
Abstract
Hematopoietic stem cells (HSCs) are the most extensively studied stem cell type in adults, and the only stem cell type with proof of clinical utility. However, the greatest challenge for the broader use of HSCs remains the true expansion of the stem cells ex vivo. The development of researches on small-molecule compounds that support the safe and efficient ex vivo expansion of HSCs would help to promote the clinical application of HSCs. In recent years, several novel small-molecule compounds have been reported to improve ex vivo HSC expansion by promoting self-renewal, delaying differentiation, increasing homing, and inhibiting apoptosis. Here, we review recent chemical developments in stem cell research and the mechanisms underlying these compounds’ effects.
Similar content being viewed by others

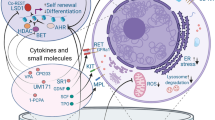
Introduction
In 1957, Dr. E. Donnall Thomas first performed the intravenous infusion of allogeneic bone marrow (later called hematopoietic stem cell transplantation, HSCT) to treat cancer [1]. It has been more than half a century since this first HSC utilization in the treatment of hematological malignant diseases. Thanks to two features of HSCs—self-renewal and multilineage differentiation—HSCs have been effectively used in the clinic. For HSCT, an HLA-match/compatible donor and a sufficient number of HSCs are crucial for the success of the procedure, avoiding high rates of rejection, graft failure, and graft versus host disease (GVHD) [2]. Allogeneic bone marrow and mobilized peripheral blood cells are the main stem cell sources for current clinical HSCT. However, HSCT use is restricted by the lack of HLA-matched donors. In recent years, umbilical cord blood (UCB) has become a new source of HSCT that allows the use of partly mismatched donors [3]. However, the low number of HSCs recovered from a single UCB unit, which is mainly used for the treatment of pediatric and young adult recipients, presents serious limitations for most adult recipient HSCT [4, 5]. Thus, ex vivo HSC expansion, which would allow one adult or even multiple transplants from a single HSC unit, has become the “Holy Grail” of stem cell expansion research [6].
The core question of ex vivo HSC expansion is how self-renewal is regulated. Recent studies have begun to shed light on the different pathways involved in this process. The self-renewal program is an integrated procedure, involving the activation and/or maintenance of cell proliferation pathways and the inhibition of differentiation and cell death pathways [7]. Gene manipulation studies have identified multiple intrinsic proteins that play important roles in HSC self-renewal regulation, including chromatin-associated factors (e.g., Bmi1 [8], Ezh2 [9], Dmnt3a, Cbx7), transcription factors (e.g., HoxB4 [10], HoxB6 [11], HoxA4 [12], HoxA9 [13], Gata2, Gfi1 [14], AML1, JunB, NF-κ), cell cycle regulators (INK4/p18 [15], WAF1/p21 [16], KIP1/p27, KIP2/p57, PTEN [17], Myc, Fbxw7), and microRNAs (miR-125a/b [18]). There are also several critical developmental regulatory pathways that have strong effects on HSC self-renewal, including wingless-type (Wnt) [19], Notch (Jagged/Delta) [20], Sonic hedgehog (Shh)/BMP [21], fibroblast growth factor (FGF) [22], IGF and Angptls [23, 24], pleiotrophin [25], and the TGFβ/smad4 signaling pathway. All the aforementioned proteins and pathways play instructive roles not only in hematopoiesis but also during ontogeny.
Previous studies first attempted to expand HSCs with hematopoietic cytokines, such as SCF, TPO, Flt3L, IL-6, IL-3, etc. [26]. However, the amplification efficiency of these cytokines remains controversial. A combination of several cytokines has also been applied to HSC expansion culture but has not yet achieved the desired expansion effect and usually causes stem cell differentiation or even exhaustion [27]. Another method is the co-culture of HSCs with stromal cells, which is intended to simulate the internal hematopoietic “niche”.
Small molecules are emerging as valuable tools for regulating stem cell fates with the advantages of easier manipulation, typically rapid and reversible effects, various and unlimited concentrations and structures, and rapid phenotype-based high-throughput screening [28–30]. Many studies have been launched to identify small molecule compounds that can control stem cells’ fates and iPS reprogramming [31]. In recent years, due to the development of computational methods, virtual design in combination with high-throughput screening has increased the potential for small molecules to manipulate cell fate and be used clinically.
Current studies attempting HSC expansion primarily focus on the following effects: promotion of self-renewal, inhibition of differentiation, inhibition of apoptosis, and promotion of homing. Here, we focused on new developments of small molecules for HSC manipulation, especially for HSC ex vivo expansion.
Increased HSC self-renewal
SR1
In 2010, an inspiring breakthrough in stem cell research was first reported by Boitano et al. [32], who showed that an aryl hydrocarbon receptor (AhR) antagonist, SR1, could promote the ex vivo expansion of human cord blood CD34+ cells with excellent repopulating activity in immunodeficient mice for up to 16 weeks. Using an unbiased screen of human cord blood primary HSCs, they identified the purine derivative SR1 and found that it could lead to a 50-fold increase in CD34+ cells and a 17-fold increase in NOD-SCID repopulating cells (SRCc), which can engraft in immunodeficient mice. Interestingly, SR1 could only affect human HSCs but not mouse HSCs which were commonly used as substitutes for human cells in biological assays. This discrepancy may indicate that some AhR-related pathways are different between human and mice.
SR1 was shown to increase the number of human cord blood CD34+ cells by directly binding to and inhibiting AhR. AhR is a ligand-activated transcription factor involved in the induction of drug-metabolizing enzymes. Several pathways, including HES-1, c-Myc, C/EBP, PU.1, β-catenin, CXCR4, and Stat5, have been reported to play important roles in regulating hematopoiesis and are thought to be related to AhR [33].
UM171
Through high-throughput screening of a chemical library, Fares et al. first discovered a pyrimidoindole derivative, UM171, which could significantly expand human cord blood HSCs ex vivo with the help of their newly developed automated and continuous medium delivery system, which is called a fed-batch culture [34]. Human CD34+CD45RA− mobilized peripheral blood (mPB) cells cultured in the presence of UM171 for 7 days significantly increased the number of CD34+38−90+45RA−49f+ cells over 100-fold compared with the control group. This expansion effect of UM171 could still be amplified when combined with SR1. Furthermore, the frequencies of LT-HSCs were 13-fold higher in the UM171-treated group compared with DMSO or uncultured controls. After treatment with UM171, these LT-HSCs remained capable of multi-lineage differentiation and long-term reconstitution. However, SR1 appeared to compromise the proliferative potential of lymphomyeloid LT-HSCs.
RNA sequencing (RNA-seq) expression profiling experiments were conducted to explore the mechanism of UM171. The most up-regulated genes in the UM171-treated group were genes encoding surface molecules such as TMEM183A and PROCR (also known as EPCR or CD201). The specific mechanism requires further study (see Table 1).
P18IN003 and P18IN011
The cell cycle is a critical point of stem cell renewal. The cell fate is largely determined in the G1 phase in most mammalian cells. Therefore, targeting the cell cycle is a new strategy for HSC manipulation. Yuan et al. [15] revealed that p18, a member of the cyclin-dependent kinase (CDK) inhibitor (CKI) INK4 family, could regulate the G1 phase cell cycle checkpoint by inhibiting CDK4/6, which makes it an ideal target for HSC ex vivo expansion. Recently, we combined computational virtual screening methods and biological validation to identify two p18 small molecular inhibitors, P18IN003 and P18IN011, which significantly increased the frequency of primitive hematopoietic cells in ex vivo culture [35]. More cobblestone areas (CAs) were generated from p18+/+ mouse bone marrow cells after 5–8 weeks culture with P18IN011 than with P18IN003 compared with DMSO control, whereas no significant difference was found with the p18−/− mouse bone marrow cells under the same conditions. Immunodeficient mouse transplantation also demonstrated that treatment with both P18IN003 and P18IN011 significantly increased the long-term repopulating ability of HSCs. Moreover, P18IN011 has a much stronger expansion effect than P18IN003. Further structure optimization for human HSC ex vivo expansion is needed in the future.
CHIR99021 + Rapamycin
CHIR99021 is an inhibitor of glycogen synthase kinase-3β (GSK3β) that increases the expression of β-galactosidase in phenotypic HSCs, activates the Wnt pathway, and supports ESC self-renewal [36] and mouse MEF cell reprogramming [37]. GSK3β inhibition would subsequently activate mTOR, which would trigger lineage commitment and HSC depletion [38]. Studies have shown that hematopoietic cytokine combinations could establish lineage commitment but could not simulate the internal “niche” that could support LT-HSCs. Huang et al. [39] recently reported a new culture method that employed cytokine-free conditions to maintain HSCs ex vivo via the combined regulation of the Wnt and mTOR pathways. They cultured mouse c-Kit+ or Lin− Sca1+ c-Kit+ (LSK) cells and human UCB CD34+ cells in serum-free, cytokine-free culture medium in the presence of the GSK-3β inhibitor CHIR99021 and the mTOR inhibitor Rapamycin (C + R) for 7 days. Functional HSCs were identified by competitive and serial transplantation into immunodeficient mice. The results showed that C + R could maintain functional LT-HSCs during cytokine-free ex vivo culture, and the chimerism of the C + R treatment group remained at a higher level in secondary recipients after 4 months compared with the control group.
Cell cycle assays revealed that C + R treatment increased the cell population in the S/G2/M phase, which indicated that C + R might expand HSCs by promoting cell cycle progression without leading to cell apoptosis or death. Knock-down mouse studies have shown that Ctnn1b is required for the C + R effect of HSC maintenance. However, further mechanistic studies are needed.
BIO
BIO is another inhibitor of GSK3β that could result in the accumulation of cells enriched with primitive progenitors with sustained reconstitution capacity. Ko et al. [40] showed that BIO could stimulate the accumulation of the slowly dividing human cord blood CD34+ cells through delayed cell cycle progression and demonstrated significantly increased engraftment and chimerism levels of the NOD/SCID repopulating cells.
Gene expression analysis suggested that this effect of BIO might be caused by the up-regulation of p57 and the down-regulation of cyclin D1. Although BIO treatment can lead to the accumulation of β-catenin, no other β-catenin target gene levels were changed. Other genes related to stem cell function were modulated by BIO treatment, including ADAMDEC1, ANGPT2, RARRES2, and HMOX.
NR-101
Thrombopoietin (TPO) is regularly applied in megakaryocyte differentiation, platelet production, and in the HSC culture medium [41]. The TPO receptor myeloproliferative leukemia virus protooncogene (c-MPL) is not only expressed in the megakaryocytic lineage but also in HSCs [42]. Previous studies showed that c-MPL plays an important role in HSC maintenance and can activate three major pathways: Janus kinase (JAK)/signal transducer and activator of transcription (STAT), Ras/mitogen-activated protein kinase (MAPK), and phosphatidylinositol-3-kinase (PI3K)/AKT [43, 44]. Nishino et al. [45] reported a novel nonpeptidyl small-molecule c-MPL agonist, NR-101, which could increase the number of CD34+CD38− hematopoietic cells and SRCs more efficiently than TPO in ex vivo culture.
HSC ex vivo culture typically results in differentiation, senescence, and eventual exhaustion [46]. NR-101 gradually increased the proportion of G0/G1 cells and activated c-MPL, which kept the expression of c-MPL and its downstream pathway proteins at high levels. There were two theories about the NR-101 mechanism. First, it could activate STAT5, which is closely related to HSC self-renewal. The second theory is that NR-101 stabilizes hypoxia-inducible factor-1α (HIF-1α), which would regulate the oxygen level in the cellular environment and the response to hypoxia and would then up-regulate the expression of VEGF. The enhanced expression of VEGF and the induction of hypoxic responses could be the mechanism by which NR-101 elicits HSC expansion.
5azaD/TSA
In the adult body, HSCs usually remain quiescent to retain their stem cell characteristics. Thus, how to activate HSCs and force them into cell division is a main challenge in expansion research. Araki et al. [47] attempted to reverse the HSC silencing status by targeting their genetic program. They chose two chromatin modifying agents, 5-aza-2-deoxycytidine (5azaD) and trichostatin A (TSA), and achieved a 3-fold increase in the CD34+CD38− cell population during expansion in the presence of either of the compounds. Nine days of ex vivo culture of human CD34+90+ cells led to much slower cell division rate and cell cycling compared with the control group. Analyzing the number of colony-forming units (CFUs-Mix) and the cobblestone area forming cells (CAFCs) also showed that 5azaD/TSA-treated CD34+90+ cells maintain much greater hematopoietic potential even while undergoing extensive cell division compared to the control group with the similar rounds of cell division. Those slowly dividing 5azaD/TSA-treated human CD34+90+ cells showed significantly greater human chimerism when injected to NOD-SCID mice compared with the control group.
Real-time quantitative PCR showed that some self-renewal related genes, such as HOXB4, Bim-1, GATA2, and cell cycle regulating genes, such as p21 and p27, were up-regulated, whereas the cell proliferation related gene C-MYC was down-regulated after 5azaD/TSA treatment. The exact roles of these genes in maintaining the self-renewal of stem cells and the effects of the chromatin modifying agents still need to be fully elucidated.
GAR
Natural products are always excellent sources for drug discovery. Nishino et al. [48] screened 92 biologically active natural products and identified a compound, garcinol (GAR), which is a plant-derived histone acetyltransferase (HAT) inhibitor; GAR expanded human hematopoietic progenitors (CD34+38− cells) by 4.5-fold in 7 days of ex vivo cytokine-supplemented culture compared with a control group. Moreover, isogarcinol (ISO), a derivative of GAR, was found to have much greater expansion effect on HSPCs. Although there was no significant difference in the number of total colonies, GAR and ISO generated more high proliferative potential colony-forming cells (HPP-CFCs) compared with the control group, which suggested that GAR or ISO treatment could yield more functional HSPCs to reconstitute normal hematology. A NOD/SCID-repopulation assay was then performed to further confirm the cultured cells’ hematopoietic function in an in vivo assay and calculate the frequency of SRCs. The frequency of SRCs in GAR treated CD34+ cells was 2.5-fold higher than that of fresh CD34+ cells and 2.2-fold higher than that of the control group. These results demonstrated that GAR and its derivative, ISO, could indeed facilitate HSPC expansion without losing the stem cells’ characteristics during ex vivo culture.
GAR was thought to be a non-specific HAT inhibitor. Consistent with this understanding, the level of cellular histone acetylation was significantly reduced in GAR-treated human cord blood CD34+ cells. Global transcriptional assays showed that GAR-treated HSPCs displayed up-regulated AMICA1, BTG2, and HLF and down-regulated IL8, PF4, and PPBP. However, some classical pathways such as HOXB4 and NOTCH1, which were reported to be involved in stem cell self-renewal, were not changed in GAR-treated HSPCs. Further mechanistic study is required.
TEPA
Metal ions, such as iron, calcium, copper, magnesium, and zinc, are thought to have critical effects on proliferation, differentiation, and other cellular function regulation. Reports have shown that copper (Cu) deficiency can lead to serious clinical manifestations, such as hematopoietic differentiation [49]. The up-take of Cu in HL-60 cells was thought to be related to their early-stage differentiation [50]. Peled et al. [51, 52] reported that the addition of a Cu chelator, tetraethylenepentamine (TEPA), could decrease the Cu pool and then promote the expansion of human CD34+38− cells compared with a control group. The short-term culture of human HSCs with TEPA could enhance the reconstitution capacity of these cells in NOD-SCID mice. Next, they synthesized a stable complex of TEPA:Cu (1:1), which showed a much more significant expansion effect on human CD34+38− cells and could increase the number of CFUs compared with the control group [53].
The Cu chelator TEPA, also known as StemEx, has been applied into a phase I/II clinical trial [54]. Although with a relatively low total cell number per kilogram infused in this study (mean 1.7 × 107 per kilogram), 9 out of 10 patients were successfully engrafted, and 9 were alive at day 100 post-transplantation. Moreover, no patients were reported to develop grades 3 or 4 acute GVHD. These data suggested a potential method of HSC ex vivo expansion utilizing chemical approaches. Further comprehensive studies and larger clinical trials are still necessary to establish whether the expanded HSCs have an effect on successful engraftment and patient survival.
Inhibition of lineage commitment differentiation
VPA
Valproic acid (VPA), a highly specific and potent HDAC inhibitor, has been used as a neurologic agent in the treatment of epilepsy, bipolar disorder, migraine headaches, and some other severe neurologic disorders for decades [55]. Recently, Felice et al. [56] reported the application of VPA onto normal human HSCs to explore its effect on the proliferation, differentiation, and survival of stem cells. After 7 days of ex vivo culture, the lower cell proliferation and higher number of G0/G1 phase VPA-treated cells showed that VPA could prolong the cell cycle duration without leading to cell death. Additionally, the increasing percentage of CD34+, CD34+90+, and CD34+38− populations and the higher number of colonies in VPA-treated cells compared with the control group revealed that VPA could maintain the hematopoietic potential of cultured cells in the short term. In long-term culture (3 weeks), VPA significantly slowed the decrease of CD34+ cells and preserved the primary HSC population (CD34+90+ and CD34+38− cells).
Mechanistic studies showed that the expression of KDR, AC133, c-kitR, GATA1, and HOXB4 (strong increase) was increased after VPA treatment. VPA especially increased the level of histone acetylation at AC133 and HOXB4 sites. However, the effects of VPA on long-term reconstitution potential require further exploration using animal models.
DEAB
Aldehyde dehydrogenase (ALDH), which plays a critical role in the conversion of retinol (vitamin A) to retinoic acids, is highly expressed in HSCs, which makes it an ideal marker for HSCs, especially human HSCs. Studies have revealed that ALDH is essential for cellular differentiation; thus, it has been applied in clinical therapy to induce the differentiation of acute promyelocytic leukemia cells. Based on these results, ALDH inhibition might be another method of maintaining HSC self-renewal. Chute et al. [57] reported that an inhibitor of ALDH, diethylaminobenzaldehyde (DEAB), significantly delayed the differentiation of HSCs and increased the percentage of human cord blood CD34+CD38− cells and created a fivefold increase in CFCs after 7 days ex vivo culture. Limiting dilution repopulation assays showed that the SRC frequency of DEAB-treated human CB lin−CD34+CD38− cells was 1 in 2200 cells, 3.4-fold higher than that of an uncultured group and 7.7-fold higher than that of a control group, which suggested that DEAB not only inhibited HSC differentiation but also promoted HSC expansion in ex vivo culture.
Mechanistic analysis indicated that ALDH might lead to HSC differentiation through intracellular retinoid production, and the addition of DEAB, as well as other RAR agonists, RXR agonists, or vitamin D could obviously maintain CD34+ cells ex vivo, which meant that ALDH could regulate HSC fate through these three ligands. Furthermore, the inhibition of ALDH subsequently up-regulated the expression of HOXB4 in HSCs. However, the interaction of these signaling pathways has yet to be elucidated.
Increased HSC homing in vivo
NAM
Another HDAC inhibitor, nicotinamide (NAM), is a well-established potent inhibitor of the sirtuin family of the NAD-dependent class III histone deacetylase (HDAC) [58]. It has been reported that hematopoietic cells derived from SIRT1-deficient mice show increased proliferation activity in ex vivo culture [59]. Peled et al. [60] reported that nicotinamide, a form of vitamin B-3 regarded as a precursor of nicotinamide adenine dinucleotide (NAD), significantly increased the percentage and absolute number of CD34+38− cells at relatively low concentrations (2.5 and 5 mM) in short-term culture. In long-term culture (3 weeks), NAM showed not only an increase in the number of CD34+ cells that displayed an early progenitor cell phenotype but also the number of CFUs, which indicated the preservation of their potential for long-term expansion. Then, they assessed the short-term SCID reconstituting capacity of NAM-treated cells, and the results showed a ninefold increase of SRC frequency compared with uncultured cells.
Cultured HSCs usually suffer from homing defects resulting from a sustained increase in adhesion receptor expression and a decrease in CXCR4 leading to the nonspecific binding of cultured cells to extramedullary endothelial surfaces [61].
PGE2
Prostaglandin E2 (PGE2), one of the most abundant eicosanoids and a mediator of numerous physiological systems [62], was reported to enhance the frequency of murine HSCs that showed a competitive reconstitution advantage during serial transplantation [63]. Previously, PGE2 was reported to stimulate the proliferation, cycling, and differentiation of quiescent bone marrow cells [64]. Hoggatt et al. [65] revealed that short-term dmPGE2 culture could improve the stable long-term enhancement of human HSCs upon serial transplantation. Twelve weeks after transplantation, human chimerism analysis of mPB showed a significant fourfold increase in the dmPGE2-treated group compared with a control group, which was used as a quantitative measure of long-term repopulating capacity. Additionally, PGE2 pulse-culture significantly enhanced the homing efficiency of human UCB CD34+ cells and mouse HSCs.
The expansion effect mediated by PGE2 was thought to be associated with an increasing level of CXCR4 mRNA, decreased apoptosis, and the elevated expression of Survivin. A twofold increase in HSC cell-cycle activity in the PGE2 group indicated that another possible mechanism might be an increasing proportion of LT-HSCs that were entering into and progressing through the cell cycle. These results suggested that homing might be critical for HSC expansion.
Inhibition of HSC apoptosis
zVADfmk/zLLYfmk
HSC ex vivo culture typically causes several cellular defects, such as stem cell exhaustion, the down-regulation of homing properties, clonogenicity reduction, and apoptosis induction [66]. In previous studies, apoptosis has been a major cause of stem cell exhaustion and eventual cell death [67]. Two cysteine proteases, caspase and calpain, have been shown to play important roles in programmed cell death [68]. Sangeetha et al. [69] found a combination of two caspase and calpain inhibitors, zVADfmk and zLLYfmk, which could significantly increase the cell yield and especially the CD34+ population. Functional HSC validation showed that cells treated with these compounds retained a higher number of long-term repopulating units compared to the control group, with an approximately threefold increase in the number of GM colonies and increased engraftment of myeloid (CD33), megakaryocytic (CD41a), and lymphoid (CD19) populations in the bone marrow.
Apoptosis was obviously reduced in the zVADfmk/zLLYfmk-treated cells. The mechanism of zVADfmk/zLLYfmk is mainly related to three aspects. First, increased bcl-2 expression is functionally relevant because bcl-2 has been reported to increase stem cell phenotype and functionality [70]. Second, the activity of Caspase 3, a cell apoptosis factor, is always decreased as the CD34+ cells number increases. Third, after zVADfmk/zLLYfmk treatment, the Notch signaling pathways is activated, which may also be the mechanism of HSC expansion.
5-HT
Serotonin, or 5-hydroxytryptamine (5-HT), is a monoamine neurotransmitter in the gastrointestinal tract and central nervous system that can regulate internal biological activities [71]. 5-HT is enzymatically metabolized to 5-hydroxyindole acetic acid and finally excreted from the body through the urine, whereas the extracellular level of serotonin is regulated by a sodium chloride-dependent membrane protein transporter (5-HTT, SERT) [72, 73]. Yang et al. [74] reported that 5-HT treatment increased the expansion of not only the total nuclear cells but also selectively expanded CD34+CD38− cells in 8 days of culture. Additionally, treatment with 5-HT significantly enhanced the formation of colony-forming unit-fibroblasts (CFU-Fs), CFU-GEMM, etc., which suggested that 5-HT could actually increase the hematopoietic potential of HSCs. Human chimerism was detected 6 weeks after 5-HT-treated cells were injected into sub-lethally irradiated NOD/SCID mice. The engraftment of the 5-HT group was significantly increased compared with the control group.
Mechanistic studies showed that 5-HT might expand HSCs through the antiapoptotic effects mediated by mitochondrial signaling, caspase activation, and the Akt/ERK1/2 pathways. The specific mechanisms of serotonin in hematopoiesis need further investigation.
Conclusion
The ex vivo expansion of UCB-derived HSCs is a sought-after technique to improve HSC transplantation therapy. However, HSCs are prone to differentiate during ex vivo culture. Small-molecule compounds are increasingly common as tools for HSC expansion. As we can see from the previously described studies, there are many types of small-molecule compounds that could increase the proportion of CD34+ cells and their subsets ex vivo and improve the transplantation level in animal models. Small molecule compounds demonstrate the potential for HSC expansion and hold great promise for making a difference in this field. Moreover, small-molecule compounds primarily affect some important signaling pathways, such as Wnt, PI3 K/AKT, Notch, etc. Strategies for regulating these key signaling pathways are important for efficient approaches to HSC expansion ex vivo.
Among all those key signaling pathways, Wnt signaling pathway is supposed to be an extremely critical one due to its highly conserved and vital in mammal development [75]. Several small molecule compounds involved in Wnt signaling pathway were continually discovered to efficiently expand HSC ex vivo, such as CHIR99021, BIO, etc. Also, small molecule Wnt regulators were found to play roles in other stem cells, like induced pluripotent stem cell. Given the various effects of small molecule Wnt regulators, we considered that small molecules targeting Wnt signaling pathway appeared promising strategies to expand HSC ex vivo. There have already been some studies involving compound combinations such as the aforementioned CHIR99021 + Rapamycin and the chemical combination of C6FZ (CHIR99021, 616452, Forskolin and DZNep) to trigger the complete reprogramming of mouse somatic cells [76]. Combination therapy can not only enhance the expansion effects of compounds but also avoid the toxic effects of these compounds. The combination of multiple pathways may become the next hotspot in stem cell expansion research.
Almost in all current expansion culture methods, cytokines are thought be indispensable in order to imitate the internal hematopoietic niche. Chemical substitutions of those cytokines were considered as potent HSC expansion strategies as well. As mentioned before, NR-101 is an ideal alternative option for TPO and even showed superior effects on HSC expansion. Therefore, further studies focused on cytokine substitutions should be addressed to achieve total chemical replacement of cytokines.
So far, chemical strategy applied into UCB-derived HSC ex vivo expansion has its successes in many clinical trials. Several small molecule compounds have already completed phase I/II and are now in phase II/III multi-center clinical trials, such as SR1 (AhR antagonist), TEPA (copper chelate), Nicord (SIRT1 inhibitor) and so on [77]. Inspiring hematopoietic reconstitution recovery and reduced graft-versus-host-disease (GVHD) were achieved in even some late-phase clinical trials. However, the expensive procedure of handling umbilical cord blood HSC ex vivo is still a major challenge in future allogeneic stem cell transplantation.
It is undeniable that even though these small-molecule compounds are considered valid, they still have yet to be approved for clinical use. The primary reason is the lack of safety associated with small molecule compounds. Small molecule compounds may cause unexpected or even unpredictable side effects when used for HSC expansion ex vivo. A detailed evaluation of the safety of small-molecule compounds, especially the confirmation that they have no carcinogenicity, is a key step in the preclinical stages of any new potential therapy. Meanwhile, the mechanism of these compounds is still not clear. Thus, experiments that can verify these compounds’ safety and effectiveness are also required. Taken together, we hope to obtain a series of small-molecule compounds that can expand human HSCs ex vivo and eventually be used clinically.
References
Thomas ED, Lochte HL Jr, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Engl J Med. 1957;257:491–6.
Krenger W, Hollander G. The role of the thymus in allogeneic hematopoietic stem cell transplantation. Swiss Med Wkly. 2010;140:w13051.
Barker JN. Umbilical cord blood (UCB) transplantation: an alternative to the use of unrelated volunteer donors? ASH Educ Program Book. 2007;2007:55–61.
Gluckman E, Ruggeri A, Volt F, Cunha R, Boudjedir K, Rocha V. Milestones in umbilical cord blood transplantation. Br J Haematol. 2011;154:441–7.
Wagner JE, Rosenthal J, Sweetman R, Shu XO, Davies SM, Ramsay N, et al. Successful transplantation of HLA-matched and HLA-mismatched umbilical cord blood from unrelated donors: analysis of engraftment and acute graft-versus-host disease. Blood. 1996;88:795–802.
Takizawa H, Schanz U, Manz MG. Ex vivo expansion of hematopoietic stem cells: mission accomplished. Swiss Med Wkly. 2011;141:w13316.
Walasek MA, van Os R, de Haan G. Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci. 2012;1266:138–50.
Rizo A, Dontje B, Vellenga E, de Haan G, Schuringa JJ. Long-term maintenance of human hematopoietic stem/progenitor cells by expression of BMI1. Blood. 2008;111:2621–30.
Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, et al. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006;107:2170–9.
Antonchuk J, Sauvageau G, Humphries RK. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109:39–45.
Fischbach NA, Rozenfeld S, Shen W, Fong S, Chrobak D, Ginzinger D, et al. HOXB6 overexpression in murine bone marrow immortalizes a myelomonocytic precursor in vitro and causes hematopoietic stem cell expansion and acute myeloid leukemia in vivo. Blood. 2005;105:1456–66.
Fournier M, Lebert-Ghali C-É, Krosl G, Bijl JJ. HOXA4 induces expansion of hematopoietic stem cells in vitro and confers enhancement of pro-B-cells in vivo. Stem Cells Dev. 2011;21:133–42.
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, et al. Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood. 2002;99:121–9.
Hock H, Hamblen MJ, Rooke HM, Schindler JW, Saleque S, Fujiwara Y, et al. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 2004;431:1002–7.
Yuan Y, Shen H, Franklin DS, Scadden DT, Cheng T. In vivo self-renewing divisions of haematopoietic stem cells are increased in the absence of the early G1-phase inhibitor, p18INK4C. Nat Cell Biol. 2004;6:436–42.
Cheng T, Rodrigues N, Shen H, Y-g Yang, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–8.
Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–22.
Guo S, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC, et al. MicroRNA miR-125a controls hematopoietic stem cell number. Proc Natl Acad Sci. 2010;107:14229–34.
Luis TC, Naber BA, Roozen PP, Brugman MH, de Haas EF, Ghazvini M, et al. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell. 2011;9:345–56.
Varnum-Finney B, Purton LE, Yu M, Brashem-Stein C, Flowers D, Staats S, et al. The Notch ligand, Jagged-1, influences the development of primitive hematopoietic precursor cells. Blood. 1998;91:4084–91.
Bhardwaj G, Murdoch B, Wu D, Baker D, Williams K, Chadwick K, et al. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat Immunol. 2001;2:172–80.
Yeoh JS, van Os R, Weersing E, Ausema A, Dontje B, Vellenga E, et al. Fibroblast growth factor-1 and-2 preserve long-term repopulating ability of hematopoietic stem cells in serum-free cultures. Stem Cells. 2006;24:1564–72.
Zhang CC, Lodish HF. Insulin-like growth factor 2 expressed in a novel fetal liver cell population is a growth factor for hematopoietic stem cells. Blood. 2004;103:2513–21.
Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C, et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006;12:240–5.
Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16:475–82.
Sauvageau G, Iscove NN, Humphries RK. In vitro and in vivo expansion of hematopoietic stem cells. Oncogene. 2004;23:7223–32.
Goff JP, Shields DS, Greenberger JS. Influence of cytokines on the growth kinetics and immunophenotype of daughter cells resulting from the first division of single CD34 + Thy-1 + lin− cells. Blood. 1998;92:4098–107.
Li W, Jiang K, Ding S. Concise review: a chemical approach to control cell fate and function. Stem Cells. 2012;30:61–8.
Xu Y, Shi Y, Ding S. A chemical approach to stem-cell biology and regenerative medicine. Nature. 2008;453:338–44.
Schugar R, Robbins P, Deasy B. Small molecules in stem cell self-renewal and differentiation. Gene Ther. 2008;15:126–35.
Li W, Ding S. Small molecules that modulate embryonic stem cell fate and somatic cell reprogramming. Trends Pharmacol Sci. 2010;31:36–45.
Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–8.
Singh KP, Casado FL, Opanashuk LA, Gasiewicz TA. The aryl hydrocarbon receptor has a normal function in the regulation of hematopoietic and other stem/progenitor cell populations. Biochem Pharmacol. 2009;77:577–87.
Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, et al. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014;345:1509–12.
Gao Y, Yang P, Shen H, Yu H, Song X, Zhang L, et al. Small-molecule inhibitors targeting INK4 protein p18INK4C enhance ex vivo expansion of haematopoietic stem cells. Nature Commun. 2015;6:6328.
Ying Q-L, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, et al. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–23.
Li W, Zhou H, Abujarour R, Zhu S, Young Joo J, Lin T, et al. Generation of human-induced pluripotent stem cells in the absence of exogenous Sox2. Stem Cells. 2009;27:2992–3000.
Chen C, Liu Y, Liu R, Ikenoue T, Guan K-L, Liu Y, et al. TSC–mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J Exp Med. 2008;205:2397–408.
Huang J, Nguyen-McCarty M, Hexner EO, Danet-Desnoyers G, Klein PS. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012;18:1778–85.
Ko KH, Holmes T, Palladinetti P, Song E, Nordon R, O’Brien TA, et al. GSK-3β inhibition promotes engraftment of ex vivo-expanded hematopoietic stem cells and modulates gene expression. Stem Cells. 2011;29:108–18.
Kaushansky K, Lok S, Holly RD, Broudy VC, Lin N, Bailey MC, et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–571.
Kaushansky K. Thrombopoietin: accumulating evidence for an important biological effect on the hematopoietic stem cell. Ann N Y Acad Sci. 2003;996:39–43.
Ezumi Y, Takayama H, Okuma M. Thrombopoietin, c-Mpl ligand, induces tyrosine phosphorylation of Tyk2, JAK2, and STAT3, and enhances agonists-induced aggregation in platelets in vitro. FEBS Lett. 1995;374:48–52.
Rojnuckarin P, Drachman JG, Kaushansky K. Thrombopoietin-induced activation of the mitogen-activated protein kinase (MAPK) pathway in normal megakaryocytes: role in endomitosis. Blood. 1999;94:1273–82.
Nishino T, Miyaji K, Ishiwata N, Arai K, Yui M, Asai Y, et al. Ex vivo expansion of human hematopoietic stem cells by a small-molecule agonist of c-MPL. Exp Hematol. 2009;37(1364–1377):e1364.
Wagner W, Ansorge A, Wirkner U, Eckstein V, Schwager C, Blake J, et al. Molecular evidence for stem cell function of the slow-dividing fraction among human hematopoietic progenitor cells by genome-wide analysis. Blood. 2004;104:675–86.
Araki H, Yoshinaga K, Boccuni P, Zhao Y, Hoffman R, Mahmud N. Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood. 2007;109:3570–8.
Nishino T, Wang C, Mochizuki-Kashio M, Osawa M, Nakauchi H, Iwama A. Ex vivo expansion of human hematopoietic stem cells by garcinol, a potent inhibitor of histone acetyltransferase. PLoS One. 2011;6:e24298.
Laughlin MJ, Eapen M, Rubinstein P, Wagner JE, Zhang M-J, Champlin RE, et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N Engl J Med. 2004;351:2265–75.
Gluckman E, Rocha V, Boyer-Chammard A, Locatelli F, Arcese W, Pasquini R, et al. Outcome of cord-blood transplantation from related and unrelated donors. N Engl J Med. 1997;337:373–81.
Peled T, Mandel J, Goudsmid R, Landor C, Hasson N, Harati D, et al. Pre-clinical development of cord blood-derived progenitor cell graft expanded ex vivo with cytokines and the polyamine copper chelator tetraethylenepentamine. Cytotherapy. 2004;6:344–55.
Peled T, Landau E, Mandel J, Glukhman E, Goudsmid NR, Nagler A, et al. Linear polyamine copper chelator tetraethylenepentamine augments long-term ex vivo expansion of cord blood-derived CD34 + cells and increases their engraftment potential in NOD/SCID mice. Exp Hematol. 2004;32:547–55.
Peled T, Glukhman E, Hasson N, Adi S, Assor H, Yudin D, et al. Chelatable cellular copper modulates differentiation and self-renewal of cord blood–derived hematopoietic progenitor cells. Exp Hematol. 2005;33:1092–100.
De Lima M, McMannis J, Gee A, Komanduri K, Couriel D, Andersson B, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transpl. 2008;41:771–8.
Göttlicher M, Minucci S, Zhu P, Krämer OH, Schimpf A, Giavara S, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001;20:6969–78.
De Felice L, Tatarelli C, Mascolo MG, Gregorj C, Agostini F, Fiorini R, et al. Histone deacetylase inhibitor valproic acid enhances the cytokine-induced expansion of human hematopoietic stem cells. Cancer Res. 2005;65:1505–13.
Chute JP, Muramoto GG, Whitesides J, Colvin M, Safi R, Chao NJ, et al. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc Natl Acad Sci. 2006;103:11707–12.
Denu JM, Vitamin B. 3 and sirtuin function. Trends Biochem Sci. 2005;30:479–83.
Narala SR, Allsopp RC, Wells TB, Zhang G, Prasad P, Coussens MJ, et al. SIRT1 acts as a nutrient-sensitive growth suppressor and its loss is associated with increased AMPK and telomerase activity. Mol Biol Cell. 2008;19:1210–9.
Peled T, Shoham H, Aschengrau D, Yackoubov D, Frei G, Rosenheimer N, et al. Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Exp Hematol. 2012;40(342–355):e341.
Denning-Kendall P, Singha S, Bradley B, Hows J. Cytokine expansion culture of cord blood CD34 + cells induces marked and sustained changes in adhesion receptor and CXCR4 expressions. Stem Cells. 2003;21:61–70.
Miller SB. Prostaglandins in health and disease: an overview. Seminars in arthritis and rheumatism. 2006;36(1):37–49.
North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447:1007–11.
Pelus L. Association between colony forming units-granulocyte macrophage expression of Ia-like (HLA-DR) antigen and control of granulocyte and macrophage production. A new role for prostaglandin E. J Clin Investig. 1982;70:568.
Hoggatt J, Singh P, Sampath J, Pelus LM. Prostaglandin E2 enhances hematopoietic stem cell homing, survival, and proliferation. Blood. 2009;113:5444–55.
Hofmeister C, Zhang J, Knight K, Le P, Stiff P. Ex vivo expansion of umbilical cord blood stem cells for transplantation: growing knowledge from the hematopoietic niche. Bone Marrow Transpl. 2007;39:11–23.
Domen J, Cheshier SH, Weissman IL. The role of apoptosis in the regulation of hematopoietic stem cells Overexpression of Bcl-2 increases both their number and repopulation potential. J Exp Med. 2000;191:253–64.
Neumar RW, Xu YA, Gada H, Guttmann RP, Siman R. Cross-talk between calpain and caspase proteolytic systems during neuronal apoptosis. J Biol Chem. 2003;278:14162–7.
Sangeetha V, Kale VP, Limaye LS. Expansion of cord blood CD34 cells in presence of zVADfmk and zLLYfmk improved their in vitro functionality and in vivo engraftment in NOD/SCID mouse. PLoS One. 2010;5:e12221.
Ogilvy S, Metcalf D, Bath ML, Harris AW, Adams JM. Constitutive Bcl-2 expression throughout the hematopoietic compartment affects multiple lineages and enhances progenitor cell survival. Proc Natl Acad Sci. 1999;96:14943–8.
Fanburg BL, Lee S-L. A new role for an old molecule: serotonin as a mitogen. Am J Physiol Lung Cell Mol Physiol. 1997;272:L795–806.
Udenfriend S, Weissbach H. Turnover of 5-hydroxytryptamine (serotonin) in tissues. Exp Biol Med. 1958;97:748–51.
Murphy DL, Lerner A, Rudnick G, Lesch K-P. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Mol Interv. 2004;4:109.
Yang M, Li K, Ng PC, Chuen CKY, Lau TK, Cheng YS, et al. Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34 + stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells. 2007;25:1800–6.
Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–77.
Hou P, Li Y, Zhang X, Liu C, Guan J, Li H, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–4.
Horwitz ME, Frassoni F. Improving the outcome of umbilical cord blood transplantation through ex vivo expansion or graft manipulation. Cytotherapy. 2015;17:730–8.
Acknowledgments
This work was funded by the National Basic Research Program of China (NO. 2012CB966600), the National Natural Science Foundation of China (NSFC 81421002, 81570100, 81500086, 81430004 and 81170465), and the key technologies R & D program of Tianjin (13RCGFSF19500).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
About this article

Cite this article
Zhang, Y., Gao, Y. Novel chemical attempts at ex vivo hematopoietic stem cell expansion. Int J Hematol 103, 519–529 (2016). https://doi.org/10.1007/s12185-016-1962-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-016-1962-x