat or vorinostat with the second generation proteasome inhibitor carfilzomib (Kyprolis) have been conducted with similar clinical efficacy [45, 46]. In addition, the efficacy of HDAC inhibitors has been examined in combination with other agents including IMiDs, based on promising preclinical anti-MM activity. Specifically, panobinostat has been combined with lenalidomide and dexamethasone [47], as well as with melphalan, thalidomide, and prednisone [48]. Similarly, vorinostat has also been examined in combination with lenalidomide and dexamethasone [49], as well as with pegylated liposomal doxorubicin and bortezomib [50].
Toward class- and isoform-selective HDAC inhibitors
Non-selective HDAC inhibitors induce potent cytotoxicity against MM cells in the preclinical setting; however, they also induce unfavorable side effects in clinical trials due to the broad range of modulation of histone and non-histone protein functions [41, 44]. To develop isoform-selective HDAC inhibitors to minimize these side effects, the biologic function of each HDAC isoform has been characterized. To date, HDAC6 inhibitors (i.e., tubacin, tubastatin-A, ricolinostat) are the only class of isoform-selective HDAC inhibitors whose significance in MM biology has well been documented. HDAC6 shows co-localization with the microtubule network, which mediates the transport of organelles within the cell. MM cells overloaded with unfolded/misfolded proteins which are degraded by both proteasomes and via lysosome through protein aggregates (aggresomes) (Fig. 4a). Specifically, HDAC6 binds to polyubiquitinated proteins and a motor protein dynein, and the unfolded protein-HDAC6-dyenin complex is ultimately processed by lysosomes. Therefore, HDAC6 inhibition results in blockade of aggresomal protein degradation and marked accumulation of ubiquitinated protein [51]. Importantly, the aggresome and proteasome pathways compensate for each other in terms of protein degradation. Hence, inhibition of both pathways leads to significant accumulation of unfolded proteins and induces cell stress, followed by cell death [51–53].

The significance of HDAC 4 and 6 inhibitions combined with proteasome inhibition in MM biology. a Unfolded or misfolded proteins are polyubiquitinated and then degraded not only by proteasome, but also processed through aggresome pathway. Polyubiquitinated proteins in association with HDAC6 bind to dynein motor protein. The polyubiquitinated protein-HDAC6-dynein complex moves to the aggresome along the microtubule. HDAC6 inhibition prevents the interaction with HDAC6 and dynein, resulting in accumulation of ubiquitinated proteins. Therefore, the dual inhibition of HDAC6 and the proteasome triggers marked accumulation of unfolded/midfolded proteins, followed by cell stress and death. b Accumulation of unfolded/misfolded proteins triggers endoplasmic reticulum (ER) stress. Excessive ER stress up-regulates activating transcription factor 4 (ATF4), followed by its translocation from the cytoplasm to nucleus. In the nucleus, ATF4 up-regulates C/EBP homologous protein (CHOP), leading to up-regulation of proapoptotic genes. HDAC4 interacts with ATF4 to prevent ATF4 translocation to the nucleus. Dual inhibition of HDAC4 and the proteasome, therefore, synergistically activate the ER stress-mediated cell death pathway
Class I HDACs are also an attractive therapeutic target in MM [54, 55]. Specifically, HDAC1 or 3, but not HDAC2, trigger apoptosis in MM cell lines. Indeed, a class I HDAC inhibitor romidepsin induces significant cytotoxicity, both alone and in combination with bortezomib. Similar results were observed after treatment with entinostat (MS-275) [56]. Interestingly, bortezomib downregulates the expression of HDAC1, 2, and 3 via activating caspases. Conversely, HDAC1 overexpression causes bortezomib resistance in both in vitro and in vivo models, suggesting that downregulation of HDAC1 mediates, at least in part, bortezomib-induced cytotoxicity in MM cells. A first-in-class selective inhibitor of HDAC3, BG45, has also been generated. BG45 shows potent MM cytotoxicity associated with downregulation of phosphorylation of STAT3. However, the underlying mechanism whereby HDAC3 inhibition induces anti-MM activity in vitro and in vivo in a mouse xenograft model has not totally been elucidated.
Recent studies have shown that class IIa HDACs are crucial transcriptional regulators of various cell developmental and differentiation processes. Class IIa HDACs are expressed in a tissue-specific manner and are strongly expressed in MM [57]. Among class IIa HDACs, HDAC4 is an attractive therapeutic target in MM because of its regulatory function of activating transcription factor 4 (ATF4). Specifically, excessive endoplasmic reticulum (ER) stress induces ATF4 and C/EBP homologous protein (CHOP), followed by downstream proapoptotic genes. Importantly, HDAC4 directly interacts with ATF4 to prevent its nuclear translocation, therefore, inhibiting ATF4 transcriptional activity [57]. Conversely, inhibition of HDAC4 under ER stress condition relieves blockade of ATF4 function, thereby triggering enhanced CHOP and apoptotic signaling. Since proteasome inhibitors are the most commonly used therapeutic agents in MM and are known to induce ER stress through accumulation of unfolded and ubiquitinated proteins (Fig. 4b), combining HDAC4 inhibitor with proteasome inhibitor has a strong preclinical rationale in MM. Indeed, a class IIa HDAC inhibitor TMP269 in combination with carfilzomib (Kyprolis) synergistically induces apoptosis. Recent studies also demonstrate that HDAC4 interacts and forms a complex with RelB and p52, which are major components of the alternative (non-canonical) NF-κB signaling pathway [58]. HDAC4-RelB-p52 complex represses pro-apoptotic genes Bim (Bcl-2 interacting mediator of cell death) and BMF (Bcl-2 modifying factor). Converesly, HDAC4 knockdown or 100aa HDAC4-mimetic polypeptide leads to apoptosis of MM cells via upregulation of Bim and BMF.
IMiDs, lenalidomide and pomalidomide, are also commonly used agents in MM treatment. Both IMiDs and HDAC inhibitors can target a transcription factor c-Myc, which regulates genes mediating proliferation, apoptosis, and metabolism in MM cells [59]. Specifically, IMiDs directly bind to cereblon (CRBN), an E3 ubiquitin ligase, followed by proteasomal degradation of IKZF1 and downregulation of c-Myc [60]. Importantly, the combination of HDAC6 inhibitor ricolinostat (ACY-1215) and lenalidomide induces synergistic cytotoxicity against MM cells, associated with marked downregulation of c-Myc [32, 33]; in contrast, high dose class I HDAC inhibitor entinostat (MS-275) shows antagonistic cytotoxic effects due to inhibition of CRBN expression [61]. These results suggest that selection of HDAC inhibitors and treatment schedules should be informed to enhance cytotoxicity, without downregulating CRBN expression.
An HDAC6 selective inhibitor ricolinostat has been examined as monotherapy or in combination with bortezomib and dexamethasone in a phase I/II study [62]. In this study, 15 patients and 22 patients were enrolled and treated with ricolinostat monotherapy and with ricolinostat in combination with bortezomib and dexamethasone, respectively. The combination therapy was well tolerated at doses of ricolinostat up to 160 mg/day. Grade 3/4 AEs were rare, and hematologic AEs were manageable. 40 % patients with monotherapy had stable disease (SD) as their best response; in addition, 60 % CBR (≥SD) was observed in patients with combination therapy.
Mechanism of resistance to HDAC inhibitors
Molecular mechanisms inducing resistance to HDAC inhibitors have not been fully delineated. However, understanding mechanisms of resistance are crucial to overcome the resistance with combination treatments and/or to develop next generation of HDAC inhibitors. A recent study shows that two signaling pathways, regulation of actin cytoskeleton and protein processing in ER, are associated with inherent resistance to HDAC inhibitors in MM cells [63]. Importantly, combination treatment of HDAC inhibitors with agents targeting those signaling pathways induces synergistic killing in MM cells.
The effect of HDAC inhibitors, like other chemotherapeutic agents, can be affected by drug efflux, target overexpression and desensitization, chromatin/epigenetic alterations, anti-apoptotic/pro-survival mechanisms, and stress response mechanisms [64]. Drug efflux is caused, at least in part, by overexpression of ATP binding cassette (ABC) transporter superfamily, which includes ABCB1 (MDR1 or P-glycoprotein), ABCC1 (MRP1), ABCG2 (BCRP or MXR). Importantly, romidepsin is a substrate for ABCB1 and ABCC1; however, vorinostat is not a substrate for either [65, 66]. It is also suggested that alterations in expression of HDACs and other epigenetic modulators can lead to resistance to HDAC inhibitors in various malignancies [64]. In addition, levels of expression of pro-apoptotic and anti-apoptotic Bcl-2 families are also important for sensitivity to HDAC inhibitors. Indeed, knockout of Bim or Bid can attenuate sensitivity to vorinostat [67]. In contrast, overexpression of anti-apoptotic Bcl-2 family members Bcl-2 and Bcl-xL attenuates the activity of vorinostat and valproic acid [68].
Other preclinical and clinical studies reveal that nuclear accumulation or persistent activation of STAT1 and 3 correlates with resistance to vorinostat in cutaneous T cell lymphoma (CTCL) [69]. It is also reported that the level of UV excision repair protein RAD23 homolog B (RAD23B; HR23B), which shuttles ubiquitinated cargo proteins to the proteasome, alters sensitivity to HDAC inhibitors in CTCL [70]. Gene expression profiling in phase I vorinostat trial shows 17 anti-oxidant genes in patients with advanced leukemia and myelodysplastic syndromes, suggesting that elevated anti-oxidant signature correlates with vorinostat resistance [71].
Conclusions and future directions
HDAC inhibitors induce cell cycle arrest and activate both intrinsic and extrinsic apoptotic pathways as single agents. Anti-MM activities of HDAC inhibitors can be further enhanced in combination with other agents including proteasome inhibitors and IMiDs. These preclinical observations have been rapidly translated to clinical trials. In 2015, the FDA approved panobinostat in combination with bortezomib and dexamethasone to treat patients with refractory/relapsed MM; however, the precise molecular mechanisms whereby this combination treatment induces anti-tumor activities in patients are not fully understood. Moreover, non-selective HDAC inhibitors show unfavorable side effects, which can be avoided by isoform and/or class selective HDAC inhibitors, which preserve significant anti-tumor activity and may improve patient outcome in MM.
References
Palumbo A, Anderson K. Multiple myeloma. N Engl J Med. 2011;364:1046–60.
Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538–48.
Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113:5412–7.
Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood. 2009;113:5418–22.
Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12:335–48.
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–20.
Gay F, Larocca A, Wijermans P, Cavallo F, Rossi D, Schaafsma R, et al. Complete response correlates with long-term progression-free and overall survival in elderly myeloma treated with novel agents: analysis of 1175 patients. Blood. 2011;117:3025–31.
Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–91.
Ocio EM, Richardson PG, Rajkumar SV, Palumbo A, Mateos MV, Orlowski R, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. 2014;28:525–42.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–9.
Yang XJ. Lysine acetylation and the bromodomain: a new partnership for signaling. BioEssays. 2004;26:1076–87.
Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31.
Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn’t fit all. Nat Rev Mol Cell Biol. 2007;8:284–95.
Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100.
Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40.
West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124:30–9.
Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84.
Smith BC, Hallows WC, Denu JM. Mechanisms and molecular probes of sirtuins. Chem Biol. 2008;15:1002–13.
Verdin E, Dequiedt F, Kasler HG. Class II histone deacetylases: versatile regulators. Trends Genet. 2003;19:286–93.
Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–67.
Parra M, Verdin E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr Opin Pharmacol. 2010;10:454–60.
Hideshima T, Anderson KC. Histone deacetylase inhibitors in the treatment for multiple myeloma. Int J Hematol. 2013;97:324–32.
Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–43.
Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): successes and challenges. Cancer Lett. 2009;280:233–41.
Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–22.
Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–62.
Khan SB, Maududi T, Barton K, Ayers J, Alkan S. Analysis of histone deacetylase inhibitor, depsipeptide (FR901228), effect on multiple myeloma. Br J Haematol. 2004;125:156–61.
Maiso P, Carvajal-Vergara X, Ocio EM, Lopez-Perez R, Mateo G, Gutierrez N, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–9.
Feng R, Oton A, Mapara MY, Anderson G, Belani C, Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007;139:385–97.
Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia. 2005;7:646–57.
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA. 2004;101:540–5.
Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D, et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794–803.
Sanchez E, Shen J, Steinberg J, Li M, Wang C, Bonavida B, et al. The histone deacetylase inhibitor LBH589 enhances the anti-myeloma effects of chemotherapy in vitro and in vivo. Leuk Res. 2011;35:373–9.
Niesvizky R, Ely S, Mark T, Aggarwal S, Gabrilove JL, Wright JJ, et al. Phase 2 trial of the histone deacetylase inhibitor romidepsin for the treatment of refractory multiple myeloma. Cancer. 2011;117:336–42.
Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008;49:502–7.
Wolf JL, Siegel D, Goldschmidt H, Hazell K, Bourquelot PM, Bengoudifa BR, et al. Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma. 2012;53:1820–3.
Badros A, Burger AM, Philip S, Niesvizky R, Kolla SS, Goloubeva O, et al. Phase I study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin Cancer Res. 2009;15:5250–7.
Weber DM, Graef T, Hussein M, Sobecks RM, Schiller GJ, Lupinacci L, et al. Phase I trial of vorinostat combined with bortezomib for the treatment of relapsing and/or refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2012;12:319–24.
Siegel DS, Dimopoulos MA, Yoon S-S, Laubach JP, Kaufman JL, Goldschmidt H, et al. Vantage 095: vorinostat in combination with bortezomib in salvage multiple myeloma patients: final study results of a global phase 2b trial. Blood. 2011;118:480.
Dimopoulos M, Siegel DS, Lonial S, Qi J, Hajek R, Facon T, et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (VANTAGE 088): a multicentre, randomised, double-blind study. Lancet Oncol. 2013;14:1129–40.
San-Miguel JF, Richardson PG, Gunther A, Sezer O, Siegel D, Blade J, et al. Phase Ib study of panobinostat and bortezomib in relapsed or relapsed and refractory multiple myeloma. J Clin Oncol. 2013;31:3696–703.
Richardson PG, Schlossman RL, Alsina M, Weber DM, Coutre SE, Gasparetto C, et al. PANORAMA 2: panobinostat in combination with bortezomib and dexamethasone in patients with relapsed and bortezomib-refractory myeloma. Blood. 2013;122:2331–7.
San-Miguel JF, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15:1195–206.
Berdeja JG, Hart LL, Mace JR, Arrowsmith ER, Essell JH, Owera RS, et al. Phase I/II study of the combination of panobinostat and carfilzomib in patients with relapsed/refractory multiple myeloma. Haematologica. 2015;100:670–6.
Vesole DH, Bilotti E, Richter JR, McNeill A, McBride L, Raucci L, et al. Phase I study of carfilzomib, lenalidomide, vorinostat, and dexamethasone in patients with relapsed and/or refractory multiple myeloma. Br J Haematol. 2015;171:52–9.
Chari A, Cho HJ, Leng S, Dhadwal A, Morgan G, La L, et al. A phase II study of panobinostat with lenalidomide and weekly dexamethasone in myeloma. ASH Annu Meet Abstr. 2015;126:#4226.
Offidani M, Polloni C, Cavallo F, Liberati AM, Ballanti S, Pulini S, et al. Phase II study of melphalan, thalidomide and prednisone combined with oral panobinostat in patients with relapsed/refractory multiple myeloma. Leuk Lymphoma. 2012;53:1722–7.
Siegel DS, Richardson P, Dimopoulos M, Moreau P, Mitsiades C, Weber D, et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J. 2014;4:e182.
Voorhees PM, Gasparetto C, Osman K, Richards KL, Ferraro M, Garcia R, et al. Vorinostat in combination with pegylated liposomal doxorubicin (PLD) and bortezomib (B) in patients with relapsed/refractory multiple myeloma (R/R MM): final results of a phase I study. ASH Annu Meet Abstr. 2011;118:#3985.
Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci USA. 2005;102:8567–72.
Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M, et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119:2579–89.
Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, et al. Ricolinostat (ACY-1215) induced inhibition of aggresome formation accelerates carfilzomib-induced multiple myeloma cell death. Br J Haematol. 2015;169:423–34.
Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K, et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406–17.
Minami J, Suzuki R, Mazitschek R, Gorgun G, Ghosh B, Cirstea D, et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia. 2014;28:680–9.
Hideshima T, Mazitschek R, Santo L, Mimura N, Gorgun G, Richardson PG, et al. Induction of differential apoptotic pathways in multiple myeloma cells by class-selective histone deacetylase inhibitors. Leukemia. 2014;28:457–60.
Kikuchi S, Suzuki R, Ohguchi H, Yoshida Y, Lu D, Cottini F, et al. Class IIa HDAC inhibition enhances ER stress-mediated cell death in multiple myeloma. Leukemia. 2015;29:1918–27.
Vallabhapurapu SD, Noothi SK, Pullum DA, Lawrie CH, Pallapati R, Potluri V, et al. Transcriptional repression by the HDAC4-RelB-p52 complex regulates multiple myeloma survival and growth. Nat Commun. 2015;6:8428.
Kuehl WM, Bergsagel PL. MYC addiction: a potential therapeutic target in MM. Blood. 2012;120:2351–2.
Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–35.
Hideshima T, Cottini F, Ohguchi H, Jakubikova J, Gorgun G, Mimura N, et al. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015;5:e312.
Raje N, Vogl DT, Hari PN, Jagannath S, Jones SS, Supko JG, et al. ACY-1215, a selective histone deacetylase (HDAC) 6 inhibitor: interim results of combination therapy with bortezomib in patients with multiple myeloma (MM). ASH Annu Meet Abstr. 2013;122:#759.
Mithraprabhu S, Khong T, Spencer A. Overcoming inherent resistance to histone deacetylase inhibitors in multiple myeloma cells by targeting pathways integral to the actin cytoskeleton. Cell Death Dis. 2014;5:e1134.
Fantin VR, Richon VM. Mechanisms of resistance to histone deacetylase inhibitors and their therapeutic implications. Clin Cancer Res. 2007;13:7237–42.
Xiao JJ, Foraker AB, Swaan PW, Liu S, Huang Y, Dai Z, et al. Efflux of depsipeptide FK228 (FR901228, NSC-630176) is mediated by P-glycoprotein and multidrug resistance-associated protein 1. J Pharmacol Exp Ther. 2005;313:268–76.
Ruefli AA, Bernhard D, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-glycoprotein-expressing cells. Int J Cancer. 2002;99:292–8.
Lindemann RK, Newbold A, Whitecross KF, Cluse LA, Frew AJ, Ellis L, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci USA. 2007;104:8071–6.
Whitecross KF, Alsop AE, Cluse LA, Wiegmans A, Banks KM, Coomans C, et al. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood. 2009;113:1982–91.
Fantin VR, Loboda A, Paweletz CP, Hendrickson RC, Pierce JW, Roth JA, et al. Constitutive activation of signal transducers and activators of transcription predicts vorinostat resistance in cutaneous T-cell lymphoma. Cancer Res. 2008;68:3785–94.
Fotheringham S, Epping MT, Stimson L, Khan O, Wood V, Pezzella F, et al. Genome-wide loss-of-function screen reveals an important role for the proteasome in HDAC inhibitor-induced apoptosis. Cancer Cell. 2009;15:57–66.
Garcia-Manero G, Yang H, Bueso-Ramos C, Ferrajoli A, Cortes J, Wierda WG, et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
T. Harada, T. Hideshima, or K.C. Anderson does not have conflict of interest.
About this article

Cite this article
Harada, T., Hideshima, T. & Anderson, K.C. Histone deacetylase inhibitors in multiple myeloma: from bench to bedside. Int J Hematol 104, 300–309 (2016). https://doi.org/10.1007/s12185-016-2008-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-2008-0