
Abstract
Histone-modifying enzymes have recently been shown to play a central role in the regulation of both normal and malignant hematopoiesis. Post-translational modifications of histones and non-histone proteins underlies a regulatory complexity affecting numerous processes including transcriptional regulation, RNA processing and DNA damage response. Insights into the functions of these enzymes as well as their role in the epigenetic alterations found in leukemia will guide the development of novel therapeutic approaches. This review discusses examples of the proteins that have been implicated in the pathogenesis of leukemia, that may serve as potential therapeutic targets.
Similar content being viewed by others

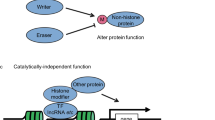
Histone-modifying enzymes catalyze the addition or removal of covalent, post-translational modifications (PTMs) in histone and non-histone proteins. These modifications include methylation, acetylation, phosphorylation, ubiquitination and sumoylation, and can regulate protein function by altering the protein’s enzymatic activity, localization within the cell, and protein–protein interactions. Histone-modifying enzymes can be classified as “writers” which add PTMs or “erasers”, proteins that can remove or alter the presence of specific PTM on histones. Another level of regulation is provided by “readers” of chromatin structure, those proteins whose domains recognize specific histone residues, generally based on the presence or absence of specific PTMs. In addition to histones, these enzymes have a broader range of substrates and as such, can regulate numerous cellular processes, including gene expression, RNA processing and the DNA damage response (DDR).
Accumulating evidence has shown that these histone-modifying enzymes play an important role in regulating virtually all aspects of hematopoiesis. Furthermore, many of these “writers, readers, or erasers” have been shown to be abnormally regulated in cancer. The epigenetic landscape is clearly altered in acute leukemia, due to a variety of acquired lesions in chromatin modifier genes, or changes in their level of expression. This provides the rationale for exploring how these abnormalities can be targeted by new therapeutic approaches. In this review, we will provide several examples of how these proteins have been implicated in the aberrant biology that characterizes malignant hematopoiesis and some suggestions as to which proteins could potentially be targeted.
Protein methylation
There are two families of histone methyltransferases, the lysine methyltransferases (PKMTs) and the protein arginine methyltransferases (PRMTs). The side chain of lysine residues can be mono-, di- or tri- methylated, while the nitrogens in arginine residues can be monomethylated or symmetrically or asymmetrically dimethylated. Unlike acetylation or phosphorylation, methylation does not change the overall charge of the molecule; however, the bulkiness of the methyl group can either promote or inhibit protein–protein interactions. These methyl marks are recognized by specific binding motifs, which include the Tudor domains, chromo domains, malignant brain tumor (MBT) domains and PHD fingers; proteins containing these motifs can distinguish the target residue (lysine vs. arginine) as well as the state of methylation. Crystal structures now exist for many of these interactions, which highlight their specificity. Recognition of combinations of PTMs dictates their output in terms of gene expression and cell behavior. Histone lysine methylation can be reversed by demethylases, which are grouped into 2 classes: (1) the amine-oxidase type lysine-specific demethylases (LSD1 and LSD2), and (2) the Jumonji C (JmJC)-domain containing histone demethylases. Not all histone lysine methyl marks appear to be susceptible to rapid reversibility, and for arginine methylation, the reversibility has not been clearly established. Rather, arginine methylation can be further chemically converted into citrulline (by the protein arginine demethylase PAD4). Such additional modifications can add further complexity to this dynamic process.
The regulation of histone methylation has been shown to be important in numerous hematopoietic processes. Alteration of a number of proteins involved in the methylation of histone and non-histone substrates has now been reported in leukemia and many other cancers [18, 81].
Protein lysine methyltransferases
The two families of protein lysine methyltransferases are characterized by the presence or absence of a SET domain: The SET domain-containing PKMTs include MLL, EZH2, NSD1 and SET7/9 (G9a) that methylate numerous substrates including histone H3 (K4, K9, K27 and K36) and H4 (K20), as well as a number of non-histone proteins, such as p53, TAF10, E2F, STAT3 and NFκB. The PKMTs that lack a SET domain include hDOT1L, a PKMT that methylates histone H3K79. Histone lysine methylation is intimately involved in gene regulation, influencing chromatin structure, a key element of the transcriptional status of a gene. Thus, H3K9me3 and H3K27me3 are typically associated with heterochromatin and gene repression, while H3K4me, H3K36me and H3K79me are associated with transcriptionally active regions, which are primarily located in the regions of euchromatin.
Histone methyltransferases are also components of large, multi-protein nuclear complexes that contain other histone-modifying enzymes and other regulatory proteins including histone acetyltransferases (HATs), histone deacetylases (HDACs), DNA methyltransferases (DNMTs) and SWI/SNF complex components. The complex nature of these interactions ensures the appropriate regulation of transcription during the execution of multiple differentiation programs that are required for normal hematopoiesis. Furthermore, methylation of key transcription factors can alter their function, and profoundly influence the expression of their target genes. Impairment at any step can promote the process of malignant transformation.
MLL
The mixed lineage leukemia (MLL) gene encodes a PKMT that is the mammalian homolog of the Drosophila trithorax (Trx) gene. The methyltransferase (SET) domain of MLL is involved in methylating H3K4, a mark usually associated with gene activation. Chromosomal rearrangements involving the MLL gene, which is located at 11q23, are seen in both AML and ALL. MLL is fused to more than 50 different partner genes. These distinct fusions are associated with unique clinical characteristics and often a poor outcome [34]. Many of these MLL fusions result in loss of the SET domain, although the fusion proteins often retain their DNA-binding domain and can positively regulate MLL target genes, including the Hox genes, a class of proteins critical for the regulation of differentiation and self-renewal. In many cases, the MLL fusion partner brings gain-of-functions, for example, the MLL-AF10 fusion, where AF10 recruits hDOT1L, a H3K79 PKMT. H3K79 methylation is generally associated with high level expression of MLL target genes such as HoxA9, which promote leukemic cell transformation [52]. Other MLL fusion partners also interact with hDOT1L including AF9 [82], AF4 [7], and ENL [34, 79], among others. In the Okuda study, the expression of an enzymatically dead form of DOT1L, or knock down of DOT1L using siRNA, abrogated the leukemia promoting activity of MLL fusions.
Several therapeutic approaches have been taken to target MLL-induced leukemia [35], and given the role of DOT1L in MLL driven leukemia, efforts to target hDOT1L have been prioritized [6, 16]. Bernt et al. [6] provided direct evidence for the essential function of DOT1L in MLL-driven leukemia, as they found a significant reduction in the in vivo transformation of dot1L −/− cells by MLL-AF9. In the study by Daigle et al., the authors identified a highly potent and selective inhibitor of DOT1L, EPZ004777 that selectively killed MLL-driven leukemic cells with minimum effect on non-MLL-rearranged cells. EPZ004777 also significantly reduced the growth of subcutaneously injected MV4-11 (MLL-AF4+) cells in tumor bearing mice, suggesting that small molecule inhibitors of DOT1L may be useful in the treatment of MLL-induced acute leukemia.
EZH2
The maintenance of gene activation promoted by the TrxG proteins is counteracted by the activity of the polycomb (PcG) proteins, which maintain gene repression. EZH2 is a catalytic component of the PcG repressive complex (PRC2), which mediates the trimethylation of H3K27 (H3K27me3) [43]. The H3K27me3 mark serves as the docking site for the Polycomb proteins, such as PC3, that are contained within PRC1, promoting the silencing of repressed target genes. EZH2 mutations are found in several hematologic malignancies, with loss-of-function mutations identified in patients with myelodysplastic syndromes (MDSs), myeloproliferative neoplasms (MPNs) [20, 49] and T-acute lymphoblastic leukemia [51, 80]. Patients with these mutations appear to have a poorer than average prognosis, and these events identify EZH2 as a tumor suppressor protein. In contrast, gain-of-function mutations in EZH2, at a single tyrosine residue (Tyr 641) in the SET domain, are found in large B-cell lymphoma patients [46], supporting the notion that EZH2 can also function as an oncogene, consistent with prior reports that EZH2 is overexpressed in breast cancer and prostate cancer [13]. Furthermore, two recent studies demonstrated a role for EZH2 in promoting MLL-AF9 driven leukemia [48, 66]. Given the potential opposing roles for EZH2 in these disorders, it will be important to assess which malignancies are dependent on EZH2 function for their maintenance.
NSD1
Another PKMT involved in AML is NSD1 (nuclear receptor-binding SET domain protein 1), which is fused to NUP98 by the cryptic t(5;11) translocation, which is seen in childhood AML [9] and adult AML [31, 67], and generally confers a poor prognosis. NSD1 methylates H3K36, which is generally an activation mark, and the NUP98–NSD1 fusion protein retains the PHD fingers and the SET domain from NSD1. Target genes of NUP98–NSD1 include the HOX and MEIS1 genes, which are normally repressed by H3K27 methylation. De-regulation of the target genes of NSD1 presumably leads to the transformation of myeloid progenitor cells and the development of AML [73].
Readers of methylation: PHD fingers
An example of a methyl lysine “reader” that is altered in cancer is the NUP98–JARID1A chimeric protein [74]. This fusion protein contains the PHD-containing domain of JARID1A, and Wang et al. demonstrated that the PHD domains of either the NUP98–JARID1A or NUP98–PHF23 chimeric proteins were essential for their ability to induce leukemia in several model systems. This effect seems to be induced by the sustained expression of several Hox genes, and also Meis1, Gata3 and Pbx1. The PHD domain in these fusion proteins recognizes the H3K4me3 mark, and when mutated there is no activation of these target genes, nor a leukemia phenotype. As these two translocations were identified in patients with AML [59, 71], this work implicates both the writers and the readers of activating histone marks in cellular transformation. These studies also support attempts to target these interactions in novel therapies for acute leukemia.
Protein lysine demethylases
Until the discovery of LSD1 (lysine-specific histone demethylase-1) and the JmJC-domain-containing histone demethylases, histone methyl marks were regarded as part of the permanent “epigenetic” signature [63]. However, the ability of demethylases to remove methyl groups from multiple histone substrates converts this mark from a stable form to a dynamic one. Recently, defining the role of these demethylases in normal and malignant hematopoiesis has triggered great scientific interest.
LSD1 (KDM1A)
LSD1/KDM1A was the first demethylase to be identified [62]. It was shown to specifically demethylate mono- and di-methyl lysine in an amine oxidation reaction that uses flavin adenine dinucleotide (FAD) as a cofactor. Targets of LSD1 include H3K4 and H3K9, and its effects on these two critical substrates give it a central role in transcriptional regulation. LSD1 is highly expressed in AML [5, 39], suggesting that it could function as an oncogene, thereby representing a potential therapeutic target. Several recent studies identified a role for LSD1 in acute leukemia, as well as suggesting that LSD1 inhibitors could be useful in its treatment [29, 39, 61]. Using two different LSD1 inhibitors (tranylcypromine—TCP and a biguanide polyamine analog), Shenk et al. showed that inhibiting LSD1 activity promoted ATRA-driven differentiation of non-APL leukemic cells. Treatment of primary AML samples with ATRA and an LSD1 inhibitor (but not ATRA or the LSD1 inhibitor alone) ex vivo diminished the potential of these cells to cause leukemia in a NOD–SCID mouse model. Harris et al. analyzed 23 MLL rearranged leukemias and found a strong correlation between LSD1 expression and clonogenic or leukemia stem cell like features. Knock down of LSD1 expression in MLL-AF9 + AML reduced the expression of MLL-AF9 target genes, which was coupled to an increase in the H3K4me2/H3K4me3 ratio and a loss of leukemic potential. The effectiveness of two TCP analog inhibitors of LSD1 was shown using both murine leukemia models, and primary human AML patient samples.
UTX (KDM6A)
UTX (Ubiquitously transcribed tetratricopeptide repeat, X chromosome) belongs to the Jumonji family of demethylases, which demethylate mono-, di- and tri-methyl lysine via a Fe2+ and α-ketoglutarate-dependent oxidative and radical attacking reaction [63]. Both UTX and Jumonji D3 (JMJD3) function as H3K27 demethylases, and both can regulate the expression of Hox genes [2]. Inactivating mutations in the UTX gene were identified in AML, CML, multiple myeloma and ALL [41, 70], implicating it in (normal and) malignant hematopoiesis. A function of UTX in leukemia was recently reported [37], as knock down of UTX expression in human and murine leukemic cells impaired their clonogenic potential and decreased their proliferation. Despite this insight, further work is needed to explore the normal functions of UTX, and determine how its demethylase activity contributes to the malignant phenotype.
IDH
While not itself a chromatin reader, writer or eraser, the recent link between metabolic processes and epigenetics has been cemented by the discovery of mutations in the isocitrate dehydrogenase (IDH) 1 and 2 enzymes in brain tumors [54] and subsequently in AML [3, 42]. These mutations generate a neomorphic enzymatic activity, which converts α-ketoglutarate (αKG) to 2-hydroxyglutarate (2-HG), the first identified oncometabolite. Reduction of αKG levels impairs the function of enzymes that require it as cofactor; these include the TET family of methylcytosine hydroxymethylases, and the JmJc-demethylases. Moreover, high levels of 2-HG competitively inhibit the catalytic activity of these dioxygenases [76]. IDH1/2 mutations were first connected to aberrant TET2 regulation in AML [22], but recently an effect of these mutations on demethylases was shown [40]. This work suggests that inhibitors of the mutant IDH1/2 enzymes could be useful in treating cancer and that blocking the effects of or production of 2-HG could similarly have a positive therapeutic effect.
Protein arginine methyltransferases
Protein arginine methyltransferases (PRMTs) catalyze the addition of one or two methyl groups from S-adenosylmethionine to the guanidino nitrogen atom of arginine (R), resulting in either mono or di-methylation, and based on their activity the PRMTs are classified into two groups: type I PRMTs, which include PRMT 1, 3, 4, 6 and 8 and catalyze the formation of asymmetric dimethylarginine, and type II PRMTs, which include PRMT 5, 7 and 11 and catalyze the formation of symmetric dimethylarginine. To date, ten mammalian PRMTs have been identified, but only eight of them have been shown to be enzymatically active. PRMTs are widely expressed and they often methylate within glycine and arginine-rich (GAR) motifs in substrates (except for CARM1/PRMT4). The cellular substrates of PRMTs include histones, and non-histone proteins such as transcription factors, heterogeneous nuclear ribonucleoproteins (hnRNP), proteins involved in DNA damage repair (Mre11), and signal transducers (e.g. growth factor receptors and their downstream effector molecules). Similarly, arginine methylation is implicated in regulation of many cellular processes including signal transduction, DNA repair, RNA processing, transcription regulation, translation and apoptosis [4]. Arginine methylation can potentially be modified or removed by protein arginine demethylating enzymes, which include peptidylarginine deiminase 4 (PAD4) and possibly Jumonji D6 (JMJD6) [12, 68].
Several studies suggest a role for PRMTs in leukemia. PRMT1 has been shown to be a critical component of the MLL-oncogenic transcriptional complex [14]. Direct fusion of MLL with PRMT1 enhanced the self-renewal capacity of primary hematopoietic cells, while knockdown of PRMT1 suppresses MLL-mediated transformation. Furthermore, during myeloid differentiation, PRMT1 methylates RUNX1 (aka AML1) within an RTAMR motif, which abrogates SIN3A binding, promoting the expression of two RUNX1 target genes, CD41 and PU.1, in primary human hematopoietic CD34+ cells [83]. This site of methylation is lost when RUNX1 is fused to ETO in t(8;21) AML. However, PRMT1 also interacts with and methylates the RUNX1/AML1–ETO fusion protein, which promotes its effects as an activator of transcription and promoter of cell proliferation [64].
PRMTs may also play a role in cancer development by regulating p53 function, and the expression or function of other tumor suppressor genes as well. p53 can be arginine methylated by PRMT5, and this may trigger p53-dependent G1 arrest, rather than apoptosis, in response to DNA damage due to specific changes in the expression of p53 target genes that are specifically required for each process[32]. A role for PRMT5 in the hematological malignancies was recently highlighted by two research groups: We showed that PRMT5 (aka JAK binding protein 1, JBP1) is a substrate of the mutant, constitutively active JAK2 kinases, such as JAK2V617F, the most common JAK2 mutation found in MPN. Phosphorylation of PRMT5 by JAK2 inhibits its enzymatic activity toward histone H4R3, and knockdown of PRMT5 promoted the erythroid differentiation of the human primary CD34+ cells [36]. This suggests that PRMT5 may block differentiation and serve as an oncogene.
In B cell lymphomas, Aggarwal et al. [1] showed that a constitutively active cyclin D kinase can phosphorylate MEP50—a regulatory factor of PRMT5 and enhance the PRMT5/MEP50 complex activity. Thus, several epigenetic enzymes appear to be oppositely affected by mutations in leukemia versus lymphoma. This likely reflects differences in their pathogenesis.
PRMT6 has been shown to antagonize the ability of MLL to methylate H3K4 [27], suggesting a potential involvement of PRMT6 in regulating hematopoietic processes. Few studies have examined the involvement of PRMTs in normal and malignant hematopoiesis; however, these histone-modifying enzymes may also contribute to the pathogenesis of human leukemia in additional ways.
Histone acetyltransferases: the writers
Lysine acetylation involves the transfer of an acetyl group from acetyl-CoA to lysine residues, to form ε-N-acetyl lysine. The lysine acetyltransferases (KAT), or histone acetyltransferases (HAT), are called writers, while the histone deacetylases (HDAC) are the erasers; the “readers” of ε-N-acetyl lysine containing motifs are the bromodomains, an evolutionary conserved, protein-interaction module. Histone acetylation is associated with a more accessible chromatin state; acetylation of lysine neutralizes its positive charge, thereby diminishing its interaction with (negatively charged) DNA. Less compact chromatin state, i.e. euchromatin, is more accessible to transcription factor binding and is generally associated with gene transcriptional activation.
Seventeen histone acetyltransferases have been identified in humans thus far; they are members of five different families of proteins: the Gcn5-related N-acetyltransferases (GNATs), the MYST acetyltransferases (MOZ, Ybf2/Sas3, Sas2, Tip60), p300 and CBP, the transcription factor TAF250, and the nuclear hormones SRC1 and SRC3. Mutations in the HATs have been identified in B-cell lymphoma and relapsed acute lymphoblastic leukemia and aberrant acetylation of cancer-related genes observed in hematological malignancies [47, 55]. Bromodomain proteins have recently been described as new therapeutic targets in acute myeloid leukemia [85]. We will focus on the MYST and p300/CBP proteins, in this review, for the role they play in hematological malignancies.
p300/CBP
The p300/CBP (CREB Binding Protein) family of HATs generally contains three cysteine-histidine rich regions, a bromodomain, HAT domain and PHD motif. p300 and CBP are distinct proteins that are usually associated with gene activation [78] and are referred to as co-activators. They interact with aspects of the basal transcriptional machinery to induce a more open chromatin conformation as a result of the acetylation of lysines within the histone tails (K12 and K15 in H2B, K14 and K18 in histone H3, and K5 and K8 in histone H4). Regulation of transcription can also be achieved through the acetylation of non-histone proteins.
Presumably inactivating mutations in the human CREBBP and EP300 genes have been identified in non-Hodgkin lymphoma, both in follicular lymphoma (FL; ~41 % of cases) and diffuse large B-cell lymphoma (DLBCL; in ~39 % of cases); these mutations primarily affect the histone acetyltransferase (HAT) domain. These missense mutations prevent p300 or CBP from acetylating the BCL6 transcriptional repressor, a PTM that diminishes its function, and from acetylating pro-apoptotic p53 molecule, a PTM that promotes its function. As a consequence, BCL6 becomes constitutively active, repressing p53 function in the germinal centers [57]; p53 activity is further reduced by the lack of its acetylation. Restoring the acetylation of BCL6 and p53, using HDAC inhibitors, may represent a promising therapy for p300/CBP mutant B-cell non-Hodgkin lymphoma [55].
In another study, mutations in the CBP gene were found in 18 % of relapsed acute lymphoblastic leukemia (ALL) patients. These mutations generally affect the HAT domain, reducing CBP’s acetyltransferase activity, which leads to impaired histone acetylation (H3K18) and diminished expression of CREB target genes, such as the glucocorticoid receptor responsive genes. Current ALL therapy includes administration of the glucocorticoid dexamethasone, and ALL cell lines harboring CBP mutations were resistant to treatment with dexamethasone. The class I/II HDAC inhibitor vorinostat was able to increase the sensitivity of these cells to dexamethasone, suggesting that HDAC inhibitor treatment may be useful for some patients with ALL [47].
Among the non-histone substrates of p300, we recently showed that acetylation of the AML1–ETO oncogene plays a crucial role in its ability to trigger the development of AML. AML1–ETO and p300 physically and functionally interact, leading to the acetylation of AML1–ETO on K24 and K43, an event that helps activate the expression of the AML1–ETO target genes involved in self-renewal. Mutation of lysine 43, but not K24, abrogates the ability of AML1–ETO to induce leukemia in vivo. Promising results were achieved after the ex vivo treatment of AML1–ETO9a, a AML1-ETO spliced variant, but not MLL–AF9, driven leukemia cells with the p300 inhibitor Lys-CoA-Tat before the cells were injected into recipient mice. It appears that the acetylation of K43 provides a docking platform for bromodomain containing proteins, which can help promote gene activation by AML1–ETO [75]. These data suggest that targeting p300 or the docking of proteins to K43 acetylated AML1–ETO could be a promising mechanism for developing novel therapeutics for some patients with AML.
MYST
The MYST family of proteins may account for as much as 30 % of all histone acetylation, making them critical for the appropriate remodeling of chromatin. The monocytic leukemia zinc finger (MOZ) protein contains two PHD domains and a HAT domain. MOZ can bind AML1 and leukemia mouse models demonstrate that Moz plays a critical role in hematopoiesis, with lack of Moz down-regulating the level of c-Mpl (a target of AML1), HoxA9 and c-Kit. Moz null mice die on embryonic day 15 from pancytopenia [33], suggesting a role for Moz in hematopoietic stem cell (HSC) maintenance. The HAT activity of Moz is required for the maintenance of HSC integrity, as mice lacking the HAT function of Moz have fewer HSCs and progenitor cells [56]. Additionally, the MOZ gene is involved in several chromosomal translocations in leukemia. The first identified translocation fused the MOZ PHD motifs and HAT domain with the entire CBP protein, leading to aberrant histone acetylation, and cell transformation [8, 15]. Another fusion protein, MOZ–TIF2, contains the zinc finger motif of MOZ, which allows it to bind nucleosomes at regulatory loci, and the CID domain of TIF2, which allow it to recruit CBP. It is this recruitment, and the HAT activity of CBP, that is thought to be at the heart of the aberrant activation of cancer-related genes seen in these cells [19]. The same mechanism of transforming activity of the MOZ–p300 fusion protein, found in two patients with AML, M5 (acute monocytic leukemia) has been postulated. These fusion proteins may have double the normal level of acetyltransferase activity [11].
TIP60
The TIP60 (Tat interacting protein 60 kDa) HAT also contains a chromodomain that is able to recognize H3K9me3. TIP60 represents the catalytic subunit of the mammalian homolog of the yeast NuA4 acetyltransferase complex. Recent evidence supports a role of TIP60 in normal hematopoiesis and in myeloproliferative neoplasms [30]. TIP60 levels are reduced in AML and lymphoma patient samples, supporting its role as a tumor suppressor [25]. TIP60 has been found to interact with c-Myb and to repress its ability to activate its target genes, such as c-Myc in murine models. The leukemogenic role of both c-Myb and c-Myc has been assessed by many investigators over the years [69]. Tip60 can induce the DDR; lack of TIP60 impairs the DDR induced by c-Myc likely affecting tumor progression. TIP60 also interacts with TEL (ETV6), a frequent fusion partner in leukemia patients, leading to repression of TEL transcriptional target genes. Further studies are needed to understand the role of TIP60 in benign hematopoiesis and in the myeloproliferative neoplasms [50, 58] (Table 1).
The bromodomains: the readers
The bromodomain (BRD) is the only protein module that can recognize and bind ε-N-acetyl lysines within histones. It is widely conserved among species, taking its name from the Drosophila Brahma protein structural domain [65]. So far 61 bromodomain containing proteins have been identified, which either have HAT activity or are factors involved in chromatin remodeling. Bromodomains are composed of 110 amino acids that are folded in a left-handed bundle of four alpha helices, with an external hydrophobic pocket that reads the acetylation mark. Flanking marks, such as acetylation or phosphorylation, can modulate the binding of bromodomains to combinations of PTMs present on ε-N-acetyl lysines and other histone marks [72].
Targeting of the epigenetic machinery has recently gained favor [23] and the bromodomains are undoubtedly attractive targets. They have a “druggable” functional pocket and they powerfully translate the histone marks they recognize by recruiting transcriptional coactivators.
Thus far, the best example of targeting a bromodomain in cancer has come from studies of BRD4, a member of the bromodomain and extra-terminal (BET) family of proteins, that is involved in a translocation in the rare NUT midline carcinoma tumor [85]. BRD4 has also been implicated in promoting the growth of MLL-AF9 driven AML [85], based on a shRNA library screen. A crucial role for Brd4 in the survival and proliferation of MLL-AF9+, NrasG12D + murine leukemic cells was demonstrated. In an important advance, recipient mice transplanted with these cells were found to have a survival advantage if treated with the BET protein inhibitor JQ1 [23]. Interestingly, leukemia cells are more sensitive to JQ1 than are the normal cells, suggesting some specificity of the drug for proliferating or malignant cells. JQ1 represents the first drug therapy to target a chromatin reader; it is a membrane soluble molecule that binds the hydrophobic pocket that recognizes acetylated lysines. Inhibition of Brd4 rapidly reduces the expression of c-Myc, and its target genes, inducing differentiation and slowing cell proliferation.
Additional studies have also demonstrated efficacy of JQ1 in the treatment of hematological malignancies. JQ1 induces the death of B-ALL cell lines, downregulating c-Myc and its target genes and the expression of IL-7R. IL-7R and CRLF2 form heterodimers, which promote cell survival via triggering of JAK/STAT5 signaling. Given the reduction in IL-7R expression, it is interesting that the most sensitive leukemia cell lines were those carrying CRLF2 rearrangements; this suggests that JQ1 could be efficacious in IL-7R-dependent diseases [53] (Table 2).
Histone deacetylases: the erasers
Histone deacetylases (HDAC) are grouped into three distinct families of proteins. Class I HDACs are widely expressed and they include HDAC1, 2, 3 and 8. Class II HDACs include HDAC 4, 5, 6, 7, 9a, 9b, and 10; they are expressed in a cell-specific manner. Class III HDACs are Sir2/Hst homologs; they are called Sirtuins (1, 2, 3, 4, 5, 6, 7) and their structure and enzymatic mechanism is totally different from class I and class II HDACs. They are NAD+ dependent and they are not inhibited by class I or class II inhibitors. HDAC proteins counteract HAT activity; they are associated with transcriptional repression, forming macromolecular complexes with corepressor molecules, such as N-COR, but also transcription factors and nuclear receptors that sit on gene regulatory elements [24]. Histone acetylation is deregulated in transformed cells, resulting in downregulation of tumor suppressor gene expression (due to promoter hypoacetylation) and oncogene over-expression (due to histone hyperacetylation at similar regulatory regions) [44].
HDAC1 has been implicated in the overexpression of the mutant c-KIT found in a subset of acute myeloid leukemias. HDAC inhibitors, particularly of HDAC class I and II, are currently under study for the treatment of AML thanks of their powerful ability to negatively affect cell proliferation and induce apoptosis [38] (Table 3; Fig. 1).

Histone modifications and the interplay between “writers”, “erasers” and “readers”. a Post-translational modifications (PTMs) of histone tails. Core histone is constituted of two of each H2A (histone 2A), H2B (histone 2B), H3 (histone 3) and H4 (histone 4). The histone tails are targeted by various PTMs including: acetylation (ac), methylation (momomethylation: me1, dimethylation: me2 and trimethylation: me3), phosphorylation (P) and ubiquitination (Ubi) at specific amino acids. b Examples of the interplay between “writers”, “erasers” and “readers”. Lysine 4 in histone 3—H3K4 is mono-, di-, and tri- methylated by “writer” MLL methyltransferases. The PHD domain in “reader” proteins recognizes the trimethyl-lysine mark. The marks can also be removed sequentially by “erasers” JARID1d (from tri- to di-methyl) and by LSD1 (removing di- and mono-methyl marks). Various lysine residues in both H3 and H4 are acetylated by p300. The bromodomain in “reader” proteins recognizes the lysine acetylation mark. The removal of the acetyl mark is mediated by “eraser” HDACs
Histone phosphorylation
Phosphorylation of serine/threonine residues in histones has been studied for all histones, especially H3S10. More recently, histone tyrosine phosphorylation has been found, for the mutant JAK2 kinase [17]. Mutant and wild type JAK2 is found not only in the cytoplasm of hematopoietic cells, but also in the nucleus, where it phosphorylates histone H3 at tyrosine 41 (H3Y41). Phosphorylation of H3Y41 prevents the binding of HP1α, thus reduces the recruitment of this repressive mark to regulatory regions of target genes, which include the LMO2 gene, resulting in an abnormal upregulation of LMO2 expression. This link between JAK2 and LMO2 expression and between signaling pathways and chromatin represents another instance of how aberrant signaling pathways can affect chromatin structure and further reinforce aberrant patterns of gene regulation. How to take advantage of this knowledge therapeutically is the challenge for the coming years.
Summary
Recent studies have found overexpression of or mutations in a number of epigenetic modifying enzymes, including those impacting histones, and those that impact on DNA itself. Learning how these abnormalities contribute to the malignant process will help guide our strategies to target the epigenetic abnormalities that characterize cancer, as these may be much more amenable to changes than the fixed genetic abnormalities. Properly designed clinical trials will help advance our knowledge of how to introduce and then combine these types of therapies.
References
Aggarwal P, Vaites LP, Kim JK, Mellert H, Gurung B, Nakagawa H, Herlyn M, Hua X, Rustgi AK, McMahon SB, et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell. 2010;18:329–40.
Agger K, Cloos PAC, Christensen J, Pasini D, Rose S, Rappsilber J, Issaeva I, Canaani E, Salcini AE, Helin K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–4.
Andersson AK, Miller DW, Lynch JA, Lemoff AS, Cai Z, Pounds SB, Radtke I, Yan B, Schuetz JD, Rubnitz JE, et al. IDH1 and IDH2 mutations in pediatric acute leukemia. Leukemia. 2011;25:1570–7.
Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13.
Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Al-Khalili Szigyarto C, Persson A, Ottosson J, Wernérus H, Nilsson P, et al. A genecentric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008;7:2019–27.
Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78.
Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106.
Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dubé I, Frischauf AM, Horsman D, Mitelman F, Volinia S, Watmore AE, Housman DE. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat Genet. 1996;14:8.
Cerveira N, Correia C, Doria S, Bizarro S, Rocha P, Gomes P, Torres L, Norton L, Borges BS, Castedo S, et al. Frequency of NUP98–NSD1 fusion transcript in childhood acute myeloid leukaemia. Leukemia. 2003;17:2244–7.
Brackmann F, Krumbholz M, Langer T, Rascher W, Holter W, Metzler M. Novel MLL2 Mutation in Kabuki syndrome with hypogammaglobulinemia and severe chronic thrombopenia. J Pediatr Hematol/Oncol 2012. [Epub ahead of print]
Chaffanet M, Gressin L, Preudhomme C, Soenen-Cornu V, Birnbaum D, Pébusque MJ. MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosom Cancer. 2000;28:6.
Chang B, Chen Y, Zhao Y, Bruick RK. JMJD6 is a histone arginine demethylase. Science. 2007;318:444–7.
Chase A, Cross NCP. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–8.
Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–15.
Crowley JA, Wang Y, Rapoport AP, Ning Y. Detection of MOZ–CBP fusion in acute myeloid leukemia with 8;16 translocation. Leukemia. 2005;19:2.
Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65.
Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1[agr] from chromatin. Nature. 2009;461:819–22.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Deguchi K, Ayton P, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML, Gilliland DG. MOZ–TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell. 2003;3:12.
Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–6.
Fabbrizio E, El Messaoudi S, Polanowska J, Paul C, Cook JR, Lee JH, Negre V, Rousset M, Pestka S, Le Cam A, et al. Negative regulation of transcription by the type II arginine methyltransferase PRMT5. EMBO Rep. 2002;3:641–5.
Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–67.
Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman T, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:6.
Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17:16.
Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, Confalonieri S, Cesaroni M, Marchesi F, Gasco M, Scanziani E, Capra M, Mai S, Nuciforo P, Crook T, Lough J, Amati B. Tip60 is a haplo-insufficient tumor suppressor required for an oncogene-induced DNA damage response. Nature. 2007;448:4.
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43.
Guccione E, Bassi C, Casadio F, Martinato F, Cesaroni M, Schuchlautz H, Luscher B, Amati B. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–7.
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8.
Harris WJ, Huang X, Lynch JT, Spencer GJ, Hitchin JR, Li Y, Ciceri F, Blaser JG, Greystoke BF, Jordan AM, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–87.
Hess JL, Bittner C, Zeisig DT, Bach C, Fuchs U, Borkhardt A, Frampton J, Slany RK. c-Myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood. 2006;108:7.
Hollink IHIM, van den Heuvel-Eibrink MM, Arentsen-Peters STCJM, Pratcorona M, Abbas S, Kuipers JE, van Galen JF, Beverloo HB, Sonneveld E, Kaspers G-JJL, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011;118:3645–56.
Jansson M, Durant ST, Cho E-C, Sheahan S, Edelmann M, Kessler B, La Thangue NB. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–9.
Katsumoto T, Aikawa Y, Iwama A, Ueda S, Ichikawa H, Ochiya T, Kitabayashi I. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006;20:9.
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33.
Liedtke M, Cleary ML. Therapeutic targeting of MLL. Blood. 2009;113:6061–8.
Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell. 2011;19:283–94.
Liu J, Mercher T, Scholl C, Brumme K, Gilliland DG, Zhu N. A functional role for the histone demethylase UTX in normal and malignant hematopoietic cells. Exp Hematol. 2012;40(487–498):e483.
Liu S, Wu L, Pang J, Santhanam R, Schwind S, Wu YZ, Hickey CJ, Yu J, Becker H, Maharry K, Radmacher MD, Li C, Whitman SP, Mishra A, Stauffer N, Eiring AM, Briesewitz R, Baiocchi RA, Chan KK, Paschka P, Caligiuri MA, Byrd JC, Croce CM, Bloomfield CD, Perrotti D, Garzon R, Marcucci G. Sp1/NFkB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:14.
Lokken AA, Zeleznik-Le NJ. Breaking the LSD1/KDM1A addiction: therapeutic targeting of the epigenetic modifier in AML. Cancer Cell. 2012;21:451–3.
Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474–8.
Mar BG, Bullinger L, Basu E, Schlis K, Silverman LB, Dohner K, Armstrong SA. Sequencing histone-modifying enzymes identifies UTX mutations in acute lymphoblastic leukemia. Leukemia. 2012;26:1881–3.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–66.
Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 2010;10:669–82.
Minucci S, Pelicci P. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev Cancer. 2006;6:13.
Miyake N, Mizuno S, Okamoto N, Ohashi H, Shiina M, Ogata K, Tsurusaki Y, Nakashima M, Saitsu H, Niikawa N, Matsumoto N. KDM6A point mutations cause Kabuki syndrome. Human Mutat. 2012. doi:10.1002/humu.22229.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5.
Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Philips L, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, Brindle PK, Dowling J. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011;471:4.
Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH, Armstrong SA. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci. 2012;109(13):5028–33.
Nikoloski G, Langemeijer SMC, Kuiper RP, Knops R, Massop M, Tonnissen ERLTM, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–7.
Nordentoft I, Jorgensen P. The acetyltransferase 60 kDa trans-acting regulatory protein of HIV type 1-interacting protein (Tip60) interacts with the translocation E26 transforming-specific leukaemia gene (TEL) and functions as a transcriptional co-repressor. Biochem J. 2003;374:8.
Ntziachristos P, Tsirigos A, Vlierberghe PV, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:298–303.
Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78.
Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120:9.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12.
Pasqualucci L, Dominguez-Sola D, Chiarenza A, Fabbri G, Grunn A, Trifonov V, Kasper LH, Lerach S, Tang H, Ma J, Rossi D, Chadburn A, Murty VV, Mullighan CG, Gaidano G, Rabadan R, Brindle PK, Dalla-Favera R. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:6.
Perez-Campo FM, Borrow J, Kouskoff V, Lacaud G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood. 2009;113:8.
Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:4.
Putnik J, Zhang C, Fröhlich Archangelo L, Tizazu B, Bartels S, Kickstein M, Greif PA, Bohlander SK. The interaction of ETV6 (TEL) and TIP60 requires a functional histone acetyltransferase domain in TIP60. Biochimica Biophys Acta. 2007;1772:13.
Reader JC, Meekins JS, Gojo I, Ning Y. A novel NUP98-PHF23 fusion resulting from a cryptic translocation t(11;17)(p15;p13) in acute myeloid leukemia. Leukemia. 2007;21:842–4.
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, Chalhoub N, Baker SJ, Huether R, Kriwacki R, Curley N, Thiruvenkatam R, Wang J, Wu G, Rusch M, Hong X, Becksfort J, Gupta P, Ma J, Easton J, Vadodaria B, Onar-Thomas A, Lin T, Li S, Pounds S, Paugh S, Zhao D, Kawauchi D, Roussel MF, Finkelstein D, Ellison DW, Lau CC, Bouffet E, Hassall T, Gururangan S, Cohn R, Fulton RS, Fulton LL, Dooling DJ, Ochoa K, Gajjar A, Mardis ER, Wilson RK, Downing JR, Zhang J, Gilbertson RJ. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–8.
Schenk T, Chen WC, Gollner S, Howell L, Jin L, Hebestreit K, Klein H-U, Popescu AC, Burnett A, Mills K, et al. Inhibition of the LSD1 (KDM1A) demethylase reactivates the all-trans-retinoic acid differentiation pathway in acute myeloid leukemia. Nat Med. 2012;18:605–11.
Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–53.
Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25:1–14.
Shia W-J, Okumura AJ, Yan M, Sarkeshik A, Lo M-C, Matsuura S, Komeno Y, Zhao X, Nimer SD, Yates JR, et al. PRMT1 interacts with AML1–ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood. 2012;119:4953–62.
Tamkun JW, Deuring R, Scott MP, Kissinger M, Pattatucci AM, Kaufman TC, Kennison JA. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator. Cell. 1992;68:11.
Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–17.
Thol F, Kolking B, Hollink IHI, Damm F, van den Heuvel-Eibrink MM, Michel Zwaan C, Bug G, Ottmann O, Wagner K, Morgan M, et al. Analysis of NUP98/NSD1 translocations in adult AML and MDS patients. Leukemia 2012. doi:10.1038/leu.2012.249.
Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1:433–41.
Uribesalgo I, Benitah S, Di Croce L. From oncogene to tumor suppressor: the dual role of Myc in leukemia. Cell Cycle. 2012;11:7.
van Haaften G, Dalgliesh GL, Davies H, Chen L, Bignell G, Greenman C, Edkins S, Hardy C, O’Meara S, Teague J, et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat Genet. 2009;41:521–3.
van Zutven LJCM, Önen E, Velthuizen SCJM, van Drunen E, von Bergh ARM, van den Heuvel-Eibrink MM, Veronese A, Mecucci C, Negrini M, de Greef GE, et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosom Cancer. 2006;45:437–46.
Vollmuth F, Geyer M. Interaction of propionylated and butyrylated histone H3 lysine marks with Brd4 bromodomains. Angew Chem Int Ed Engl. 2010;49:4.
Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98–NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9:804–12.
Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo J-L, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459:847–51.
Wang L, Gural A, Sun XJ, Zhao X, Perna F, Huang G, Hatlen M, Vu L, Liu F, Xu H, Asai T, Xu H, Deblasio T, Menendez S, Voza F, Jiang Y, Cole PA, Zhang J, Melnick A, Roeder RG, Nimer SD. The leukemogenicity of AML1–ETO is dependent on site-specific lysine acetylation. Science. 2011;333:4.
Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim S-H, Ito S, Yang C, Wang P, Xiao M-T, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30.
Yan J, Diaz J, Jiao J, Wang R, You J. Perturbation of BRD4 protein function by BRD4–NUT protein abrogates cellular differentiation in NUT midline carcinoma. J Biol Chem. 2011;286:12.
Yang XJ, Ogryzko V, Nishikawa J, Howard BH, Nakatani Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:5.
Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar M-P, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005;24:5525–32.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Zhang K, Dent SYR. Histone modifying enzymes and cancer: going beyond histones. J Cell Biochem. 2005;96:1137–48.
Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a–AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J Biol Chem. 2006;281:18059–68.
Zhao X, Jankovic V, Gural A, Huang G, Pardanani A, Menendez S, Zhang J, Dunne R, Xiao A, Erdjument-Bromage H, et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008;22:640–53.
Zhao Q, Rank G, Tan YT, Li H, Moritz RL, Simpson RJ, Cerruti L, Curtis DJ, Patel DJ, Allis CD, et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat Struct Mol Biol. 2009;16:304–11.
Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:6.
Acknowledgments
We thank the members of the Nimer lab, especially Sarah Greenblatt and Guoyan Cheng, for their helpful comments and thoughtful discussions on this manuscript. The work has been supported by an LLS SCOR grant and an NCI R01 grant to SDN.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Vu, L.P., Luciani, L. & Nimer, S.D. Histone-modifying enzymes: their role in the pathogenesis of acute leukemia and their therapeutic potential. Int J Hematol 97, 198–209 (2013). https://doi.org/10.1007/s12185-012-1247-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-012-1247-y