
Abstract
Encouraged by the wide spectrum of novel applications of gas hydrates, e.g., energy recovery, gas separation, gas storage, gas transportation, water desalination, and hydrogen hydrate as a green energy resource, as well as CO2 capturing, many scientists have focused their attention on investigating this important phenomenon. Of course, from an engineering viewpoint, the mathematical modeling of gas hydrates is of paramount importance, as anticipation of gas hydrate stability conditions is effective in the design and control of industrial processes. Overall, the thermodynamic modeling of gas hydrate can be tackled as an equilibration of three phases, i.e., liquid, gas, and solid hydrate. The inseparable component in all hydrate systems, water, is highly polar and non-ideal, necessitating the use of more advanced equation of states (EoSs) that take into account more intermolecular forces for thermodynamic modeling of these systems. Motivated by the ever-increasing number of publications on this topic, this study aims to review the application of associating EoSs for the thermodynamic modeling of gas hydrates. Three most important hydrate-based models available in the literature including the van der Waals–Platteeuw (vdW–P) model, Chen–Guo model, and Klauda–Sandler model coupled with CPA and SAFT EoSs were investigated and compared with cubic EoSs. It was concluded that the CPA and SAFT EoSs gave very accurate results for hydrate systems as they take into account the association interactions, which are very crucial in gas hydrate systems in which water, methanol, glycols, and other types of associating compounds are available. Moreover, it was concluded that the CPA EoS is easier to use than the SAFT-type EoSs and our suggestion for the gas hydrate systems is the CPA EoS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
1.1 Gas hydrates
The framework of water molecules formed by hydrogen bonds may cause the formation of vacant cavities or cages in which small molecules (< 0.9 nm) like small paraffin, CO2, H2S, etc. can be hold (“trapped”) and creates crystalline compounds resemble ice named as gas hydrates (Sarshar et al. 2010c; Esmaeilzadeh 2006; Sun et al. 2005). To date, more than 130 gas molecules are known to form hydrate. Indeed, the stabilization of the gas hydrate depends on the van der Waals intermolecular forces between the gas and water molecules. Despite the ice, gas hydrates can be stable at temperatures higher than 273.15 K (Talaghat et al. 2009b; Kvenvolden 1998; Milkov 2004; Taylor and Kwan 2004). Originally, the word “clathrate” stems from the Greek word “Khlatron” which means barrier (Chatti et al. 2005).
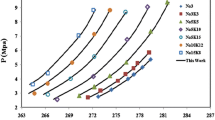
Based on previous publications, to compare hydrate stability regions for a variety of hydrate formers, as shown in Fig. 1 the optimum conditions for hydrate formation are investigated. As can be seen, the formation of hydrate is more likely to occur at high pressures and low temperatures. Actually, the left side of each line in Fig. 1 presents the conditions in which the hydrate can be formed, an area with high pressure and/or low temperature (Sloan 2003; Lee et al. 2012).

Phase diagram of pure methane (Sloan 1998), propane (Strobel et al. 2009), ethylene (Ma et al. 2001), carbon dioxide (Sabil et al. 2010b), mixture of hydrogen and methane hydrate (Zhang et al. 2000), and refrigerants (R14b, R13, R23, and R32) (Kubota et al. 1984; Akiya et al. 1999; Hashimoto et al. 2010; Liang et al. 2001)
Figure 1 demonstrates that methane needs extremely higher pressures to form hydrate with respect to propane. Therefore, it can be concluded that according to the type (size) of gust molecules, the nature of the guest molecules, the pressure–temperature conditions, and the number of water molecules involved in the cavities, three different structures of gas hydrate are considered: structure I (sI), structure II (sII), and structure H (sH) (Pauling and Marsh 1952; Claussen 1951; Ripmeester et al. 1987; Talaghat et al. 2009a). The detailed explanations of these structures can be found in previous studies (Sloan 2003; Sloan 2005; Sloan and Koh 2007).
1.2 Effect of inhibitors and promoters
Some problems are arising by hydrate formation, such as obstacles in pipelines, waste of money, safety risks in oil and gas production, transportation, and processing (Afzal et al. 2007; Mohammadi et al. 2009b, 2010; Sarshar et al. 2008, 2010c). Therefore, there is a considerable need in the industry to prevent hydrate formation. Alongside various hydrate inhibition methods like heating, pressure reduction (Esmaeilzadeh et al. 2011), and water removal, one of the most useful and applicable ways is to apply gas hydrate inhibitors (Sarshar et al. 2010a). Gas hydrate inhibitors (GHIs) are classified into two big categories: thermodynamic hydrate inhibitors (THIs) and low-dosage hydrate inhibitors (LDHIs). THIs prevent hydrate formation by reducing the activity of water in the aqueous system through intermolecular interactions with water molecules (Mech et al. 2015; Esmaeilzadeh and Fathikalajahi 2009). Some common THIs are sodium chloride, magnesium chloride, methanol, ethylene glycol, etc. (Mohammadi et al. 2009b; Mohammadi and Richon 2010). While THIs affect the thermodynamic conditions of hydrate formation (e.g., temperature and pressure) and make the hydrate stability region smaller, LDHIs influence the induction and nucleation stages by decreasing the hydrate nucleation and growth rate through increasing the gas hydrate formation induction time. Induction time is the time interval between reaching the hydrate formation condition and the formation of the hydrate. As a result, they delay hydrate crystal agglomeration (Bakhtyari et al. 2018; Moeini et al. 2017). During the past years, many polymer chemicals like polyvinylpyrrolidone (PVP) and polyvinylcaprolactam (PVCap) have been investigated, among which biodegradable and environmentally friendly ones are of great importance (Daraboina et al. 2011; Kelland 2006; Ohno et al. 2010; Al-Adel et al. 2008).
On the other hand, hydrate can be taken into account as a useful phase in many applications (Sarshar et al. 2010b). Indeed, it can be used in gas storage (Sun et al. 2003b; Khokhar et al. 1998; Gudmundsson et al. 1994; Ohgaki et al. 1996; Sun et al. 2003a; Lee et al. 2005; Veluswamy and Linga 2013; Aliabadi et al. 2015), transmission (Sun et al. 2003b), separation technology (Eslamimanesh et al. 2012b; Kamata et al. 2004; Arjmandi et al. 2007; Nagata et al. 2009; Shiojiri et al. 2004; Tang et al. 2013), energy resource (Collett 2002; Kvenvolden 1993; Makogon et al. 2007; Collett 2004; Chong et al. 2016), CO2 capturing (Kang and Lee 2000; Zhong et al. 2016; Spencer and Currier 2002; Duc et al. 2007; Dickens 2003; Ma et al. 2016; Babu et al. 2016; Zhong et al. 2015; Yang et al. 2015; Sarshar et al. 2009), and solving geohazard problems (Maslin et al. 2010; Kvenvolden 1999; Milkov et al. 2000; Ruppel et al. 2008; Yamamoto et al. 2015). As mentioned, the conditions of hydrate formation are difficult (high pressure and low temperature), and in practical applications, the conditions need to be moderated (Eslamimanesh et al. 2012a). Therefore, promoters appear to be significantly useful chemicals moderating the formation conditions of hydrates (Papadimitriou et al. 2011; Illbeigi et al. 2011; Sloan Jr and Koh 2007; Partoon and Javanmardi 2013; Shahnazar and Hasan 2014; Sabil et al. 2010a; Aliabadi et al. 2015). Overall, one can classify promoters into two essential classifications: thermodynamic and kinetic promoters. As the names suggest, thermodynamic promoters affect the equilibrium conditions of liquid water, hydrate, and vapor (Lw–H–V) and shift it to a higher temperature and lower pressure like tetrahydrofuran (THF), dimethyl cyclohexane, and cyclopentane; however, kinetic promoters cause the hydrate formation to become faster through accelerating the nucleation and growth steps like biosurfactants (Sloan Jr and Koh 2007; Partoon and Javanmardi 2013).
1.3 Equation of states for associating fluids
Simple molecules, either organic (e.g., toluene, methyl chloride) or inorganic (O2, CO, N2, N2O, etc.), have a long history of being thermodynamically modeled by many commonly used EoSs, namely Peng–Robinson (PR) (Peng and Robinson 1976), Soave–Redlich–Kwong (SRK) (Soave 1972), and Esmaeilzadeh-Roshanfekr (ER) (Esmaeilzadeh and Roshanfekr 2006). These EoSs only take into account the van der Waals attractions as well as weak electrostatic forces, resulting from dipoles, quadruples, etc. The aforementioned cubic EoSs have two parts: the attraction intermolecular force and the repulsion intermolecular force.
On the other hand, many real-world fluids have Columbic, strong polar forces, along with forces together with chain flexibility, induction forces, acid–base interactions, electrolyte solutions, etc. To deal with the associating compounds, the term “chemical theory” has been coined, which means the associating complexes are acted as unique new chemical species. Taking into account the chemical equilibria between the initial components and these complexes leads to new EoSs, reflecting the effect of non-ideal structures of associating fluids (Müller and Gubbins 2001). This idea is the foundation of the development of several molecular-based EoSs from statistical thermodynamics such as different versions of statistical associating fluid theory (SAFT) (Gil-Villegas et al. 1997; Tan et al. 2008; Economou 2002) and cubic plus association (CPA) (Kontogeorgis et al. 1999, 2006a, b). The inevitable existence of water or alcohol-based hydrate inhibitors in any hydrate system is a significant challenge to model these systems using conventional EoSs, which neglect any association interactions. Therefore, the application of newly developed statistical EoSs has been becoming more and more substantial for the thermodynamic modeling of hydrate systems, which is the focus of this study to propose a suitable thermodynamic package for modeling of gas hydrate equilibrium conditions.
1.4 Objective
Whether as a negative phenomenon or a useful application in chemical processes, the thermodynamic modeling and phase behavior of gas hydrate are a vital engineering necessity. Indeed, the prediction of dissociation conditions of the hydrate phase plays a major role in the design of industrial applications regarding gas hydrate. This study aims to address the statistical EoS application for the thermodynamic modeling of gas hydrates and come to a conclusion about the advantages and shortcomings of each approach. Since a variety of models were presented to compute the chemical potential of components in the solid and fluid phases, adequate knowledge on the advantages and shortcomings of each model is of interest and significance. The water vapor pressures at the hydrate equilibrium temperatures (below 350 K) are much lower than the pure or mixed gas vapor pressures. Therefore, the vapor phase primarily consists of gas molecules. Hence, the fugacity of the vapor phase can be simply computed using common cubic EoS and mixing rules. However, the bottleneck of the hydrate equilibrium conditions modeling is to computing the chemical potential or fugacity of the solid and liquid phases. The objective of this work is to introduce several popular novel models that are used for hydrate modeling of systems containing associating compounds. Three important models are reviewed for the hydrate phase, while for the fluid phases the SAFT and CPA EoS along with the cubic EoSs are discussed.
2 Thermodynamic modeling of gas hydrate
The basic deterministic idea to enhance a reliable thermodynamic model anticipating gas hydrate dissociation conditions is the equality of the chemical potential of the components in the three involved phases (i.e., hydrate, gas, and fluid phases). Reviewing the previous publications, the thermodynamic approaches of hydrate systems modeling are categorized into three sections. The equality of the chemical potential and the fugacity, as well as the type of the component (water or other components), are the basis of categorization. More details of the different approaches for gas hydrate modeling can be found in the following subsections.
2.1 The van der Waals and Platteeuw (vdW–P) solid solution theory
The most common and well-known model to calculate the behavior of the hydrate phase is the van der Waals and Platteeuw (vdW–P) (Van der Waals and Platteeuw 2007; Platteeuw and Van der Waals 1959). This statistically based, thermodynamic model is the starting point for many further types of research regarding the hydrate thermodynamic model. In the original work, the equalization of the water chemical potential in all the coexisting phases is considered:
Since the vapor pressures of water in the hydrate formation temperature ranges are very low, the contribution of water in the vapor phase is neglected. The chemical potential of water in the hydrate phase is calculated as follows:
where \(\vartheta_{m}\) and \(\theta_{mi},\) respectively, denote the number of cages of type “m” per water molecules in a hydrate unit cell and the fractional occupancy of the hydrate cavity type “m” by the guest molecule of type “i”. The latter is specified as follows (Sloan Jr and Koh 2007; Van der Waals and Platteeuw 2007):
where \(f_{i}\) stands for the fugacity of the guest component “i”. The parameter \(C_{mi}\) represents the Langmuir constant of component “i” and simulates the occupation of the cavity by the guest molecule like the ideal adsorption of gas molecules on the solid surfaces and is formulated as follows:
where \(R^{\prime}\) is the radius of the spherical cavity and \(W\left(r \right)\) specifies the appropriate potential function for calculation of the intermolecular forces between the gas and water molecules. McKoy and Sinanoğlu (1963) used the three-parameter Kihara potential function and developed the formula for calculation of \(W\left(r \right)\) to take into account all the interactions between the gas molecule on the cavity and all the water molecules on the cavity wall, which can be seen in the following equation:
where
Apart from the aforementioned equations, Parrish and Prausnitz (1972) developed an empirical correlation for \(C_{mi}\) calculation. Their equation, which made the process of \(C_{mi}\) calculation much easier, is valid for temperatures between 260 and 300 K. This equation can be shown by the following equation:
In Eq. (7), \(A_{mi}\) and \(B_{mi}\) are optimized constants, which can be found in Parrish and Prausnitz (1972). They also improved the vdW–P model by applying the fugacity of hydrate former instead of its partial pressure to take into account the non-ideality of the vapor phase. It is worth mentioning that the constants were found for most of the components ranging from hydrocarbons to noble gases. Moreover, they introduced a procedure to calculate the hydrate equilibrium conditions for the gas mixtures.
Apart from the aforementioned equation, the water chemical potential difference between the liquid/ice phase and vacant lattice, \({\Delta}\mu_{w}^{\beta - L/I}\), is calculated based on the following equation:
In Eq. (8), \({\Delta}\mu_{w}^{\beta - L/I}\) denotes the chemical potential difference between the vacant lattice and liquid water or ice at reference condition (101.325 kPa, \(T_{0}\)). \({\Delta}h_{w}^{\beta - L/I}\) and \({\Delta}v_{w}^{\beta - L/I}\), respectively, represent the volume and enthalpy difference between vacant lattice and liquid water or ice. \(P\) is pressure, and the term \(x_{w} \gamma_{w}\) represents the activity coefficient of water in the aqueous solution. Also, \(T_{0}\) represents the temperature at which ice appears and depends on the guest molecules. \({\Delta}h_{w}^{\beta - L/I}\) can be measured using the following equation:
Table 1 presents the previous studies on the gas hydrate equilibrium modeling using the vdW–P model.
2.2 The Chen–Guo hydrate model
The Chen–Guo hydrate model is in accordance with a two-stage mechanism: first, the formation of hydrate empty cages via a quasi-chemical reaction and second, the adsorption of some gases (with relatively small dimensions) into the cavities, accounting for the non-stoichiometric hydrate properties. As opposed to the vdW–P thermodynamic model, which considers the equality of water in the hydrate and fluid phases, the Chen–Guo hydrate model balances the hydrate former fugacity in the fluid phase and that in hydrate phase as follows (Chen and Guo 1996):
In Eq. (13), \(\lambda_{1}\) and \(\lambda_{2}\) denote the number of small cavities per water molecule and the number of hydrate formers (salt molecules) encaged in the basic hydrate (large cavities) per water molecule, respectively. \(f_{i}\) stands for the fugacity of hydrate former “i” in the gas/liquid phase computed by an equation of state, and \(x_{i}\) stands for the mole fraction of gas component “i” in the large cavities. \(\theta_{k}\) represents the fraction of small voids occupied by the gas component. It is formulated as:
\(C_{k}\) stands for the Langmuir constant, which represents the interactions between the guest and host molecules, and in the Chen–Guo hydrate model, the Antonie-type equation was considered for it:
Here, \(X_{k}\), \(Y_{k}\), and \(Z_{k}\) represent the constants of component “k,” which is optimized using the gas hydrate equilibrium data and the values of them have been given in the literature (Chen and Guo 1998).
The symbol \(f_{i}^{H0}\) in Eq. (11) denotes the hydrate former fugacity in an equilibrium state with the unfilled pure basic hydrate “i,” manipulated as:
In Eq. (16), \(a_{w}\) is the activity of water that can be calculated using an appropriate relation for water activity coefficient. It is worth mentioning that, in most cases without a thermodynamic inhibitor or promoter in water, the activity of water is assumed to be equal to unity and for the hydrate inhibitors has the value less than unity.
In Eq. (17), \(A_{ij}\) is the binary interaction parameters expressing the interaction between the guest molecules in the small voids and in the large voids; \(A_{i}^{\prime}\), \(B_{i}^{\prime}\), and \(C_{i}^{\prime}\) are the constants of component “i.” The values of them can be optimized or found in the literature (Chen and Guo 1998). Table 2 presents a review of the studies that used Chen–Guo model for modeling of hydrate equilibrium conditions.
2.3 The equalization of water fugacity in the hydrate and fluid phases
Another common approach in hydrate modeling is the equality of the water fugacities in all of the phases. Usually, three phases, i.e., hydrate, hydrocarbon (either gas or liquid), and liquid water, are considered, at the same temperature and pressure. This thermodynamic problem can be formulized as follows (Klauda and Sandler 2000):
where the subscript “w” represents the water.
As stated, the vdW–P model assumes the equality of the chemical potential of water in the hydrate and fluid phases. Using the vdW–P expression of chemical potential, the fugacity of water in the hydrate phase is calculated as follows:
In Eq. (20), \(f_{w}^{\beta}\) is the fugacity of empty hydrate lattice. The fugacity of water in the gas and liquid phases is computed using the EoS.
Based on phase equilibrium and by using different methods predicting fugacities, many more-or-less complex modeling approaches have been developed (Table 3).
3 Results
Chocked up pipelines by hydrate were the main reasons for starting researches about hydrate formation. Afterward, its applications, such as gas storage and CO2 capturing, were discovered, and researchers became more interested in investigating the hydrate formation and dissociation conditions. Indeed, in some cases hydrate formation is very beneficial, whereas sometimes it has disadvantages; either way, this research topic is of great importance (Shahnazar and Hasan 2014; Chen and Guo 1996; Parrish and Prausnitz 1972). Inspired by Sloan (2005), Fig. 2 shows the increasing rate of the number of publications in the twentieth century. As can be seen, the number of publications each year had an increasing manner until 2010, when it started to oscillate.

Number of publications related to hydrate from 2000 until 2019
As mentioned, thermodynamic modeling of gas hydrates is one of the most important engineering topics, with lots of applications in oil, gas, and chemical industries. The first statistical EoS used to model these systems was the SAFT EoS. Li et al. (2006) suggested the SAFT in conjunction with the vdW–P to predict the behavior of single hydrates (C1–C3, and CO2) in the presence of methanol and glycerol as hydrate inhibitors. However, CPA application for the vapor–liquid equilibria (VLE) (Folas et al. 2006) and liquid–liquid equilibria (LLE) (Oliveira et al. 2007) of natural gas hydrate composition has been investigated before 2007; Kontogeorgis et al. (2007) were the first ones to combine CPA EoS with the vdW–P model and suggested reliable models for the phase behavior of water, alcohols, and natural gas components.
Tavasoli et al. (2016) investigated the influence of cyclic hydrocarbons (i.e., benzene and cyclohexane) on the methane and CO2 hydrates. They implemented the fugacity-based model, in which Valderrama–Patel–Teja (VPT) EoS [with non-density-dependent (NDD) mixing rule] (Valderrama 1990) and Statistical Associating Fluid Theory EoS presented by Huang and Radosz (SAFT-HR) (Huang and Radosz 1990, 1991) were compared. They investigated the four systems of methane + benzene + water, methane + cyclohexane + water, CO2 + benzene + water, and CO2 + cyclohexane + water and, respectively, reported the error of 8.09, 8.42, 6.18, and 13.25 for the SAFT-HR EoS, corresponding to 9.37, 6.95, 3.99, and 15.35 for VPT EoS. Their obtained outcomes are shown in Fig. 3. As can be seen, the two EoSs generally led to almost the same results; however, the SAFT-HR superiority at high pressures is quite visible. It is worth mentioning that due to the consideration of associating term in SAFT-HR, it is expected to result in more reliable predictions; nonetheless, in some cases, VPT EoS even provided more accurate results. This can be attributed to the fact that the introduction of the fitting parameters in the mixing rule (non-density-dependent) of VPT EoS compensated its weaknesses.

Comparison of the experimental data (Sun et al. 2002; Tohidi et al. 1996; Tavasoli and Feyzi 2016; Danesh et al. 1993; Mohammadi et al. 2009a; Tavasoli and Feyzi 2016; Mooijer-van den Heuvel et al. 2001) and the predictions of SAFT-HR and VPT EoSs (Tavasoli and Feyzi 2016) for the effect of cyclic hydrocarbons on the methane and CO2 gas hydrates
In order to compare the error of the SAFT EoS with that of some non-associating EoSs, the value of errors corresponding to various EoSs for systems of pure natural gas hydrates (C1–C4, CO2, H2S), their combination with alcohols and electrolytes are tabulated in Table 4. As can be seen, for the pure gas system, the SAFT EoS is not the most accurate one. However, it results in a lower error in the cases of the existence of alcohols and electrolytes.
Several factors influence the hydrate equilibrium calculations including: the hydrate model selection, the appropriate EoS selection, type of hydrate former, existence of inhibitors and promoters in the system, and choosing the proper statistical or empirical relations for calculation of the Langmuir constants. In the thermodynamic modeling of hydrate equilibrium conditions, two approaches are used in the literature. The first approach is the chemical potential-based approach introduced with the van der Waals-Platteeuw (vdW–P) model, and the other one is the fugacity-based approach proposed by Klauda–Sandler and Chen–Guo. There is also a significant difference between the fugacity-based model proposed by Klauda and Sandler and that obtained by Chen–Guo. The basis of the Klauda–Sandler model is the equality of water chemical potential in all coexisting phases, while the basis of the Chen–Guo model is the equality of the hydrate formers fugacities in all phases. Moreover, the vdW–P ignores the contribution of water in the vapor phase because of its low vapor pressures at the hydrate formation temperature range. Therefore, several models with various assumptions are available and the sensitivity analysis can be applied for one model, for example, the vdW–P model. In Table 5, as a case study, we compared the average absolute relative deviation in calculated pressures of the PC-SAFT EoS and the PR EoS for the same gases, the same hydrate model (vdW–P), and the same Kihara parameters (Sloan Jr and Koh 2007).
To verify the inhibition effect of monoethylene glycol, Boesen et al. (2017) suggested a fugacity-based model for C1, CO2, and H2S hydrate systems. They compared the results of CPA and SRK EoSs and reached almost the same results. Moreover, chapoy et al. (Chapoy et al. 2012a) conducted an experimental and modeling assessment to investigate the phase behavior of CO2 + water system. Considering the fugacity-based model, they compared the performance of CPA, SRK [with Huron–Vidal (HV) (Huron and Vidal 1979) mixing rules], and VPT [with NDD (Valderrama 1990; Avlonitis et al. 1994; Wong and Sandler 1992) mixing rule]. They came to the conclusion that VPT + NDD model resulted in the highest accuracy, followed by CPA, and SRK + HV models. Implementing the fugacity-based model, Karamoddin and Varaminian (Karamoddin and Varaminian 2013) addressed the capability of three EoSs, namely SRK, VPT, and CPA, for the prediction of refrigerants hydrate dissociation condition. Figure 4 provides a visual comparison of the performance of these three EoSs for HCFC22 hydrate. As can be seen, the three approaches led to acceptable errors. Indeed, the average error of SRK, VPT, and CPA was reported to be 2.8, 3.2, and 3.0 percent, respectively. This implies that the associating term of CPA was not able to provide the most accurate results.

In order to compare the popularity of the CPA and SAFT EoSs, Fig. 5 is depicted. In general, the number of studies related to CPA is higher. As can be seen, in some of the years (e.i. 2009, 2010, and 2012) the SAFT was not the case of study at all. Moreover, the highest number of publications about CPA was published in 2019, whereas the SAFT EoS was not considered in any publication in 2019. One can consider the complexity of the SAFT EoS for this observation. To fully investigate the reason for this, we need to assess the type of gas hydrates along with the chemical formula of the promoters and inhibitors. Thus, Fig. 6 is plotted.

Comparison between the number of publications related to CPA and SAFT

a Percentage of different types of gas hydrate, for modeling of which statistical EoS have been used, b comparison of the number of publications related to CPA and SAFT for natural gas hydrate
As Fig. 6 exhibits, most of the applications of statistical EoSs are related to natural gas hydrate. It is worth mentioning that usually a mixture of C1–C5, CO2, N2, Ar, H2S, O2, CO is considered as a synthetic natural gas. Figure 6b demonstrates the number of publications using the CPA and SAFT EoSs for different types of components including ILs, electrolytes, surfactants, alcohols, and other hydrocarbons. The number of publications implementing CPA is more significant in almost all of the cases, even for electrolyte mixtures. Kontogeorgis et al. (2007) stated that CPA and SAFT result in similar predictions for mixtures of water and alcohols (methanol, MEG, and TEG). Also, using the CPA EoS leads to negligible errors for electrolyte mixtures (Chapoy et al. 2012b; Ngema et al. 2019a, b).
4 Conclusions
In this study, different approaches using statistical EoSs for predicting hydrate dissociation conditions have been reviewed. According to the fact that hydrate has many novel, promising applications, its modeling has gained much attention. Indeed, as the models get developed, they are more sophisticated in order to more accurately predict the phase behavior of hydrates. Moreover, because of the existence of water along with promoters, inhibitors, or even impurities in the system, applying statistical thermodynamic equations of states is of great importance. According to the previous publications, CPA is more popular than SAFT. This can be attributed to the fact that it is more facile and yet completely reliable. In addition, this study reveals that even though using a complex associating EoS, such as SAFT or CPA, contributes to slightly better results (e.g., for systems containing alcohols or electrolytes), it does not necessarily guarantee more accurate predictions in all of the cases. Indeed, the introduction of adjustable parameters in the mixing rule of non-associating EoSs overcomes their weaknesses, making them proper options for thermodynamically model such polar systems.
Abbreviations
- PR:
-
Peng–Robinson
- PT:
-
Patel–Teja
- ANNs:
-
Artificial neural networks
- ANFIS:
-
Adaptive neuro-fuzzy inference system
- vdW–P:
-
Van der Waals and Platteeuw
- PRSV2:
-
Stryjek and Vera modification of Peng–Robinson
- BiMSA:
-
Binding mean spherical approximation
- NRTL:
-
Non-random two-liquid
- \(A^{\text{id}}\) :
-
Free energy of an ideal gas
- \(A^{\text{hs}}\) :
-
Free energy of a hard-sphere fluid relative to the ideal gas
- \(A^{\text{chain}}\) :
-
Free energy when chains are formed from hard spheres
- \(A^{\text{disp}}\) :
-
Contributions to the free energy of dispersion
- \(A^{\text{assoc}}\) :
-
Contributions to the free energy of association
- \(\rho_{n}\) :
-
Total number density of molecules in solution
- \(d_{ii}\) :
-
Hard sphere diameter of segment i
- \(\sigma_{ii}\) :
-
Soft sphere diameter of segment i
- \(\varepsilon_{ii}\) :
-
Energy parameter
- \(\sigma_{ij}\) :
-
Cross parameter between different segments
- \(\varepsilon_{ij}\) :
-
Cross parameter between different segments
- \(M_{i}\) :
-
Number of associating sites
- \(X^{{A_{i}}}\) :
-
Mole fraction of molecule i, not bonded at site A, in mixtures with other components
- \(\sum\limits_{{B_{j}}}\) :
-
Summation over all sites on molecule j
- \(\sum\limits_{j}\) :
-
Summation over all components
- \(\rho\) :
-
Total molar density of molecules in the solution
- \(\Delta^{{A_{i} B_{j}}}\) :
-
Associating strength
- \(\kappa^{AB}\) :
-
Bonding volume
- \(k_{ij}^{AB}\) :
-
Binary associating interaction parameter
- T c :
-
Critical temperature
- P c :
-
Critical pressure
- P :
-
Pressure
- V :
-
Specific volume
- R :
-
Universal gas constant
- T :
-
Temperature
- P :
-
Pressure
- W :
-
Acentric factor
- F :
-
Fugacity
- ϕ :
-
Fugacity coefficient
- v :
-
Molar volume
- \(P_{w}^{MT}\) :
-
Vapor pressure of water in empty hydrate lattice
- \(\phi_{w}^{MT}\) :
-
Fugacity coefficient of water in empty hydrate lattice
- \(v_{w}^{MT}\) :
-
Partial molar volume of water in the empty hydrate lattice
- \(v_{i}\) :
-
Number of cages of type i per water molecule in a unit hydrate cell
- \(Y_{ki}\) :
-
Fractional occupancy of the hydrate cavity i by guest molecule type k
- x :
-
Mole fraction in the aqueous phase
- \(\gamma\) :
-
Activity coefficient
- \(x_{P}^{L}\) :
-
Mole fraction of promoter in the aqueous phase
- \(\gamma_{P}\) :
-
Activity coefficient of promoter in the aqueous phase
- \(P_{P}^{sat}\) :
-
Promoter vapor pressure
- W:
-
Water
- L:
-
Liquid
- H:
-
Hydrate phase
- G:
-
Gas phase
References
Abolala M, Varaminian F. Thermodynamic model for predicting equilibrium conditions of clathrate hydrates of noble gases + light hydrocarbons: combination of Van der Waals-Platteeuw model and sPC-SAFT EoS. J Chem Thermodyn. 2015;81:89–94. https://doi.org/10.1016/j.jct.2014.09.013.
Abolala M, Karamoddin M, Varaminian F. Thermodynamic modeling of phase equilibrium for gas hydrate in single and mixed refrigerants by using sPC-SAFT equation of state. Fluid Phase Equilib. 2014;370:69–74. https://doi.org/10.1016/j.fluid.2014.02.013.
Afzal W, Mohammadi AH, Richon D. Experimental measurements and predictions of dissociation conditions for carbon dioxide and methane hydrates in the presence of triethylene glycol aqueous solutions. J Chem Eng Data. 2007;52(5):2053–5. https://doi.org/10.1021/je700170t.
Ahmadian S, Mohammadi M, Ehsani MR. Thermodynamic modeling of methane hydrate formation in the presence of imidazolium-based ionic liquids using two-step hydrate formation theory and CPA EoS. Fluid Phase Equilib. 2018;474:32–42. https://doi.org/10.1016/j.fluid.2018.07.004.
Akiya T, Shimazaki T, Oowa M, Matsuo M, Yoshida Y. Formation conditions of clathrates between HFC alternative refrigerants and water. Int J Thermophys. 1999;20(6):1753–63. https://doi.org/10.1023/A:1022614114505.
Al-Adel S, Dick JA, El-Ghafari R, Servio P. The effect of biological and polymeric inhibitors on methane gas hydrate growth kinetics. Fluid Phase Equilib. 2008;267(1):92–8. https://doi.org/10.1016/j.fluid.2008.02.012.
Aliabadi M, Rasoolzadeh A, Esmaeilzadeh F, Alamdari A. Experimental study of using CuO nanoparticles as a methane hydrate promoter. J Nat Gas Sci Eng. 2015;27:1518–22. https://doi.org/10.1016/j.jngse.2015.10.017.
Arjmandi M, Chapoy A, Tohidi B. Equilibrium data of hydrogen, methane, nitrogen, carbon dioxide, and natural gas in semi-clathrate hydrates of tetrabutyl ammonium bromide. J Chem Eng Data. 2007;52(6):2153–8. https://doi.org/10.1021/je700144p.
Avlonitis D, Danesh A, Todd AC. Prediction of VL and VLL equilibria of mixtures containing petroleum reservoir fluids and methanol with a cubic EoS. Fluid Phase Equilib. 1994;94:181–216. https://doi.org/10.1016/0378-3812(94)87057-8.
Babu P, Ong HWN, Linga P. A systematic kinetic study to evaluate the effect of tetrahydrofuran on the clathrate process for pre-combustion capture of carbon dioxide. Energy. 2016;94:431–42. https://doi.org/10.1016/j.energy.2015.11.009.
Bakhtyari A, Fayazi Y, Esmaeilzadeh F, Fathikaljahi J. Experimental determination of the temperature suppression in formation of gas hydrate in water based drilling mud. J Pet Sci Technol. 2018;8(1):16–31. https://doi.org/10.22078/jpst.2017.2103.1372.
Boesen RR, Herslund PJ, Sørensen H. Loss of monoethylene glycol to CO2 and H2S-rich fluids: modeled using Soave–Redlich–Kwong with the Huron and Vidal mixing rule and Cubic-Plus-Association equations of state. Energy Fuels. 2017;31(4):3417–26. https://doi.org/10.1021/acs.energyfuels.6b02365.
Burgass R, Chapoy A, Li X. Gas hydrate equilibria in the presence of monoethylene glycol, sodium chloride and sodium bromide at pressures up to 150 MPa. J Chem Thermodyn. 2018;118:193–7. https://doi.org/10.1016/j.jct.2017.10.007.
Chapoy A, Haghighi H, Burgass R, Tohidi B. Gas hydrates in low water content gases: experimental measurements and modelling using the CPA equation of state. Fluid Phase Equilib. 2010;296(1):9–14. https://doi.org/10.1016/j.fluid.2009.11.026.
Chapoy A, Haghighi H, Burgass R, Tohidi B. On the phase behaviour of the (carbon dioxide + water) systems at low temperatures: experimental and modelling. J Chem Thermodyn. 2012a;47:6–12. https://doi.org/10.1016/j.jct.2011.10.026.
Chapoy A, Mazloum S, Burgass R, Haghighi H, Tohidi B. Clathrate hydrate equilibria in mixed monoethylene glycol and electrolyte aqueous solutions. J Chem Thermodyn. 2012b;48:7–12. https://doi.org/10.1016/j.jct.2011.12.031.
Chapoy A, Alsiyabi I, Gholinezhad J, Burgass R, Tohidi B. Clathrate hydrate equilibria in light olefins and mixed methane–olefins systems. Fluid Phase Equilib. 2013;337:150–5. https://doi.org/10.1016/j.fluid.2012.09.015.
Chapoy A, Burgass R, Tohidi B, Alsiyabi I. Hydrate and phase behavior modeling in CO2-rich pipelines. J Chem Eng Data. 2015;60(2):447–53. https://doi.org/10.1021/je500834t.
Chatti I, Delahaye A, Fournaison L, Petitet J-P. Benefits and drawbacks of clathrate hydrates: a review of their areas of interest. Energy Convers Manag. 2005;46(9):1333–43. https://doi.org/10.1016/j.enconman.2004.06.032.
Chen G-J, Guo T-M. Thermodynamic modeling of hydrate formation based on new concepts. Fluid Phase Equilib. 1996;122(1):43–65. https://doi.org/10.1016/0378-3812(96)03032-4.
Chen G-J, Guo T-M. A new approach to gas hydrate modelling. Chem Eng J. 1998;71(2):145–51. https://doi.org/10.1016/S1385-8947(98)00126-0.
Chin H-Y, Hsieh M-K, Chen Y-P, Chen P-C, Lin S-T, Chen L-J. Prediction of phase equilibrium for gas hydrate in the presence of organic inhibitors and electrolytes by using an explicit pressure-dependent Langmuir adsorption constant in the van der Waals-Platteeuw model. J Chem Thermodyn. 2013;66:34–43. https://doi.org/10.1016/j.jct.2013.06.014.
Chong ZR, Yang SHB, Babu P, Linga P, Li XS. Review of natural gas hydrates as an energy resource: prospects and challenges. Appl Energy. 2016;162:1633–52. https://doi.org/10.1016/j.apenergy.2014.12.061.
Claussen W. A second water structure for inert gas hydrates. J Chem Phys. 1951;19(11):1425–6. https://doi.org/10.1063/1.1748079.
Collett TS. Energy resource potential of natural gas hydrates. AAPG Bull. 2002;86(11):1971–92. https://doi.org/10.1306/61EEDDD2-173E-11D7-8645000102C1865D.
Collett TS. Gas hydrates as a future energy resource. Geotimes. 2004;49:24–7.
Danesh BT, Burgass RW, Todd AC. Benzene can form gas hydrates. Chem Eng Res Des. 1993;71:457–9.
Daraboina N, Linga P, Ripmeester J, Walker VK, Englezos P. Natural gas hydrate formation and decomposition in the presence of kinetic inhibitors. 2. Stirred reactor experiments. Energy Fuels. 2011;25(10):4384–91. https://doi.org/10.1021/ef200813v.
Delavar H, Haghtalab A. Prediction of hydrate formation conditions using GE-EoS and UNIQUAC models for pure and mixed-gas systems. Fluid Phase Equilib. 2014;369:1–12. https://doi.org/10.1016/j.fluid.2014.02.009.
Dickens GR. Rethinking the global carbon cycle with a large, dynamic and microbially mediated gas hydrate capacitor. Earth Planet Sci Lett. 2003;213(3):169–83. https://doi.org/10.1016/S0012-821X(03)00325-X.
Duc NH, Chauvy F, Herri J-M. CO2 capture by hydrate crystallization—a potential solution for gas emission of steelmaking industry. Energy Convers Manag. 2007;48(4):1313–22. https://doi.org/10.1016/j.enconman.2006.09.024.
Economou IG. Statistical associating fluid theory: a successful model for the calculation of thermodynamic and phase equilibrium properties of complex fluid mixtures. Ind Eng Chem Res. 2002;41(5):953–62. https://doi.org/10.1021/ie0102201.
El Meragawi S, Diamantonis N, Tsimpanogiannis IN, Economou I. Hydrate–fluid phase equilibria modeling using PC-SAFT and Peng–Robinson equations of state. Fluid Phase Equilib. 2015;413:209–19. https://doi.org/10.1016/j.fluid.2015.12.003.
Eslamimanesh A, Gharagheizi F, Illbeigi M, Mohammadi AH, Fazlali A, Richon D. Phase equilibrium modeling of clathrate hydrates of methane, carbon dioxide, nitrogen, and hydrogen + water soluble organic promoters using Support Vector Machine algorithm. Fluid Phase Equilib. 2012a;316:34–45. https://doi.org/10.1016/j.fluid.2011.11.029.
Eslamimanesh A, Mohammadi AH, Richon D, Naidoo P, Ramjugernath D. Application of gas hydrate formation in separation processes: a review of experimental studies. J Chem Thermodyn. 2012b;46:62–71. https://doi.org/10.1016/j.jct.2011.10.006.
Esmaeilzadeh F. Simulation examines ice, hydrate formation in Iran separator centers. Oil Gas J. 2006;104(11):46–52.
Esmaeilzadeh F, Fathikalajahi J. Prediction of gas consumption during hydrate formation with or without the presence of inhibitors in a batch system using the Esmaeilzadeh-Roshanfekr Equation of State. Chem Biochem Eng Q. 2009;23:23.
Esmaeilzadeh F, Roshanfekr M. A new cubic equation of state for reservoir fluids. Fluid Phase Equilib. 2006;239(1):83–90. https://doi.org/10.1016/j.fluid.2005.10.013.
Esmaeilzadeh F, Zeighami M, Kaljahi J. 1-D mathematical modeling of hydrate decomposition in porous media by depressurization and thermal stimulation. J Porous Media. 2011;14:1–16. https://doi.org/10.1615/JPorMedia.v14.i1.10.
Ferrari PF, Guembaroski AZ, Marcelino Neto MA, Morales REM, Sum AK. Experimental measurements and modelling of carbon dioxide hydrate phase equilibrium with and without ethanol. Fluid Phase Equilib. 2016;413:176–83. https://doi.org/10.1016/j.fluid.2015.
Folas GK, Kontogeorgis GM, Michelsen ML, Stenby EH. Vapor–liquid, liquid–liquid and vapor–liquid–liquid equilibrium of binary and multicomponent systems with MEG: modeling with the CPA EoS and an EoS/GE model. Fluid Phase Equilib. 2006;249(1):67–74. https://doi.org/10.1016/j.fluid.2006.08.021.
Fouad WA, Song KY, Chapman WG. Experimental measurements and molecular modeling of the hydrate equilibrium as a function of water content for pressures up to 40 MPa. Ind Eng Chem Res. 2015;54(39):9637–44. https://doi.org/10.1021/acs.iecr.5b02240.
Gil-Villegas A, Galindo A, Whitehead PJ, Mills SJ, Jackson G, Burgess AN. Statistical associating fluid theory for chain molecules with attractive potentials of variable range. J Chem Phys. 1997;106(10):4168–86. https://doi.org/10.1063/1.473101.
Gudmundsson JS, Parlaktuna M, Khokhar A. Storage of natural gas as frozen hydrate. SPE Prod Facil. 1994;9(01):69–73. https://doi.org/10.2118/24924-PA.
Haghighi H. Methane and water phase equilibria in the presence of single and mixed electrolyte solutions using the Cubic-Plus-Association Equation of State. Oil Gas Sci Technol. 2009;64:141–54. https://doi.org/10.2516/ogst:2008043.
Haghighi H, Chapoy A, Burgess R, Mazloum S, Tohidi B. Phase equilibria for petroleum reservoir fluids containing water and aqueous methanol solutions: experimental measurements and modelling using the CPA equation of state. Fluid Phase Equilib. 2009a;278(1):109–16. https://doi.org/10.1016/j.fluid.2009.01.009.
Haghighi H, Chapoy A, Burgess R, Tohidi B. Experimental and thermodynamic modelling of systems containing water and ethylene glycol: application to flow assurance and gas processing. Fluid Phase Equilib. 2009b;276(1):24–30. https://doi.org/10.1016/j.fluid.2008.10.006.
Hashimoto S, Miyauchi H, Inoue Y, Ohgaki K. Thermodynamic and Raman spectroscopic studies on Difluoromethane (HFC-32) + Water binary system. J Chem Eng Data. 2010;55(8):2764–8. https://doi.org/10.1021/je9009859.
Hejrati Lahijani MA, Xiao C. SAFT modeling of multiphase equilibria of methane–CO2–water–hydrate. Fuel. 2017;188:636–44. https://doi.org/10.1016/j.fuel.2016.10.008.
Herslund PJ, Thomsen K, Abildskov J, von Solms N. Phase equilibrium modeling of gas hydrate systems for CO2 capture. J Chem Thermodyn. 2012;48:13–27. https://doi.org/10.1016/j.jct.2011.12.039.
Herslund PJ, Daraboina N, Thomsen K, Abildskov J, von Solms N. Measuring and modelling of the combined thermodynamic promoting effect of tetrahydrofuran and cyclopentane on carbon dioxide hydrates. Fluid Phase Equilib. 2014a;381:20–7. https://doi.org/10.1016/j.fluid.2014.08.015.
Herslund PJ, Thomsen K, Abildskov J, von Solms N. Modelling of cyclopentane promoted gas hydrate systems for carbon dioxide capture processes. Fluid Phase Equilib. 2014b;375:89–103. https://doi.org/10.1016/j.fluid.2014.04.039.
Herslund PJ, Thomsen K, Abildskov J, von Solms N. Modelling of tetrahydrofuran promoted gas hydrate systems for carbon dioxide capture processes. Fluid Phase Equilib. 2014c;375:45–65. https://doi.org/10.1016/j.fluid.2014.04.031.
Hsieh M-K, Yeh Y-T, Chen Y-P, Chen P-C, Lin S-T, Chen L-J. Predictive method for the change in equilibrium conditions of gas hydrates with addition of inhibitors and electrolytes. Ind Eng Chem Res. 2012;51(5):2456–69. https://doi.org/10.1021/ie202103a.
Huang SH, Radosz M. Equation of state for small, large, polydisperse, and associating molecules. Ind Eng Chem Res. 1990;29(11):2284–94. https://doi.org/10.1021/ie00107a014.
Huang SH, Radosz M. Equation of state for small, large, polydisperse, and associating molecules: extension to fluid mixtures. Ind Eng Chem Res. 1991;30(8):1994–2005. https://doi.org/10.1021/ie00056a050.
Huron MJ, Vidal J. New mixing rules in simple equations of state for representing vapour-liquid equilibria of strongly non-ideal mixtures. Fluid Phase Equilib. 1979;3(4):255–71. https://doi.org/10.1016/0378-3812(79)80001-1.
Illbeigi M, Fazlali A, Mohammadi AH. Thermodynamic model for the prediction of equilibrium conditions of clathrate hydrates of methane + water-soluble or-insoluble hydrate former. Ind Eng Chem Res. 2011;50(15):9437–50. https://doi.org/10.1021/ie200442h.
Javanmardi J, Moshfeghian M, Maddox RN. An accurate model for prediction of gas hydrate formation conditions in mixtures of aqueous electrolyte solutions and alcohol. Can J Chem Eng. 2001;79(3):367–73. https://doi.org/10.1002/cjce.5450790309.
Javanmardi J, Ayatollahi S, Motealleh R, Moshfeghian M. Experimental measurement and modeling of R22 (CHClF2) hydrates in mixtures of Acetone + Water. J Chem Eng Data. 2004;49(4):886–9. https://doi.org/10.1021/je034198p.
Jiang H, Adidharma H. Hydrate equilibrium modeling for pure alkanes and mixtures of alkanes using statistical associating fluid theory. Ind Eng Chem Res. 2011;50:12815–23. https://doi.org/10.1021/ie2014444.
Jiang H, Adidharma H. Thermodynamic modeling of aqueous ionic liquid solutions and prediction of methane hydrate dissociation conditions in the presence of ionic liquid. Chem Eng Sci. 2013;102:24–31. https://doi.org/10.1016/j.ces.2013.07.049.
Kamata Y, Oyama H, Shimada W, Ebinuma T, Takeya S, Uchida T, et al. Gas separation method using tetra-n-butyl ammonium bromide semi-clathrate hydrate. Jpn J Appl Phys. 2004;43(1R):362. https://doi.org/10.1143/JJAP.43.362.
Kang S-P, Lee H. Recovery of CO2 from flue gas using gas hydrate: thermodynamic verification through phase equilibrium measurements. Environ Sci Technol. 2000;34(20):4397–400. https://doi.org/10.1021/es001148l.
Karakatsani EK, Kontogeorgis GM. Thermodynamic modeling of natural gas systems containing water. Ind Eng Chem Res. 2013;52(9):3499–513. https://doi.org/10.1021/ie302916h.
Karamoddin M, Varaminian F. Experimental measurement of phase equilibrium for gas hydrates of refrigerants, and thermodynamic modeling by SRK, VPT and CPA EoSs. J Chem Thermodyn. 2013;65:213–9. https://doi.org/10.1016/j.jct.2013.06.001.
Kelland MA. History of the development of low dosage hydrate inhibitors. Energy Fuels. 2006;20(3):825–47. https://doi.org/10.1021/ef050427x.
Khokhar A, Gudmundsson J, Sloan E. Gas storage in structure H hydrates. Fluid Phase Equilib. 1998;150:383–92. https://doi.org/10.1016/S0378-3812(98)00338-0.
Khosravani E, Moradi G, Sajjadifar S. Application of PRSV2 equation of state and explicit pressure dependence of the Langmuir adsorption constant to study phase behavior of gas hydrates in the presence and absence of methanol. Fluid Phase Equilib. 2012;333:63–73. https://doi.org/10.1016/j.fluid.2012.07.023.
Klauda JB, Sandler SI. A fugacity model for gas hydrate phase equilibria. Ind Eng Chem Res. 2000;39(9):3377–86. https://doi.org/10.1021/ie000322b.
Klauda JB, Sandler SI. Phase behavior of clathrate hydrates: a model for single and multiple gas component hydrates. Chem Eng Sci. 2003;58(1):27–41. https://doi.org/10.1016/S0009-2509(02)00435-9.
Kondori J, Zendehboudi S, James L. Evaluation of gas hydrate formation temperature for gas/water/salt/alcohol systems: utilization of extended UNIQUAC model and PC-SAFT equation of state. Ind Eng Chem Res. 2018;57(41):13833–55. https://doi.org/10.1021/acs.iecr.8b03011.
Kontogeorgis GM, Yakoumis IV, Meijer H, Hendriks E, Moorwood T. Multicomponent phase equilibrium calculations for water–methanol–alkane mixtures. Fluid Phase Equilib. 1999;158–160:201–9. https://doi.org/10.1016/S0378-3812(99)00060-6.
Kontogeorgis GM, Michelsen ML, Folas GK, Derawi S, von Solms N, Stenby EH. Ten years with the CPA (Cubic-Plus-Association) Equation of State. Part 1. Pure compounds and self-associating systems. Ind Eng Chem Res. 2006a;45(14):4855–68. https://doi.org/10.1021/ie051305v.
Kontogeorgis GM, Michelsen ML, Folas GK, Derawi S, von Solms N, Stenby EH. Ten years with the CPA (Cubic-Plus-Association) Equation of State. Part 2. Cross-Associating and multicomponent systems. Ind Eng Chem Res. 2006b;45(14):4869–78. https://doi.org/10.1021/ie051306n.
Kontogeorgis GM, Folas GK, Muro-Suñé N, von Solms N, Michelsen ML, Stenby EH. Modelling of associating mixtures for applications in the oil & gas and chemical industries. Fluid Phase Equilib. 2007;261(1):205–11. https://doi.org/10.1016/j.fluid.2007.05.022.
Kubota H, Shimizu K, Tanaka Y, Makita T. Thermodynamic properties of R13 (CClF3), R23 (CHF3), R152a (C2H4F2), and propane hydrates for desalination of sea water. J Chem Eng Jpn. 1984;17(4):423–9. https://doi.org/10.1252/jcej.17.423.
Kvenvolden KA. Gas hydrates as a potential energy resource—a review of their methane content. U. S. Geol Surv. 1993;1570:555–61.
Kvenvolden K. A primer on the geological occurrence of gas hydrate. Spec Publ Geol Soc Lond. 1998;137(1):9–30. https://doi.org/10.1144/GSL.SP.1998.137.01.02.
Kvenvolden KA. Potential effects of gas hydrate on human welfare. Proc Natl Acad Sci. 1999;96(7):3420–6.
Lee H, Lee J, Park J, Seo Y-T, Zeng H, Moudrakovski IL, et al. Tuning clathrate hydrates for hydrogen storage. Nature. 2005;434(7034):743–6. https://doi.org/10.1142/9789814317665_0042.
Lee Y-J, Kawamura T, Yamamoto Y, Yoon J-H. Phase equilibrium studies of tetrahydrofuran (THF) + CH4, THF + CO2, CH4 + CO2, and THF + CO2 + CH4 hydrates. J Chem Eng Data. 2012;57(12):3543–8. https://doi.org/10.1021/je300850q.
Li X-S, Wu H-J, Englezos P. Prediction of gas hydrate formation conditions in the presence of methanol, glycerol, ethylene glycol, and triethylene glycol with the statistical associating fluid theory equation of state. Ind Eng Chem Res. 2006;45(6):2131–7. https://doi.org/10.1021/ie051204x.
Li X-S, Wu H-J, Li Y-G, Feng Z-P, Tang L-G, Fan S-S. Hydrate dissociation conditions for gas mixtures containing carbon dioxide, hydrogen, hydrogen sulfide, nitrogen, and hydrocarbons using SAFT. J Chem Thermodyn. 2007;39(3):417–25. https://doi.org/10.1016/j.jct.2006.07.028.
Li L, Zhu L, Fan J. The application of CPA-vdWP to the phase equilibrium modeling of methane-rich sour natural gas hydrates. Fluid Phase Equilib. 2016;409:291–300. https://doi.org/10.1016/j.fluid.2015.10.017.
Li L, Fan S, Chen Q, Yang G, Zhao J, Wei N, et al. Experimental and modeling phase equilibria of gas hydrate systems for post-combustion CO2 capture. J Taiwan Inst Chem Eng. 2019;96:35–44. https://doi.org/10.1016/j.jtice.2018.11.007.
Liang D, Guo K, Wang R, Fan S. Hydrate equilibrium data of 1, 1, 1, 2-tetrafluoroethane (HFC-134a), 1, 1-dichloro-1-fluoroethane (HCFC-141b) and 1, 1-difluoroethane (HFC-152a). Fluid Phase Equilib. 2001;187:61–70. https://doi.org/10.1016/S0378-3812(01)00526-X.
Ma C-F, Chen G-J, Wang F, Sun C-Y, Guo T-M. Hydrate formation of (CH4 + C2H4) and (CH4 + C3H6) gas mixtures. Fluid Phase Equilib. 2001;191(1):41–7. https://doi.org/10.1016/S0378-3812(01)00610-0.
Ma Q-L, Chen G-J, Sun C-Y, Yang L-Y, Liu B. Predictions of hydrate formation for systems containing hydrogen. Fluid Phase Equilib. 2013;358:290–5. https://doi.org/10.1016/j.fluid.2013.08.019.
Ma ZW, Zhang P, Bao HS, Deng S. Review of fundamental properties of CO2 hydrates and CO2 capture and separation using hydration method. Renew Sustain Energy Rev. 2016;53:1273–302. https://doi.org/10.1016/j.rser.2015.09.076.
Maeda K, Katsura Y, Asakuma Y, Fukui K. Concentration of sodium chloride in aqueous solution by chlorodifluoromethane gas hydrate. Chem Eng Process. 2008;47(12):2281–6. https://doi.org/10.1016/j.cep.2008.01.002.
Mahabadian MA, Chapoy A, Burgass R, Tohidi B. Development of a multiphase flash in presence of hydrates: experimental measurements and validation with the CPA equation of state. Fluid Phase Equilib. 2016;414:117–32. https://doi.org/10.1016/j.fluid.2016.01.009.
Makogon Y, Holditch S, Makogon T. Natural gas-hydrates—a potential energy source for the 21st century. J Pet Sci Eng. 2007;56(1):14–31. https://doi.org/10.1016/j.petrol.2005.10.009.
Martín Á, Peters CJ. Hydrogen storage in sH clathrate hydrates: thermodynamic model. J Phys Chem B. 2009;113(21):7558–63. https://doi.org/10.1021/jp8074578.
Maslin M, Owen M, Betts R, Day S, Jones TD, Ridgwell A. Gas hydrates: past and future geohazard? Philos Trans R Soc A. 2010;368(1919):2369–93.
McKoy V, Sinanoğlu O. Theory of dissociation pressures of some gas hydrates. J Chem Phys. 1963;38(12):2946–56. https://doi.org/10.1063/1.1733625.
Mech D, Pandey G, Sangwai JS. Effect of NaCl, methanol and ethylene glycol on the phase equilibrium of methane hydrate in aqueous solutions of tetrahydrofuran (THF) and tetra-n-butyl ammonium bromide (TBAB). Fluid Phase Equilib. 2015;402:9–17. https://doi.org/10.1016/j.fluid.2015.05.030.
Menezes DÉSD, Ralha TW, Franco LFM, Pessôa Filho PDA, Fuentes MDR. Simulation and experimental study of methane-propane hydrate dissociation by high pressure differential scanning calorimetry. Braz J Chem Eng. 2018;35:403–14. https://doi.org/10.1590/0104-6632.20180352s20160329.
Milkov AV. Global estimates of hydrate-bound gas in marine sediments: how much is really out there? Earth-Sci Rev. 2004;66(3):183–97. https://doi.org/10.1016/j.earscirev.2003.11.002.
Milkov AV, Sassen R, Novikova I, Mikhailov E. Gas hydrates at minimum stability water depths in the Gulf of Mexico: significance to geohazard assessment. 2000.
Moeini H, Bonyadi M, Esmaeilzadeh F, Rasoolzadeh A. Experimental study of sodium chloride aqueous solution effect on the kinetic parameters of carbon dioxide hydrate formation in the presence/absence of magnetic field. J Nat Gas Sci Eng. 2017;50:231–9. https://doi.org/10.1016/j.jngse.2017.12.012.
Mohammadi AH, Richon D. Gas hydrate phase equilibrium in the presence of ethylene glycol or methanol aqueous solution. Ind Eng Chem Res. 2010;49(18):8865–9. https://doi.org/10.1021/ie100908d.
Mohammadi AH, Belandria V, Richon D. Can toluene or xylene form clathrate hydrates? Ind Eng Chem Res. 2009a;48(12):5916–8. https://doi.org/10.1021/ie900362v.
Mohammadi AH, Kraouti I, Richon D. Methane hydrate phase equilibrium in the presence of NaBr, KBr, CaBr2, K2CO3, and MgCl2 aqueous solutions: experimental measurements and predictions of dissociation conditions. J Chem Thermodyn. 2009b;41(6):779–82. https://doi.org/10.1016/j.jct.2009.01.004.
Mohammadi AH, Martínez-López JF, Richon D. Determining phase diagrams of tetrahydrofuran + methane, carbon dioxide or nitrogen clathrate hydrates using an artificial neural network algorithm. Chem Eng Sci. 2010;65(22):6059–63. https://doi.org/10.1016/j.ces.2010.07.013.
Mooijer-van den Heuvel MM, Witteman R, Peters CJ. Phase behaviour of gas hydrates of carbon dioxide in the presence of tetrahydropyran, cyclobutanone, cyclohexane and methylcyclohexane. Fluid Phase Equilib. 2001;182(1):97–110. https://doi.org/10.1016/S0378-3812(01)00384-3.
Müller EA, Gubbins KE. Molecular-based equations of state for associating fluids: a review of SAFT and related approaches. Ind Eng Chem Res. 2001;40(10):2193–211. https://doi.org/10.1021/ie000773w.
Nagata T, Tajima H, Yamasaki A, Kiyono F, Abe Y. An analysis of gas separation processes of HFC-134a from gaseous mixtures with nitrogen—Comparison of two types of gas separation methods, liquefaction and hydrate-based methods, in terms of the equilibrium recovery ratio. Sep Purif Technol. 2009;64(3):351–6. https://doi.org/10.1016/j.seppur.2008.10.023.
Ngema PT, Naidoo P, Mohammadi AH, Ramjugernath D. Phase stability conditions for clathrate hydrate formation in (fluorinated refrigerant + water + single and mixed electrolytes + cyclopentane) systems: experimental measurements and thermodynamic modelling. J Chem Thermodyn. 2019a;136:59–76. https://doi.org/10.1016/j.jct.2019.04.012.
Ngema PT, Naidoo P, Mohammadi AH, Ramjugernath D. Phase stability conditions for clathrate hydrates formation in CO2 + (NaCl or CaCl2 or MgCl2) + Cyclopentane + Water systems: experimental measurements and thermodynamic modeling. J Chem Eng Data. 2019b;64:4638–46. https://doi.org/10.1021/acs.jced.8b00872.
Nikbakht F, Izadpanah AA, Varaminian F, Mohammadi AH. Thermodynamic modeling of hydrate dissociation conditions for refrigerants R-134a, R-141b and R-152a. Int J Refrig. 2012;35(7):1914–20. https://doi.org/10.1016/j.ijrefrig.2012.07.004.
Ohgaki K, Takano K, Sangawa H, Matsubara T, Nakano S. Methane exploitation by carbon dioxide from gas hydrates. Phase equilibria for CO2-CH4 mixed hydrate system. J Chem Eng Jpn. 1996;29(3):478–83. https://doi.org/10.1252/jcej.29.478.
Ohno H, Susilo R, Gordienko R, Ripmeester J, Walker VK. Interaction of antifreeze proteins with hydrocarbon hydrates. Chem Eur J. 2010;16(34):10409–17. https://doi.org/10.1002/chem.200903201.
Oliveira MB, Coutinho JAP, Queimada AJ. Mutual solubilities of hydrocarbons and water with the CPA EoS. Fluid Phase Equilib. 2007;258(1):58–66. https://doi.org/10.1016/j.fluid.2007.05.023.
Pahlavanzadeh H, Khanlarkhani M, Rezaei S, Mohammadi AH. Experimental and modelling studies on the effects of nanofluids (SiO2, Al2O3, and CuO) and surfactants (SDS and CTAB) on CH4 and CO2 clathrate hydrates formation. Fuel. 2019;253:1392–405. https://doi.org/10.1016/j.fuel.2019.05.010.
Papadimitriou NI, Tsimpanogiannis IN, Stubos AK, Martín A, Rovetto LJ, Florusse LJ, et al. Experimental and computational investigation of the sII binary He–THF hydrate. J Phys Chem B. 2011;115(6):1411–5. https://doi.org/10.1021/jp105451m.
Parrish WR, Prausnitz JM. Dissociation pressures of gas hydrates formed by gas mixtures. Ind Eng Chem Process Des Dev. 1972;11(1):26–35. https://doi.org/10.1021/i260041a006.
Partoon B, Javanmardi J. Effect of mixed thermodynamic and kinetic hydrate promoters on methane hydrate phase boundary and formation kinetics. J Chem Eng Data. 2013;58(3):501–9. https://doi.org/10.1021/je301153t.
Pauling L, Marsh RE. The structure of chlorine hydrate. Proc Natl Acad Sci. 1952;38(2):112–8. https://doi.org/10.1073/pnas.38.2.112.
Peng DY, Robinson DB. A new two-constant equation of state. Ind Eng Chem Fundam. 1976;15(1):59–64. https://doi.org/10.1021/i160057a011.
Platteeuw J, Van der Waals J. Thermodynamic properties of gas hydrates II: phase equilibria in the system H2S–C3H3-H2O at − 3°C. Recl Trav Chim Pays-Bas. 1959;78(2):126–33. https://doi.org/10.1002/recl.19590780208.
Ripmeester JA, John ST, Ratcliffe CI, Powell BM. A new clathrate hydrate structure. Nature. 1987;325(6100):135–6.
Rod Burgass AC, Bahman Tohidi, editor. Experimental and modelling low temperature water content in multicomponent gas mixtures. In: 7th International Conference on Gas Hydrates July 2011; Edinburgh, Scotland, United Kingdom; 2011.
Ruppel C, Boswell R, Jones E. Scientific results from Gulf of Mexico gas hydrates Joint Industry Project Leg 1 drilling: introduction and overview. Mar Pet Geol. 2008;25(9):819–29. https://doi.org/10.1016/j.marpetgeo.2008.02.007.
Sabil KM, Duarte ARC, Zevenbergen J, Ahmad MM, Yusup S, Omar AA, et al. Kinetic of formation for single carbon dioxide and mixed carbon dioxide and tetrahydrofuran hydrates in water and sodium chloride aqueous solution. Int J Greenhouse Gas Control. 2010a;4(5):798–805. https://doi.org/10.1016/j.ijggc.2010.05.010.
Sabil KM, Witkamp G-J, Peters CJ. Estimations of enthalpies of dissociation of simple and mixed carbon dioxide hydrates from phase equilibrium data. Fluid Phase Equilib. 2010b;290(1):109–14. https://doi.org/10.1016/j.fluid.2009.07.006.
Sarshar M, Esmaeilzadeh F, Fathikaljahi J. Predicting the induction time of hydrate formation on a water droplet. Oil Gas Sci Technol. 2008;63:657–67. https://doi.org/10.2516/ogst:2008032.
Sarshar M, Esmaeilzadeh F, Fathikalajahi J. Study of capturing emitted CO2 in the form of hydrates in a tubular reactor. Chem Eng Commun. 2009;196(11):1348–65. https://doi.org/10.1080/00986440902900832.
Sarshar M, Esmaeilzadeh F, Fathikalajahi J. Induction time of hydrate formation in a flow loop. Theor Found Chem Eng. 2010a;44(2):201–5. https://doi.org/10.1134/s0040579510020119.
Sarshar M, Fathikalajahi J, Esmaeilzadeh F. Experimental and theoretical study of gas hydrate formation in a high-pressure flow loop. Can J Chem Eng. 2010b;88(5):751–7. https://doi.org/10.1002/cjce.20332.
Sarshar M, Fathikalajahi J, Esmaeilzadeh F. Kinetic of hydrate formation of propane and its mixture with methane in a circulating flow reactor. Fluid Phase Equilib. 2010c;298(1):38–47. https://doi.org/10.1016/j.fluid.2010.06.016.
Sfaxi IBA, Belandria V, Mohammadi AH, Lugo R, Richon D. Phase equilibria of CO2 + N2 and CO2 + CH4 clathrate hydrates: experimental measurements and thermodynamic modelling. Chem Eng Sci. 2012;84:602–11. https://doi.org/10.1016/j.ces.2012.08.041.
Shahnazar S, Hasan N. Gas hydrate formation condition: review on experimental and modeling approaches. Fluid Phase Equilib. 2014;379:72–85. https://doi.org/10.1016/j.fluid.2014.07.012.
Shiojiri K, Okano T, Yanagisawa Y, Fujii M, Yamasaki A, Tajima H, et al. A new type separation process of condensable greenhouse gases by the formation of clathrate hydrates. Stud Surf Sci Catal. 2004;153:507–12.
Sinehbaghizadeh S, Javanmardi J, Roosta A, Mohammadi AH. Estimation of the dissociation conditions and storage capacities of various sH clathrate hydrate systems using effective deterministic frameworks. Fuel. 2019;247:272–86. https://doi.org/10.1016/j.fuel.2019.01.189.
Sirino TH, Marcelino Neto MA, Bertoldi D, Morales REM, Sum AK. Multiphase flash calculations for gas hydrates systems. Fluid Phase Equilib. 2018;475:45–63. https://doi.org/10.1016/j.fluid.2018.07.029.
Sloan ED. Gas hydrates: review of physical/chemical properties. Energy Fuels. 1998;12(2):191–6. https://doi.org/10.1021/ef970164+.
Sloan ED. Fundamental principles and applications of natural gas hydrates. Nature. 2003;426(6964):353–63. https://doi.org/10.1038/nature02135.
Sloan ED. A changing hydrate paradigm—from apprehension to avoidance to risk management. Fluid Phase Equilib. 2005;228:67–74. https://doi.org/10.1016/j.fluid.2004.08.009.
Sloan ED Jr, Koh C. Clathrate hydrates of natural gases. Boca Raton: CRC Press; 2007.
Soave G. Equilibrium constants from a modified Redlich-Kwong equation of state. Chem Eng Sci. 1972;27(6):1197–203. https://doi.org/10.1016/0009-2509(72)80096-4.
Spencer DF, Currier RP. Methods of selectively separating CO2 from a multicomponent gaseous stream using CO2 hydrate promoters. Google Patents; 2002.
Strobel TA, Koh CA, Sloan ED. Thermodynamic predictions of various tetrahydrofuran and hydrogen clathrate hydrates. Fluid Phase Equilib. 2009;280(1):61–7. https://doi.org/10.1016/j.fluid.2009.02.012.
Sun Z-G, Fan S-S, Guo K-H, Shi L, Guo Y-K, Wang R-Z. Gas hydrate phase equilibrium data of cyclohexane and cyclopentane. J Chem Eng Data. 2002;47(2):313–5. https://doi.org/10.1021/je0102199.
Sun Z-G, Ma R, Wang R-Z, Guo K-H, Fa S-S. Experimental studying of additives effects on gas storage in hydrates. Energy Fuels. 2003a;17(5):1180–5. https://doi.org/10.1021/ef020191m.
Sun Z, Wang R, Ma R, Guo K, Fan S. Natural gas storage in hydrates with the presence of promoters. Energy Convers Manage. 2003b;44(17):2733–42. https://doi.org/10.1016/S0196-8904(03)00048-7.
Sun X, Nanchary N, Mohanty KK. 1-D modeling of hydrate depressurization in porous media. Transp Porous Media. 2005;58(3):315–38. https://doi.org/10.1007/s11242-004-1410-x.
Talaghat M, Esmaeilzadeh F, Fathikaljahi J. Experimental and theoretical investigation of simple gas hydrate formation with or without presence of kinetic inhibitors in a flow mini-loop apparatus. Fluid Phase Equilib. 2009a;279(1):28–40. https://doi.org/10.1016/j.fluid.2009.01.017.
Talaghat MR, Esmaeilzadeh F, Fathikaljahi J. Experimental and theoretical investigation of double gas hydrate formation in the presence or absence of kinetic inhibitors in a flow mini-loop apparatus. Chem Eng Technol. 2009b;32(5):805–19. https://doi.org/10.1002/ceat.200800601.
Tan SP, Adidharma H, Radosz M. Recent advances and applications of statistical associating fluid theory. Ind Eng Chem Res. 2008;47(21):8063–82. https://doi.org/10.1021/ie8008764.
Tang J, Zeng D, Wang C, Chen Y, He L, Cai N. Study on the influence of SDS and THF on hydrate-based gas separation performance. Chem Eng Res Des. 2013;91(9):1777–82. https://doi.org/10.1016/j.cherd.2013.03.013.
Tavasoli H, Feyzi F. Four phase hydrate equilibria of methane and carbon dioxide with heavy hydrate former compounds: experimental measurements and thermodynamic modeling. Korean J Chem Eng. 2016;33(8):2426–38. https://doi.org/10.1007/s11814-016-0110-x.
Taylor CE, Kwan JT. Advances in the study of gas hydrates. Berlin: Springer; 2004.
Tohidi B, Danesh A, Burgass RW, Todd AC. Equilibrium data and thermodynamic modelling of cyclohexane gas hydrates. Chem Eng Sci. 1996;51(1):159–63. https://doi.org/10.1016/0009-2509(95)00253-7.
Valderrama JO. A generalized Patel–Teja equation of state for polar and nonpolar fluids and their mixtures. J Chem Eng Jpn. 1990;23(1):87–91. https://doi.org/10.1252/jcej.23.87.
Van der Waals J, Platteeuw J. Clathrate solutions. Adv Chem Phys. 2007;2:1–57. https://doi.org/10.1002/9780470143483.ch1.
Veluswamy HP, Linga P. Macroscopic kinetics of hydrate formation of mixed hydrates of hydrogen/tetrahydrofuran for hydrogen storage. Int J Hydrogen Energy. 2013;38(11):4587–96. https://doi.org/10.1016/j.ijhydene.2013.01.123.
Waseem MS, Alsaifi NM. Prediction of vapor-liquid-hydrate equilibrium conditions for single and mixed guest hydrates with the SAFT-VR Mie EoS. J Chem Thermodyn. 2018;117:223–35. https://doi.org/10.1016/j.jct.2017.09.032.
Wong DSH, Sandler SI. A theoretically correct mixing rule for cubic equations of state. AIChE J. 1992;38(5):671–80. https://doi.org/10.1002/aic.690380505.
Yamamoto K, Nakatsuka Y, Sato R, Kvalstad T, Qiu K, Birchwood R. Geohazard risk evaluation and related data acquisition and sampling program for the methane hydrate offshore production test. Frontiers in Offshore Geotechnics III. 2015;1:173.
Yang M, Jing W, Wang P, Jiang L, Song Y. Effects of an additive mixture (THF + TBAB) on CO2 hydrate phase equilibrium. Fluid Phase Equilib. 2015;401:27–33. https://doi.org/10.1016/j.fluid.2015.05.007.
Youssef Z, Barreau A, Mougin P, Jose J, Mokbel I. Measurements of hydrate dissociation temperature of methane, ethane, and CO2 in the absence of any aqueous phase and prediction with the cubic plus association equation of state. Ind Eng Chem Res. 2009;48(8):4045–50. https://doi.org/10.1021/ie801351e.
ZareNezhad B, Ziaee M. Accurate prediction of H2S and CO2 containing sour gas hydrates formation conditions considering hydrolytic and hydrogen bonding association effects. Fluid Phase Equilib. 2013;356:321–8. https://doi.org/10.1016/j.fluid.2013.07.055.
Zhang S-X, Chen G-J, Ma C-F, Yang L-Y, Guo T-M. Hydrate formation of hydrogen + hydrocarbon gas mixtures. J Chem Eng Data. 2000;45(5):908–11. https://doi.org/10.1021/je000076a.
Zhang Y, Debenedetti PG, Prud’homme RK, Pethica BA. Accurate prediction of clathrate hydrate phase equilibria below 300 K from a simple model. J Pet Sci Eng. 2006;51(1):45–53. https://doi.org/10.1016/j.petrol.2005.11.008.
Zhang L, Burgass R, Chapoy A, Tohidi B. Measurement and modeling of water content in low temperature hydrate–methane and hydrate–natural gas systems. J Chem Eng Data. 2011;56(6):2932–5. https://doi.org/10.1021/je2001655.
Zhong DL, Li Z, Lu YY, Wang JL, Yan J. Evaluation of CO2 removal from a CO2 + CH4 gas mixture using gas hydrate formation in liquid water and THF solutions. Appl Energy. 2015;158:133–41. https://doi.org/10.1016/j.apenergy.2015.08.058.
Zhong DL, Wang JL, Lu YY, Li Z, Yan J. Precombustion CO2 capture using a hybrid process of adsorption and gas hydrate formation. Energy. 2016;102:621–9. https://doi.org/10.1016/j.energy.2016.02.135.
Zuo YX, Gommesen S, Guo TM. Equation of state based hydrate model for natural gas systems containing brine and polar inhibitor. Chin J Chem Eng. 1996;4(3):189–202.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Xiu-Qiu Peng
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Esmaeilzadeh, F., Hamedi, N., Karimipourfard, D. et al. An insight into the role of the association equations of states in gas hydrate modeling: a review. Pet. Sci. 17, 1432–1450 (2020). https://doi.org/10.1007/s12182-020-00471-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-020-00471-9