
Abstract
Gold-tube pyrolysis experiments were performed on two Athabasca oil sand bitumens at 300 °C to 525 °C with 2 °C/h rate and 25 °C step under 50 MPa. Pyrolysis temperature of 425 °C is critical for weight loss of bulk bitumen and hydrocarbon generation and destruction. Polar compounds are the main source of saturated and aromatic hydrocarbon, gas and coke fractions. Molecular compositions in pyrolyzates vary systematically with increasing pyrolysis temperatures. High molecular weight n-alkanes (C26+) are gradually destructed during pyrolysis due to thermal cracking. Moderate molecular weight n-alkanes (C21–C25) show the highest thermal stability in designed pyrolysis temperatures. The loss of low molecular weight n-alkanes (C20−) might be caused by volatilization during pyrolysis, which may alter commonly used molecular parameters such as ∑n-C20−/∑n-C21+, Pr/n-C17 and Ph/n-C18. Aromatic hydrocarbons were generated from 300 to 425 °C, then condensation and dealkylation have been initiated at 425 °C as evidenced by decreased summed alkylnaphthalenes to alkylphenanthrenes ratios and increased unsubstituted aromatics to substituted homologs ratios in higher temperatures. The occurrence of anthracene and benz[a]anthracene in pyrolysates indicates pyrogenic origin, while fluoranthene shows unexpected behaviors during pyrolysis. Ratios derived from them are not always reliable for pyrogenic source input diagnosis in environmental samples.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Oil sand bitumen (OSB) is one of the most important energy resources in the world. The largest OSB deposit in the Western Canada Sedimentary Basin contains about 1.7 trillion barrels of heavy oil and bitumen. Three major deposits are situated at Athabasca, Cold Lake and Peace River in Alberta, which are hosted in the reservoirs of varying age, ranging from the Devonian (Grosmont Formation) to Early Cretaceous (Mannville Group) (ST98-2018). These bitumens are originated from matured source rocks with initial API gravity about 28° in the southwest Alberta and migrated to the north and east for over 100 km during the Laramide Orogeny (Larter et al. 2006; Yin et al. 2013). Biodegradation is the main alteration process to transform conventional oil to current API gravity about of 8°‒12° bitumen by preferentially removal of light components (Head et al. 2003; Zhou et al. 2008). OSB is commonly enriched in heavy and base metals, and polar components such as oxygen, sulfur and nitrogen bearing compounds (Connan 1984). The geologic characteristics of the major bitumen and heavy oil occurrences have been recently reviewed by Hein (2017).
In order to elucidate oil and gas generation potentials and their genetic origins, numerous pyrolysis experiments have been performed on kerogens, oil sand bitumens and asphaltenes (Gong et al. 2004; Jia et al. 2004; Tian et al. 2006; Li et al. 2018). Pyrolysis for the Athabasca OSBs has been extensively applied for thermal behavior investigation during in situ upgrading and refinery processes. Speight (1970) examined structural changes in the Athabasca bitumen during thermal cracking. n-Alkanes from C9 to C26 have been detected by gas chromatography analysis after heating treatment. Pyrolysis of asphaltenes and OSBs from Athabasca region has been thoroughly investigated to obtain oil-like materials, n-alkanes, biomarkers (Rubinstein et al. 1979; Strausz et al.1992). Rubinstein and Strausz (1979) noticed that saturated hydrocarbon fraction is more stable than aromatic hydrocarbon fraction during thermal treatment of the Athabasca OSB. Significant cracking and polymerization mainly occur in the aromatics. However, Fowler and Brooks (1987) found some significant differences between pyrolyzates of the Athabasca asphaltenes isolated from biodegraded bitumen and undegraded oils and cautioned against directly correlate asphaltene pyrolyzates with oils, whereas Jones et al. (1988) shown the genetic link between the Athabasca asphaltenes separated from bitumens and oils. Our recent study illustrated that such difference might be caused by the biodegradation of bound biomarkers rather than different pyrolysis conditions (Li et al. 2020). Peng et al. (1997) applied molecular compositions derived from pyrolysis for maturity assessment of different fractions in the Athabasca OSB. In addition to liquid products, gas has been produced by thermolysis of the Athabasca OSB. Both hydrocarbon and non-hydrocarbon gases are originated from thermal cracking of OSB rather than bacterial alteration (Strausz et al. 1977; Xu et al. 2019). Pyrolysis studies have also been applied for understanding the in situ upgrading or developing reaction schemes for use in numerical simulators and kinetics (Kapadia et al. 2015; Jia et al. 2016; Gai et al. 2016; Eshraghian and Husein 2017). However, the chemical aspects of the thermal behavior of industrial bitumens are still not well documented (Schlepp et al. 2001; Peng et al. 2018). The lack of pyrolysis data on systematic changes of molecular compositions in severely biodegraded bitumen introduces a strong limitation in kinetic parameter extraction, heavy oil and bitumen transformation modeling and exploitation optimization (Sonibare et al. 2009). The purpose of the present study is to record sequential changes of bulk and molecular compositions of OSB during pyrolysis experiments and to deepen our understanding of reaction processes during in situ upgrading and/or refinery processes.
2 Samples and methods
Two severely biodegraded oil sand bitumen samples from drilling core of the McMurray Formation in the Athabasca region were pyrolyzed in the present study. Sample A was selected from the top of oil sand column at depth 239.0 m, and sample B was selected from the bottom of the same column at depth 271.5 m. Confined pyrolysis was performed in gold-tubes (length 10 cm, diameter 5 mm and wall thickness 0.5 mm) using the same methods as those described by Xu et al. (2019) and Li et al. (2020). Briefly, the gold tubes were welded at one end and 50–200 mg oil sand was introduced into the gold tube then sealed under argon atmosphere. The samples were heated to temperatures of 300–525 °C with a 25 °C step at heating rates of 2 °C/h under a pressure of 50 MPa. Each sample has a duplicate run to ensure the reliability. After gas collection, the oil sand residual was extracted by Soxhlet extraction with dichloromethane and methanol (93:7 v:v) to obtain the extractable organic matter (EOM). Total EOM was loaded on a packed silica gel and neutral alumina column. The saturated hydrocarbon fraction was eluted by n-hexane and the aromatic hydrocarbon faction was eluted by a mixture of n-hexane/dichloromethane (1:1, v/v). The polar (resin + asphaltene) fraction retained in the column has not been recovered, and its weight is a subtraction of hydrocarbon fractions from the EOM.
The GC–MS analyses were performed on an Agilent 7890A gas chromatograph (GC) coupled with 5975C mass spectrometer (MS) system. An Agilent HP-5 (60 m × 0.25 mm inner diameter × 0.25 μm) fused silica capillary column was used. The temperature program for the saturated hydrocarbon fraction is the same as that described by Li et al. (2020). For the aromatic hydrocarbon fraction, the GC oven was held at 50 °C for 1 min, increased to 120 °C at 15 °C/min, then to 300 °C at 3 °C/min and held constant at 300 °C for 35 min. The mass spectrometer was equipped with an electron impact source at 70 eV and select ion monitoring mode was applied. The carrier gas was helium at 1 mL/min, and the ion source temperature was maintained at 250 °C. Due to limited sample size, no internal standards have been added in the analyzed fraction. Peak area was used for integration and component ratio calculation, and no response factor calibration has been performed.
3 Results and discussions
3.1 Bulk compositions
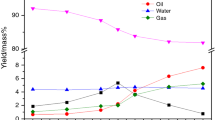
Weights of sample loaded in each gold tube, bitumen content, EOM yield, saturated hydrocarbons, aromatic hydrocarbons, polars (resins + asphaltenes), gas and coke fraction after pyrolysis were listed in Table 1. The original bitumen contents in sample A and B are 14.5 and 16.0 wt%, respectively. After pyrolysis experiments, the residual bitumen (EOM) contents decrease gradually with increasing heating temperatures. At 300 °C, the residual bitumen yields are 13.9 and 15.4 wt%, respectively, while both samples contain < 5 wt% of residual bitumen after pyrolysis at 525 °C (Fig. 1a). No extractable organic matter can be obtained once pyrolysis temperature reaches to 550 °C where gas and coke rise continuously (Fig. 1b, c). Subtle difference between two samples is caused by biodegradation influence which alters the molecular structure of the raw bitumen. Sample A has suffered relatively less intensive biodegradation than sample B which was situated near oil water contact (Li et al. 2020). The weight loss during the pyrolysis can be regarded as neoformed non-extractable fraction (coke) and volatile fraction (gas). Significant weight loss occurs around 425‒450 °C, implying substantial thermal cracking has been initiated.

Bulk composition variations of the Athabasca oil sand bitumen during gold-tube pyrolysis experiments. a Original bitumen content and residual bitumen (EOM) yield; b, c Proportions of saturated hydrocarbons, aromatic hydrocarbons, polars (resins + asphaltenes), gas and coke in samples A and B, respectively
The EOM has been separated into saturated hydrocarbons, aromatic hydrocarbons and polars (Table 1). The saturated hydrocarbons content increases from 16.1% at 300 °C to 27.1% at 400 °C then decreases gradually to about 12.0% at 475–525 °C in sample A. The highest saturated hydrocarbons content of 28.1% occurs at 350 °C in sample B and a sudden drop occurs at 450 °C where saturated hydrocarbons content is only 5.5%. After 450 °C, a small increment of saturated hydrocarbons content occurs at higher temperature range (Fig. 1b, c). The aromatic hydrocarbons fraction exhibits a similar trend as the saturated hydrocarbons but has slightly different patterns. The highest proportion of 27.1% aromatic hydrocarbons occurs at 350 °C in sample A whereas the highest proportion of 25.3% occurs at 425 °C in sample B. At 525 °C, the aromatic hydrocarbons contents in sample A and B fall to 11.4% and 6.1%, respectively (Fig. 1b, c). The polars content shows a general decrease trend with increasing pyrolysis temperatures, decreasing from about 60% at 300 °C to around 10% at 525 °C. This indicates that polars serve as main source for hydrocarbon and coke generation. The amount of total gas increases from 3.8% to 40.9% in sample A, while it increases from 1.6% to 42.7% in sample B. While no obvious difference in gas yields between two studied samples at the studied temperature range, the final gas generation potential is biodegradation related with sample B having lower yield than sample A (see more gas data from Xu et al. 2019). The amount of coke was direct subtraction of EOM from the bitumen plus gas content. Almost no coke has been formed at the initial pyrolysis temperature from sample A, while small amount of coke has been detected from sample B. The highest amount of coke was 30% in sample A formed at 475 °C, while the highest amount of coke reached to 38% in B at 450 °C (Fig. 1b, c). Higher proportion of coke in sample B further verifies its more intensive biodegradation influence, which not only affects the free hydrocarbons but bound biomarkers as well (Li et al. 2020). The most important weight loss was observed at 425‒475 °C where intermolecular associations and chemical bond scissions are likely intensive. Above 50% of the mass loss occurs at 475 °C in sample A, while the same mass loss occurs at 450 °C in sample B, indicating subtle structure difference in original bitumens. The opposite evolution trend between polars and coke plus gas fraction implies that polars contribute the most to the later even though the hydrocarbon fractions have also been converted to coke and gas.
Three evolution stages can be identified on the basis of bulk composition variations. At low temperature range of 300‒350 °C, both saturated and aromatic hydrocarbon contents increase with pyrolysis temperatures, which can be attributed to the release of occluded hydrocarbons and neoformed hydrocarbons from polar fraction. The polar fraction decreases significantly while only small amount of coke and gas has been generated. This is a liquid hydrocarbon generation stage derived from the decomposition of polars. The second stage spans a temperature range of 350‒450 °C, where both saturated and aromatic hydrocarbons contents decrease together with polars, while the coke and gas fraction increases substantially. This is a main condensation and polymerization stage. The third one in the temperature range of 450‒525 °C is the thermal cracking of polars and gas generation stage. The hydrocarbon fractions remain constant or increase slightly, but the polar fraction decreases significantly accompanied by coke formation.
3.2 Aliphatic hydrocarbon fraction
Total ion chromatograms (TIC) of the saturated hydrocarbons fraction in original bitumen and representative TICs of pyrolyzates at 300 and 500 °C of pyrolysis were illustrated in Fig. 2. As bacterial consumed all n-alkanes and isoprenoids and most of hopanes and steranes, the TIC of saturated hydrocarbons in the original bitumen consists of large hump of unresolved complex mixture (UCM) superimposed by a few biodegradation-resistant components such as pregnanes, tricyclic terpanes and C2917α(H)21β(H) hopane (C29H). C30H is normally higher than C29H in normal oil but is more susceptible to biodegradation (Peters and Moldowan 1993) leaving the dominance of C29H in the biomarker region. Saturated hydrocarbons from the pyrolyzates of OSB show systematic changes with increasing temperatures. Large quantity of n-alkanes, which are absent in the original bitumen due to their prior removal by biodegradation, have been released at 300 °C and becomes the dominant compound class at 500 °C. The size of UCM hump decreases gradually with increasing pyrolysis temperatures, while various hydrocarbon classes were released and/or generated from asphaltenes. Ratios between these compound classes suggested that the adsorbed and occluded biomarkers were released at intermediate-temperature pyrolysis (< 425 °C), while the covalently bonded biomarkers were mainly released at high temperature (≥ 425 °C). Biodegradation exerts more intensive influence on sample B than sample A especially for the bound phase biomarkers (Li et al. 2020).

Representative total ion chromatograms of the saturated hydrocarbon fraction in original bitumen and pyrolyzates at 300 and 500 °C of pyrolysis. 17–28: carbon number of n-alkanes; Pr, pristine; Ph, phytane; 23T, C23 tricyclic terpane; 29H, C29 αβ 30-norhopane; 30H, C30 αβ hopane
n-Alkanes and isoprenoids are the major compound class in OSB pyrolyzates, which accounts for about half of mass weight in non-biodegraded oil. The generation and destruction of n-alkanes and isoprenoids reflect most of oil and gas generation processes. The distribution of n-alkanes and isoprenoids can be extracted from m/z 85 mass chromatogram in GC–MS analysis of the saturated hydrocarbon fraction. n-Alkanes from C11 to C35 were present in the pyrolyzates. The n-alkanes obtained at 300 °C from sample A is marked by the predominance of heavy n-alkanes eluted between C21 and C31 and maximized at C29 (Fig. 3a), while heavy end predominance does not occur in sample B whose n-alkanes was maximized at C21 (Fig. 3b). Different n-alkane generation characteristics cannot clarify such difference as the higher pyrolysis temperatures did not yield the same n-alkane distribution pattern. Biodegradation of bound n-alkanes is likely involved (Li et al. 2020), while detailed impact requires further investigation. From 325 to 375 °C, more low molecular weight n-alkanes were generated from sample A and the maximum carbon was shifted to C24. The maximum carbon was shifted to C23 or C25 in the same temperature interval in sample B. Significant change of n-alkanes distribution patterns occurs at 400 °C in sample A where the maximum carbon was shifted to C18, while such change happens at 425 °C in sample B with the maximum carbon shifted to C19. After this temperature, the maximum carbon was moved to around C24 in the higher temperature range (Fig. 3).

The m/z 85 mass chromatograms showing distribution variations of n-alkanes and isoprenoids with increasing pyrolysis temperatures. a Sample A; b Sample B. 17‒29: carbon number of n-alkanes; Pr, pristine; Ph, phytane
The change of relative abundance of light and heavy molecular weight n-alkanes can be illustrated by ratio of Ʃn-C20−/Ʃn-C21+. This ratio is commonly applied to indicate source input and thermal maturation. The terrestrial plant originated organic matter is dominated by high molecular weight n-alkanes, while marine algal formed organic matter is enriched in low molecular weight n-alkanes. With increasing thermal maturation, high molecular weight n-alkanes will crack to form low molecular weight n-alkanes (Peters and Moldowan 1993). The Ʃn-C20−/Ʃn-C21+ ratio in pyrolyzate of sample A at 300 °C is only 0.06 but it increases to 0.98 at 450 °C, then the ratio decreases to 0.03 at 525 °C. Relative abundance of light versus heavy n-alkanes does not change significantly at temperature range of 300‒400 °C in sample B with Ʃn-C20−/Ʃn-C21+ ratios fluctuated between 0.26 and 0.34. A sharp increase of Ʃn-C20−/Ʃn-C21+ ratio from 0.29 to 0.79 occurs at 425 °C, then it decreases continuously to 0.15 at 525 °C (Fig. 4a).

Variations of relative abundance of n-alkanes with different carbon numbers during pyrolysis experiments. a Ratios Ʃn-C20−/Ʃn-C21+; b ratios of Ʃn-C21-25/Ʃn-C15-33
The generation of low molecular weight n-alkanes during pyrolysis is a well-known phenomenon. The lengths of the carbon chains bias toward the short carbon chains with increasing pyrolysis temperatures can be demonstrated by increased Ʃn-C20−/Ʃn-C21+ ratios. However, this trend was inverted in the high temperature range. A marked change of the n-alkanes distributions occurs at 425‒450 °C, where the low molecular weight n-alkanes reach the maximum proportion, then gradually lost in the system with further increasing temperatures. The increase in the lower-molecular weight n-alkanes can be attributed to the cracking of high molecular weight compounds by the scission of alkyl groups located in peripheral sites of asphaltene structure. This process seems continue in the entire pyrolysis temperatures as indicated by almost disappearing of C30+ n-alkanes after 425 °C. The cracking of long-chain alkanes becomes more intensive at higher temperatures, which should generate more short-chained alkanes. The depletion of low molecular weight n-alkanes in pyrolysis experiments has not been reported in the literature. Our data suggested that C20− n-alkanes are more vaporized than C21+ n-alkanes during pyrolysis. Thermal cracking of C26+ n-alkanes and volatilization of C20− n-alkanes lead to concentrated C21-C25 n-alkanes as evidenced by continuous increment of Ʃn-C21-25/Ʃn-C15-33 ratios with increasing temperatures (Fig. 4b). The evolution of this ratio can be divided into at least two stages. At temperature between 300 and 375 °C, the intensive cracking of hydrocarbons may be not available, and this ratio did not increase apparently. In the second stage when pyrolysis temperatures are ≥ 400 °C, the cracking of C26+ in addition to the loss of C20− may occur and resulted in the increase of Ʃn-C21-25/Ʃn-C15-33 ratio in our experiments.
While acyclic isoprenoids are minor constituents in the saturated hydrocarbon fraction, they have been widely used in studies for characterization and correlation of source rock and oil depositional environment. Ratios of pristane (Pr)/n-C17 and phytane (Ph)/n-C18 can be applied for depositional phase, maturity and biodegradation influence assessment because of their different source original, thermal stability and biodegradation susceptibility. Values of Pr/n-C17 are generally high in oxidized depositional environment, while values of Ph/n-C18 are high in reducing depositional environment. Both of Pr/n-C17 and Ph/n-C18 ratios increase with increasing levels of biodegradation, but decrease with increasing levels of maturity (Peters and Moldowan 1993). In our pyrolysis sample suite, ratios of Pr/n-C17 and Ph/n-C18 show three stage evolution trajectories (Fig. 5). Slightly incremental of Pr/n-C17 and Ph/n-C18 occurs in the initial pyrolyzates of sample A from 300 to 325 °C, while the increasing ratio values continue to 400 °C in sample B. Difference between two samples is again likely related to biodegradation influence on bound molecular moieties. The second stage reflects typical maturation impact where Pr/n-C17 and Ph/n-C18 ratios decrease with increasing pyrolysis temperatures. This trend is reversed at 450 °C, where Pr/n-C17 and Ph/n-C18 ratios increase with further increasing pyrolysis temperatures (Fig. 5a, b). The third stage is incompatible with compound thermal stability as isoprenoids are thermally less stable than n-alkanes. Our preliminary interpretation for such unusual phenomenon is caused by evaporative loss during experiments. Alternatively, more bound isoprenoids are likely to be released from covalently bonded phase, while n-alkanes are mainly resided in the occluded phase. However, such hypothesis requires more investigations.

Variation of ratios a Pr/n-C17; b Ph/n-C18 and c Pr/Ph in residual bitumen after pyrolysis experiments
Pristane and phytane are mainly derived from phytol, which is an important part of phytoplanktonic chlorophylls. Large proportion of phytol will be oxidized to phytenic acid under relatively oxidized conditions, then to pristine after decarboxylation, while it will be reduced to phytane under relatively anoxic conditions after dehydration. Therefore, the ratio of Pr/Ph may provide a measure of redox conditions during organic deposition. Pr/Ph ratio of < 1.0 indicates anoxic conditions during early diagenesis, and values between 1.0 and 3.0 were interpreted as reflecting sub-oxic environments (Didyk et al. 1978). While a subtle decrease trend with increasing pyrolysis may occur in studied samples, the Pr/Ph ratio of consistently < 1.0 indicates highly reducing depositional environment during the formation bitumen precursors (Fig. 5c).
3.3 Aromatic hydrocarbon fraction
The aromatic hydrocarbons in raw bitumen have been severely altered by biodegradation, resulted in the absence of alkylbenzenes, alkylnaphthalenes and alkyldibenzothiophenes. The alkylphenanthrenes were heavily altered, while the tetraromatic hydrocarbons (pyrene, chrysene) were less affected and mono- and triaromatic steroids were largely intact in raw bitumen. No significant change can be observed from pyrolyzate in sample B at 300 °C, which shows basically the same pattern as unheated bitumen, while unusually high abundance of unsubstituted dibenzothiophene, phenanthrenes, fluoranthene and pyrene has been observed in sample A (Fig. 6). These compounds did not occur in the higher pyrolysis temperatures and their unexpected occurrence likely reflect the release of adsorbed aromatic components, but the mechanisms and processes of such releasement require further investigation. Above 350 °C, the generation of aromatic hydrocarbons becomes more important. Alkylnaphthalenes are the most abundant compounds detected in the aromatic fractions at 425 °C. The enrichment of alkylnaphthalenes during pyrolysis, reflecting thermal cracking processes of polars. The distributions of alkylnaphthalenes, alkylphenanthrenes and alkyldibenzothiophenes in pyrolyzates at 425 °C are very similar to these in non-biodegraded oils. Above 425 °C, distributions of alkylnaphthalenes, alkyldibenzothiophenes, alkylphenanthrenes and alkyldibenzothiophenes changed drastically. The TIC at 500 °C is characterized by the predominance of alkylphenanthrenes and a few tetracyclic aromatics, while alkylnaphthalenes and triaromatic steroids are depleted (Fig. 6).

Representative total ion chromatograms of the aromatic hydrocarbon fraction in initial bitumen and pyrolyzates at 300, 425 and 500 °C of pyrolysis. MN, methylnaphthalenes; C2N, dimethylnaphthalenes and ethylnapthalenes; C3N, trimethylnaphthalenes; C4N, tetramethylnaphthalenes; DBT, dibenzothiophene; P, phenanthrene; An: anthracene; MP, methylphenanthrenes; C2P, dimethylphenanthrenes and ethylphenanthrenes; Fl, fluoranthene; Py, pyrene; C3P, trimethylphenanthrenes; BAn, benz[a]anthracene; Ch, chrysene; MCh, methylchrysene; TAS, triaromatic steroids
The change of relative importance of aromatic rings can be illustrated by summed alkylnaphthalenes to alkylphenanthrenes ratio (ƩC1-5N/ƩC1-3P). The ratio values are 0.14 and 0.24 at 300 °C, raised to 1.81 and 0.79 at 425 °C in samples A and B, respectively (Fig. 7a). The most dramatic changes in the distribution of PAH distributions occurred at the 425 °C. After 425 °C heating step, aromatic hydrocarbons exhibit contrasting behaviors toward thermal cracking. Alkylnaphthalenes seem more easily condensed to form other PAHs and coke than alkylphenanthrenes with increases in temperature. The condensation of 2-ring aromatics caused the ƩC1-5N/ƩC1-3P ratios dropping to 0.11 and 0.03 at 525 °C in samples A and B, respectively. Our experimental results can account for the general ring buildup with increasing pyrolysis temperatures at high temperature regime. Lower ƩC1-5N/ƩC1-3P ratios in sample B than sample A of most pyrolyzates reflects higher condensed nature in sample B, which is likely related to more severe biodegradation influence (Li et al. 2020). However, the depletion of light aromatics has not been observed in natural evolution cases where the alkylnaphthalenes are generally more enriched than alkylphenanthrenes with increasing maturity levels (Zhang et al. 2005). It is likely controlled by thermal maturity regime while further investigation is still essential.

a Ratios of summed alkylnaphthalenes to alkylphenanthrenes ratio (ƩC1-5N/ƩC1-3P) reflecting variation of condensation degree during pyrolysis experiments; b Ratios of phenanthrene/C3-alkylphenanthrene ratios (P/C3P) indicating change of alkylation degree during pyrolysis experiments
The degree of alkylation of aromatic hydrocarbons is generally controlled by organic source inputs and maturity levels. Generally, terrestrial originated aromatic hydrocarbons are enriched in unsubstituted component, while marine derived aromatic hydrocarbons contain more substituted homologs. Thermal maturation reduces the proportion of highly alkylated homologs with increasing thermal stress due to the loss of methyl groups (Garrigues et al. 1988). The changes seen in nature can also be observed from the heating experiments in more dramatic way. Here, use phenanthrene/C3-alkylphenanthrene ratios (P/C3P) as an illustration to account for unsubstituted and substituted proportion variation within an aromatic hydrocarbon homolog. From 300 to 425 °C, phenanthrene homolog is dominated by highly alkylated components and P/C3P ratios decrease slightly from about 0.5–0.2 with increasing pyrolysis temperatures, indicating an alkylation process. From 425 to 525 °C, the trend is reversed with unsubstituted phenanthrene being the dominated component in whole homolog and the P/C3P ratios increase drastically to 8.1 and 1.3 for sample A and B, respectively (Fig. 7b).
A few pyrogenic origin marker compounds are commonly applied for hydrocarbon source diagnosis (Yunker et al. 2002) but whether there are universally occurred in pyrolysis process has not been well recorded. Anthracene (An) is one of the common pyrogenic origin components, which is typically low or absent in normal oils. A relative high abundance of anthracene has been reported previously for high temperature coal tars (Radke et al. 1982), hydrous pyrolysates of sedimentary organic matter (Rushdi and Simoneit 2002) or igneous intrusion pyrolysis-related oils (Huang et al. 2015). Petrogenic input influenced samples, have ratios of anthracene to anthracene plus phenanthrene, [An/(An + P)], generally < 0.10, while typically, pyrogenic input-influenced samples have An/(An + P) ≥ 0.10 (Yunker et al. 2002). There is no anthracene in raw bitumen and the ratios of An/(An + P) in pyrolyzates vary from 0.03 to 0.37 in sample A and from 0.06 to 0.4 in sample B with the highest peak ratio occurring at 350 and 400 °C pyrolysis temperatures, respectively. Majority samples have their An/(An + P) ratios > 0.15, clearly indicating a pyrogenic process (Fig. 8a). The An/(An + P) ratio increase with temperatures at early pyrolysis stage reflects a gradual generation process, while the decrease of An/(An + P) ratio with further increasing of temperatures at late pyrolysis stage suggests that thermal stability of anthracene is lower than phenanthrene. The An/(An + P) ratio drops to below 0.1 at 525 °C implying that the An/(An + P) ratio is not always robust diagnosis of combustion input in the environmental samples.

The occurrence of pyrogenic markers and their related diagnostic ratio variations during pyrolysis experiments. a Anthracene to anthracene plus phenanthrene, [An/(An + P)]; b benz[a]anthracene to benz[a]anthracene plus chrysene, [BAn/(Ch + BAn)]; c fluoranthene to fluoranthene plus pyrene, Fl/(Fl + Py)
Benz[a]anthracene represents a combustion derived PAH component in modern geological settings. Similar to anthracene, benz[a]anthracene has been detected in very low concentrations in conventional crude oils (Tobiszewski and Namieśnik 2012). Yunker et al. (2002) proposed that for recent sediments, ratios of benz[a]anthracene to the sum of benz[a]anthracene and chrysene [BAn/(Ch + BAn)] < 0.2 indicate petrogenic inputs, from 0.20 to 0.35 indicate either petroleum or combustion and > 0.35 imply combustion. No benz[a]anthracene has been identified from the raw bitumen. In the pyrolyzates, both samples show very similar evolution trend. Slightly high BAn/(Ch + BAn) ratio around 0.2 has been observed at 300 °C for unknown reason. Then, the BAn/(Ch + BAn) ratios increase gradually from 0.14 and 0.11 at 325 °C to 0.38 and 0.4 at 525 °C in samples A and B, respectively (Fig. 8b). The 0.35 threshold of combustion marker occurs at 475 °C and BAn/(Ch + BAn) ratio clearly indicates pyrogenic process once pyrolysis temperatures are higher than 475 °C.
The relative abundance of fluoranthene (Fl) and pyrene (Py) has also been applied for source input diagnosis. Ratios of Fl/(Fl + Py) < 0.4 generally signify a predominance of petrogenic PAHs, between 0.4 and 0.5 signify petroleum combustion, and > 0.5 signify grass, wood or coal combustion (Yunker et al. 2002). Pyrene is one of the major individual aromatic hydrocarbon components with the Fl/(Fl + Py) ratio of < 0.1 in raw bitumen. Unusually high amount of fluoranthene and pyrene has been released from sample A at 300 °C and the Fl/(Fl + Py) ratio was elevated to 0.35. In contrast to the BAn/(Ch + BAn) ratio, the Fl/(Fl + Py) ratio decreases progressively with pyrolysis temperatures down to 0.09 at 525 °C. Generally decrease but slightly different trend occurs in sample B. From 300 to 350 °C, the Fl/(Fl + Py) ratio drops from 0.2 to 0.13 then increases to the highest value of 0.24 at 400 °C. After that, the Fl/(Fl + Py) ratio decreases continuously to 0.08 at 525 °C. Nevertheless, all Fl/(Fl + Py) ratios are < 0.4 in pyrolyzates suggesting the petrogenic process without combustion interference. Our experimental results raise an issue of legitimacy of Fl/(Fl + Py) ratio as diagnosis tool for pyrogenic input in environmental samples. While some combustion-related processes may produce more fluoranthene than pyrene, the Fl/(Fl + Py) ratio is not robust in all cases. This can explain why the Fl/(Fl + Py) ratio is only about 0.15 in northwestern Saskatchewan lake sediments east of the Athabasca oil sands where a petrogenic input can be ruled out (Ahad et al. 2015).
4 Conclusions
Athabasca oil sand bitumen has suffered severe biodegradation influence, which consumes most of light saturated and aromatic hydrocarbons. Gold-tube pyrolysis experiments performed on two Athabasca oil sand bitumens from one drilling core illustrate systematic changes of bulk and molecular compositions with temperatures. Both saturated and aromatic hydrocarbons increase their content before 425‒450 °C then decrease in the higher temperature range. Polars (resins + asphaltenes) and coke plus gas fractions vary in the opposite direction suggesting that most coke plus gas was derived from polar fraction with minor contribution from hydrocarbons. High molecular weight n-alkanes were initially generated then the maximum carbon was shifted to C18 or C19 at 425 °C, however, the enrichment of low molecular weight n-alkanes (C20−) was ended at 425 °C, but the moderate weight n-alkanes (C21‒C25) become the dominated components from 425 to 525 °C. In the aromatic hydrocarbons fraction, alkylnaphthalenes are gradually enriched from 300 to 425 °C then depleted afterward, while alkylphenanthrenes become the dominated components from 425 to 525 °C. The loss of low molecular weight n-alkanes and alkylnaphthalenes in high pyrolysis temperature range has not been observed in nature but likely caused by vaporization during pyrolysis while the mechanism still requires further investigation. The degree of alkylation in aromatic homologs was initially increased from 300 to 425 °C, implying alkylation process, then decreased dramatically from 425 to 525 °C, indicating demethylation process. Pyrogenic origin diagnosis ratios have very different response to pyrolysis temperatures. Anthracene was gradually generated at low pyrolysis temperature but destructed at high pyrolysis temperature. Benz[a]anthracene increased continuously against chrysene but fluoranthene decreased against pyrene during pyrolysis. Caution should be applied when ratios between them were applied for source input diagnosis as they are not always robust.
References
Ahad JM, Jautzy JJ, Cumming BF, et al. Sources of polycyclic aromatic hydrocarbons (PAHs) to northwestern Saskatchewan lakes east of the Athabasca oil sands. Org Geochem. 2015;80:35–45. https://doi.org/10.1016/j.orggeochem.2015.01.001.
Connan J. Biodegradation of crude oils in reservoirs. In: Brooks J, Welte D, editors. Advances in petroleum geochemistry I. London: Academic Press; 1984. p. 299–335.
Didyk BM, Simoneit BRT, Brassell ST, et al. Organic geochemical indicators of palaeoenvironmental conditions of sedimentation. Nature. 1978;272:216–22. https://doi.org/10.1038/272216a0.
Eshraghian A, Husein M. Thermal cracking of Athabasca VR and bitumen and their maltene fraction in a closed reactor system. Fuel. 2017;190:396–408. https://doi.org/10.1016/j.fuel.2016.10.111.
Fowler M, Brooks P. Organic geochemistry of Western Canada Basin bitumen and heavy oils. 2. Correlation of bitumen by using hydrous-pyrolysis of asphaltenes. Energy Fuels. 1987;1:459–67. https://doi.org/10.1021/ef00006a002.
Gai X, Arano H, Lu P, et al. Catalytic bitumen cracking in sub-and supercritical water. Fuel Process Technol. 2016;142:315–8. https://doi.org/10.1016/j.fuproc.2015.10.032.
Garrigues P, De Sury R, Angelin ML, et al. Relation of the methylated aromatic hydrocarbon distribution pattern to the maturity of organic matter in ancient sediments from the Mahakam delta. Geochim Cosmochim Acta. 1988;52(2):375–84. https://doi.org/10.1016/0016-7037(88)90093-2.
Gong S, Peng PA, Lu YH, et al. Simulation experiments on secondary thermal stress of biodegraded bituminous sandstones. Chin Sci Bull. 2004;49:50–9. https://doi.org/10.1007/BF02890453.
Head IM, Jones DM, Larter SR. Biological activity in the deep subsurface and the origin of heavy oil. Nature. 2003;426:344–53. https://doi.org/10.1038/nature02134.
Hein F. Geology of bitumen and heavy oil: an overview. J Pet Sci Eng. 2017;154:551–63. https://doi.org/10.1016/j.petrol.2016.11.025.
Huang HP, Zhang SC, Su J. Pyrolytically derived polycyclic aromatic hydrocarbons in marine oils from the Tarim Basin, NW China. Energy Fuel. 2015;29(9):5578–86. https://doi.org/10.1021/acs.energyfuels.5b01007.
Jia H, Liu PG, Pu WF, et al. In situ catalytic upgrading of heavy crude oil through low-temperature oxidation. Pet Sci. 2016;13:476–88. https://doi.org/10.1007/s12182-016-0113-6.
Jia WL, Shuai YH, Peng PA, et al. Kinetic study of hydrocarbon generation of oil asphaltene from Lunnan area, Tabei uplift. Chin Sci Bull. 2004;49(1):83–8. https://doi.org/10.1007/BF02890458.
Jones D, Douglas A, Connan J. Hydrous pyrolysis of asphaltenes and polar fractions of biodegraded oils. Org Geochem. 1988;13:981–93. https://doi.org/10.1016/0146-6380(88)90280-X.
Kapadia PR, Kallos MS, Gates ID. A review of pyrolysis, aquathermolysis, and oxidation of Athabasca bitumen. Fuel Process Technol. 2015;131:270–89. https://doi.org/10.1016/j.fuproc.2014.11.027.
Larter S, Huang HP, Adams J, et al. The controls on the composition of biodegraded oils in the deep subsurface: part II—geological controls on subsurface biodegradation fluxes and constraints on reservoir-fluid property prediction. AAPG Bull. 2006;90(6):921–38. https://doi.org/10.1306/01270605130.
Li J, Ma W, Wang YF, et al. Modeling of the whole hydrocarbon-generating process of sapropelic source rock. Pet Explor Dev. 2018;45(3):461–71. https://doi.org/10.1016/S1876-3804(18)30051-X.
Li Z, Huang H, Zhang S. The effect of biodegradation on bound biomarkers released from intermediate-temperature gold-tube pyrolysis of severely biodegraded Athabasca bitumen. Fuel. 2020;263:116669. https://doi.org/10.1016/j.fuel.2019.116669.
Peng PA, Morales-Izquierdo A, Hogg A, et al. Molecular structure of Athabasca asphaltene: sulfide, ether, and ester linkages. Energy Fuels. 1997;11:1171–87. https://doi.org/10.1021/ef970027c.
Peng WL, Hu GY, Liu QY, et al. Research status on thermal simulation experiment and several issues of concern. J Nat Gas Geosci. 2018;3(5):283–93. https://doi.org/10.1016/j.jnggs.2018.11.006.
Peters KE, Moldowan JM. The biomarker guide: interpreting molecular fossils in petroleum and ancient sediments. Englewood Cliffs: Prentice Hall; 1993. p. 363.
Radke M, Willsch H, Leythaeuser D, et al. Aromatic components of coal: relation of distribution pattern to rank. Geochim Cosmochim Acta. 1982;46(10):1831–48. https://doi.org/10.1016/0016-7037(82)90122-3.
Rubinstein I, Strausz OP. Thermal treatment of the Athabasca oil sand bitumen and its component parts. Geochim Cosmochim Acta. 1979;43(12):1887–93. https://doi.org/10.1016/0016-7037(79)90002-4.
Rubinstein I, Spyckerelle C, Strausz OP. Pyrolysis of asphaltenes: a source of geochemical information. Geochim Cosmochim Acta. 1979;43(1):1–6. https://doi.org/10.1016/0016-7037(79)90041-3.
Rushdi AI, Simoneit BRT. Hydrothermal alteration of organic matter in sediments of the Northeastern Pacific Ocean: Part 1. Middle Valley, Juan de Fuca Ridge. Appl Geochem. 2002;17(11):1401–28. https://doi.org/10.1016/s0883-2927(02)00109-9.
Schlepp L, Elie M, Landais P, et al. Pyrolysis of asphalt in the presence and absence of water. Fuel Process Technol. 2001;74(2):107–23. https://doi.org/10.1016/S0378-3820(01)00207-7.
Sonibare OO, Snape CE, Meredith W, et al. Geochemical characterisation of heavily biodegraded tar sand bitumens by catalytic hydropyrolysis. J Anal Appl Pyrol. 2009;86(1):135–40. https://doi.org/10.1016/j.jaap.2009.05.003.
Speight JG. Thermal cracking of Athabasca bitumen, Athabasca asphaltenes, and Athabasca deasphalted heavy oil. Fuel. 1970;49(2):134–45. https://doi.org/10.1016/0016-2361(70)90034-7.
ST98-2018: Alberta’s energy reserves& supply/demand outlook (https://www.aer.ca/documents/sts/ST98/ST98-2018_Executive_Summary.pdf).
Strausz OP, Jha KN, Montgomery DS. Chemical composition of gases in Athabasca bitumen and in low-temperature thermolysis of oil sand, asphaltene and maltene. Fuel. 1977;56(2):114–20. https://doi.org/10.1016/0016-2361(77)90128-4.
Strausz OP, Mojelsky TW, Lown EM. The molecular structure of asphaltene: an unfolding story. Fuel. 1992;71(12):1355–63. https://doi.org/10.1016/0016-2361(92)90206-4.
Tian H, Wang ZM, Xiao ZY, et al. Oil cracking to gases: kinetic modeling and geological significance. Chin Sci Bull. 2006;51(22):2763–70. https://doi.org/10.1007/s11434-006-2188-8.
Tobiszewski M, Namieśnik J. PAH diagnostic ratios for the identification of pollution emission sources. Environ Pollut. 2012;162:110–9. https://doi.org/10.1016/j.envpol.2011.10.025.
Xu X, Huang HP, Zhang SC, et al. Gas generation potential and processes of Athabasca oil sand bitumen from gold-tube pyrolysis experiments. Fuel. 2019;239:804–13. https://doi.org/10.1016/j.fuel.2018.11.077.
Yin PF, Liu GD, Liu YQ, et al. Evaluation of oil sands resources—a case study in the Athabasca Oil Sands, NE Alberta, Canada. Pet Sci. 2013;10(1):30–7. https://doi.org/10.1007/s12182-013-0246-9.
Yunker MB, Macdonald RW, Vingarzan R, et al. PAHs in the Fraser River basin: a critical appraisal of PAH ratios as indicators of PAH source and composition. Org Geochem. 2002;33(4):489–515. https://doi.org/10.1016/S0146-6380(02)00002-5.
Zhang SC, Huang HP, Xiao ZY, et al. Geochemistry of Palaeozoic marine petroleum from the Tarim Basin, NW China. Part 2: Maturity assessment. Org Geochem. 2005;36(8):1215–25. https://doi.org/10.1016/j.orggeochem.2005.01.014.
Zhou S, Huang H, Liu Y. Biodegradation and origin of oil sands in the Western Canada Sedimentary Basin. Pet Sci. 2008;5:87–94. https://doi.org/10.1007/s12182-008-0015-3.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant Number 41573035, 41873049) and the Mitacs project at University of Calgary. Dr Jingkui Mi from Research Institute of Petroleum Exploration and Development, PetroChina was acknowledged for gold –tube experiments. Profs. Steve Larter, Lloyd Snowdon from University of Calgary, three reviewers and associated editor are gratefully acknowledged for the constructive comments that substantially improved the quality of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Jie Hao
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, ZX., Huang, HP. Bulk and molecular composition variations of gold-tube pyrolysates from severely biodegraded Athabasca bitumen. Pet. Sci. 17, 1527–1539 (2020). https://doi.org/10.1007/s12182-020-00484-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-020-00484-4