
Abstract
Retinal neurons are highly vulnerable to a diverse array of neurotoxic stimuli that leads to their degeneration, which is a major contributor to blindness. This review summarizes the role of epigenetic factors in mediating neuronal homeostasis and survival to protect against cell death and neurodegenerative conditions. Studies in human patients and mouse models implicate numerous chromatin modifications in neuroprotective processes including histone protein acetylation and methylation, DNA methylation, and ATP-dependent nucleosome remodeling. Recent research has begun to uncover specific epigenetic mechanisms invoked by neurotoxic stimuli. Continued investigation in this area will be the key to the generation of therapeutic strategies for the intervention of retinal neurodegenerative diseases.
Similar content being viewed by others

Introduction
The development of the nervous system from primordial structures occurs through a highly organized and intricately regulated series of events involving cell proliferation, differentiation, migration and maturation, culminating in the establishment of numerous neuronal cell types that participate in complex neural circuits and networks. Once fully differentiated, neurons are required to carry out their functions for the lifespan of the organism as most are considered irreplaceable if lost. While adult neurogenesis does occur in select regions of the central nervous system (CNS), the vast majority of neuronal cell types cannot be regenerated naturally from existing cellular pools. This implies that neurons are required to remain intact and functional in order for proper neuronal communication to persist and to avoid deficits in neural activity that may occur through accumulated losses over time. In order to overcome the wide range of insults and stressors encountered over a lifetime, neurons must be able to rapidly react to changing conditions and appropriately modify gene expression to maintain cellular integrity and functionality.
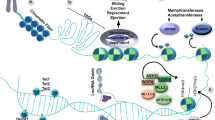
To quickly respond to extracellular cues and signals, the cell relies on epigenetic mechanisms of transcriptional regulation. Changes in gene expression occur as a result of alterations in the structure of chromatin that either allow or restrict access to the regulatory regions that control transcriptional activity. The basic subunit of chromatin is the nucleosome, consisting of 146 base pairs of DNA wrapped around an octamer of two copies of each of the core histone proteins H2A, H2B, H3, and H4 [1]. The linker histone H1 binds to the DNA between nucleosomes to enable further chromatin compaction. It is through the dynamic reorganization of nucleosomes and higher-order chromatin structure that genetic regulatory elements are made accessible to transcription factors to modify the expression of specific genes [2–4]. Covalent modifications of DNA and histone proteins disrupt chromatin compaction and also serve as landmarks that are recognized by chromatin-binding proteins that initiate downstream genetic processes. The enzymes that catalyze DNA and histone modification as well as the factors that interact with these marks are collectively termed chromatin remodeling proteins. The finding that mutations in a growing number of these proteins cause developmental and neurodegenerative defects is evidence of their critical function in the CNS from the embryo right through adulthood.
The development of the retina follows defined genetic programs that have been extensively characterized [5, 6]. An increasing number of studies describe interactions between the transcription factors that control the development of the retina and various chromatin remodeling proteins that may contribute to the execution of critical developmental programs [7]. However, much less attention has focussed on the epigenetic regulation of retinal neuron maintenance and homeostasis. This review describes the role of chromatin remodeling proteins in mediating survival or loss of neuronal cells in the retina under various conditions, including during developmental stages, in response to neurotoxic stimuli, and in heritable retinal degenerations. Neuroprotective mechanisms and strategies that follow from these findings may enable the development of novel therapeutics for treating human retinal degenerative conditions.
Histone modifications impact retinal neuron survival
Histone proteins can be post-translationally modified by a variety of enzyme activities, resulting in their acetylation, methylation, phosphorylation, sumoylation, ubiquitination, ADP-ribosylation, deimination, and β-N-acetylglucosamination [8]. These covalent modifications can alter the association of histones with DNA and can also serve as a scaffold for the recognition and binding of other proteins that ultimately affect chromatin compaction and transcriptional regulation [9, 10]. Histone acetylation and methylation and the enzymatic activities that create or remove these marks have been implicated in the survival of CNS neurons.
Histone acetylation
The transfer of an acetyl group to the lysine side chains of histone proteins causes the neutralization of the positive charge on the lysine residues and can therefore disrupt the interactions between histones and DNA within nucleosomes. Acetylation has been described for all of the core histone proteins and is generally associated with chromatin relaxation and transcriptional activation, while deacetylation favors chromatin condensation and transcriptional repression. The extent of post-translational acetylation is determined by the enzymatic activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). It is the net effect of both HAT and HDAC activities that determines the overall acetylation level of histones and impacts the transcriptional status of particular genes.
The acetylation of histones has been the most extensively studied chromatin modification in the context of neurodegenerative processes. Despite the different pathophysiologies associated with various neurodegenerative diseases, in vivo models demonstrate that a common feature of the degenerative process in the affected neurons is a reduction in the global levels of histone acetylation [11–13]. Consistent with this observation is the association of reduced or absent acetyltransferase activity mediated by CREB-binding protein (CBP)/p300 in these models [11, 12]. In the retina, the CBP and p300 HATs can form complexes with a variety of transcription factors that play critical roles in gene expression programs that direct retinal cell differentiation and maturation, including Crx, Pax6, Prox1, Mash1, NeuroD1, Sox9, RXRγ, and pRb [7]. Transcriptional activation of target genes by these factors can in turn be facilitated through the CBP/p300-mediated acetylation of histones to create permissive chromatin configurations that promote gene expression. Indeed, such is the case for the homeodomain transcription factor Crx, whose inactivation results in a retinal degenerative phenotype [14]. Moreover, mutations in Crx target genes also cause retinal degeneration [15]. Crx binds to the regulatory regions of its target photoreceptor genes, where it recruits HATs that acetylate histone H3, to enable chromatin remodeling and subsequent binding of transcriptional coactivators and RNA polymerase II for transcription to occur [16]. Since loss of Crx leads to retinopathy, it may follow that loss of the HAT activity and H3 acetylation on its target gene promoters may also result in photoreceptor degeneration. In Crx knockout mice, binding of HATs to target promoters and the levels of acetylated histone H3 on these promoters was reduced [16], and expression of the target genes was concomitantly reduced [14].
Similar to CBP and p300, the GCN5-HAT component of the multi-subunit SPT3-TAF9-ADA-GCN5 acetyltransferase (STAGA) transcription coactivator complex can interact with Crx and is associated with histone H3 acetylation on its target gene promoters [16]. The interaction of GCN5 and Crx is mediated by the ataxin-7 protein, another component of the STAGA complex [17]. Mutation of ataxin-7 results in inhibition of HAT function, reduced H3 acetylation and reduced Crx occupancy of target genes [17], and causes neurodegeneration of the retina and brain [18, 19]. In addition to altered transcription profiles in photoreceptor cells, ataxin-7 mutant mice also exhibit dramatic reorganization of chromatin correlated with reduced expression and abnormal distribution of the linker histone H1c in rod photoreceptor nuclei; however, global histone acetylation levels are unchanged [20]. It is possible that aberrant ataxin-7 function leads to altered targeting of HAT complexes, causing inappropriate acetylation and activation of otherwise repressed genes, and contributing to the upregulation of some photoreceptor genes that are observed in these mice [21]. Nevertheless, it appears that the survival of retinal neurons is impacted by multiple mechanisms involving changes in histone processing and dynamics.
This is further supported by the important role of HDACs that has been demonstrated in various mouse models of retinal neurodegeneration. In the rd1 mouse model of retinitis pigmentosa, reduced histone acetylation levels were detected in photoreceptors but not other retinal cell types that do not undergo degeneration [22]. This was related to elevated HDAC activity predominantly attributed to class I and II HDACs. Furthermore, protein hypoacetylation and increased HDAC activity occurred in rd1 photoreceptor cells undergoing apoptosis which could be protected from cell death by exposure to HDAC I/II inhibitors [22]. HDAC inhibition was also shown to promote retinal ganglion cell survival in optic nerve crush-induced neurodegeneration [23]. However, inhibition of class I/II HDACs can lead to broadly distributed apoptotic cell death in wild-type retinas [24], indicating that disruption of normal levels of protein acetylation can be cytotoxic, while reduction of HDAC overactivity occurring in retinal degeneration may result in the normalization of pathophysiological acetylation levels and subsequent neuroprotection. Additional studies in which resting levels of histone acetylation in neuronal cells under normal conditions are altered by either exposure to HDAC inhibitors [25, 26] or elevation of HATs [27, 28] further suggest that hyperacetylation of histones is toxic for neurons and that disturbing the sensitive balance between HAT and HDAC activities in either direction can trigger neuronal cell death.
Class III HDACs are also involved in mediating neuronal survival. A neuroprotective role for the Sirt1 histone deacetylase has been demonstrated in various neurodegenerative disease conditions [29–32]. Consistent with this, Sirt1 protein distribution is altered in degenerating retinas of rd10 mice, where it co-localizes with apoptotic photoreceptors as well as pro-apoptotic proteins in the outer nuclear layer of the retina at the peak of cell death, and after which, its retinal expression is dramatically reduced [33]. It is hypothesized that the neuroprotective effects of Sirt1 are lost in the rd10 photoreceptors due to its cellular mislocalization and reduced level of expression, therefore resulting in the degeneration of these cells.
Individual HDACs also appear to function in distinct neuronal survival pathways. Specific inactivation of HDAC1 in post-mitotic primary neurons results in significant cell death, while increased HDAC1 activity is protective against neurotoxicity in vivo [34]. HDAC4 has a neuroprotective role in the retina, as overexpression causes reduced levels of naturally occurring bipolar cell death during development and also rescues rod and cone photoreceptor cell loss in rd1 mice [35]. Accordingly, inhibition of HDAC4 function in wild-type retinas by RNA interference induces significant cell loss due to apoptosis, indicating that HDAC4 is required for retinal neuron survival [35]. In contrast, HDAC5 or HDAC6 was unable to mediate rescue of the photoreceptors in the rd1 mice, despite the ability of HDAC6 to rescue neurodegeneration in a Drosophila model of spinobulbar muscular atrophy [36]. In optic nerve injury-induced retinal neurodegeneration, apoptotic retinal ganglion cells (RGC) exhibit increased HDAC activity, reduced levels of acetylated histone H4, and downregulation of RGC marker and survival genes [37]. Only HDAC3 was shown to translocate to the nuclei of apoptotic RGC and exhibited increased and sustained expression that was consistent with the time course of acetylated H4 reduction following optic nerve crush in these mice [37]. This series of studies demonstrates that the use of broad HDAC inhibitors as potential therapeutic agents for the impaired histone acetylation levels and/or reduced HAT activities associated with transcriptional dysregulation and neuronal death must take into account the roles of specific HDACs in mediating retinal cell survival in physiological and pathological contexts.
Histone methylation
Histone methylation is a non-charge neutralizing modification that occurs on specific lysine and arginine residues of histones H3 and H4. Methylation of particular lysine side chains can act as either permissive (e.g., H3K4, H3K36, and H3K79) or repressive (e.g., H3K9, H3K27, and H4K20) marks in the context of transcription. However, the extent of methylation of these residues can also affect their function in transcriptional activation or repression, with differential effects mediated by mono-, di- or trimethylation, thereby adding another layer of complexity [38, 39].
Similar to histone acetylation, histone methylation appears to be involved in the maintenance of different retinal cell types. The Purkinje cell degeneration (pcd) mouse is characterized by postnatal loss of several neuronal cell types including retinal photoreceptors [40, 41]. Neurodegeneration in these mice is associated with profound chromatin disorganization, an increase in the levels of trimethylated histone H4 at lysine 20, and transcriptional silencing [42]. While only Purkinje cells were examined in this study, the mechanisms underlying cell death may be relevant to the rods and cones of the retina that are also lost in these mice.
Specific methyltransferase activities are important for retinal ganglion cell survival, as inhibitors of the Ezh2 and G9a histone methyltransferases can induce apoptosis of these neurons in vitro [43]. Other methyltransferases have also been implicated in neurodegenerative processes. The association of the Suv39H1 histone methyltransferase with p130, the Rb protein family member that is required for the survival of neurons in vitro, is lost in response to apoptotic stimuli, and this is accompanied by a substantial reduction in histone H3 methyltransferase activity [44]. Furthermore, expression of a p130 mutant that fails to interact with Suv39H1, or mutant Suv39H1 that lacks histone methyltransferase activity, leads to death of cultured neuronal cells [44]. Suv39H1 functions as a chromatin-modifying transcriptional silencer and, in this study, was found to cause derepression of pro-apoptotic genes when its interaction with p130 on the promoters of these genes was lost. The mutation underlying adult-onset cerebellar Purkinje cell degeneration occurring in the robotic mouse mutant lies within the gene that encodes Af4, a component of a chromatin remodeling complex which contains the DOT1 histone H3 lysine 79 methyltransferase [45]. The Af4 mutation causes a gain-of-function resulting in protein stabilization and accumulation, and increased levels of H3K79 methylation were correspondingly detected in cerebellar homogenates, which in turn is presumed to lead to transcriptional dysregulation in Purkinje neurons [46]. Therefore, the specific methylation of particular histones, as catalyzed by specific enzymes, is important for directing pro-survival gene expression in the retina as well as in other regions of the CNS. These studies also demonstrate that both elevated and reduced levels of histone methylation can contribute to altered transcription that leads to neuronal cell death, further underscoring the importance of tight regulation of histone post-translational modification.
DNA methylation defects lead to neurodegeneration
DNA methylation is generally associated with transcriptional repression, either through direct physical inhibition of transcription factor binding to methylated promoters, or by the association of methyl-CpG-binding domain (MBD) protein-containing repressive complexes with gene promoters [47]. Two DNA methyltransferases (Dnmts), Dnmt3a and Dnmt3b, function to establish de novo DNA methylation patterns during mammalian embryonic development, while a third enzyme, Dnmt1, acts as a maintenance methyltransferase and preferentially methylates hemi-methylated DNA generated following DNA replication [48].
A number of studies involving CNS-specific knockouts of Dnmts implicate altered DNA methylation in neurodegenerative phenotypes. Conditional deletion of Dnmt1 in mouse CNS precursor cells in vivo leads to global DNA hypomethylation and postnatal death of neurons in multiple brain regions [49]. Dnmt1 deletion targeted to the dorsal forebrain causes severe and progressive degeneration of cortical and hippocampal neurons due to hypomethylation-induced apoptotic cell death occurring both pre- and postnatally [50]. This was accompanied by electrophysiological deficits, neurobehavioural defects in learning and memory, and deregulation of neuronal gene expression [50, 51]. These studies demonstrate the importance of Dnmt1-mediated maintenance methylation for proper function and survival of CNS neurons at multiple developmental stages. In mice with specific deletion of Dnmt3a in the CNS, global DNA methylation was unperturbed; however, mutant mice exhibited loss of hypoglossal motor neurons located in the brainstem, abnormal neuromuscular junctions, motor defects, and premature death [52]. This may result from changes in gene-specific DNA methylation and/or altered function of Dnmt3a-associated chromatin remodeling proteins that may cause misregulation of target genes that are critical for motor neuron survival and function. The phenotype of these mice was similar to transgenic mouse models of amyotrophic lateral sclerosis (ALS) [53–57], further implicating Dnmt3a in the pathogenesis of motor neuron degeneration. Animal models of Dnmt3b loss in the CNS have not been described to date; however, the identification of DNMT3b mutations in human mental retardation [58, 59] indicate the essential function of this protein for proper brain function which may include the preservation of neuronal integrity.
These effects of impaired Dnmt activity and resulting reduction in DNA methylation levels have been extended to primate models of neurodegeneration. In postmortem analyses of human brains, the promoter region of the Alzheimer’s disease (AD)-causing amyloid precursor protein (APP) gene was shown to be hypomethylated in older individuals, suggesting a link between enhanced transcriptional potential of a pathogenic gene and increased susceptibility to neurodegeneration with age [60]. Indeed, reduced DNA methyltransferase activity as well as decreased cortical levels of Dnmt1, Dnmt3a, and MeCP2 proteins were associated with increased expression of the AD-related genes APP and beta-site APP cleaving enzyme 1 (BACE1) in aged monkeys with AD-like neuropathology [61, 62]. Additional examination of brain tissues from AD patients revealed global DNA hypomethylation along with diminished immunoreactivity for DNMT1 and multiple components of the MeCP1/MBD2 methylation complex [63, 64], further supporting a neuroprotective role of DNA methylation in AD etiology.
Increased DNA methyltransferase levels and activity, as well as consequent hypermethylation of DNA are also associated with neuronal cell death. Elevated methyltransferase activity and DNA methylation were detected upon ischemic brain injury in mice, and either genetic or pharmacological inhibition of Dnmt1 was found to be neuroprotective in this mild stroke model [65]. Similarly, in a motor neuron degeneration model, Dnmt1 and Dnmt3a protein levels and total Dnmt enzyme activities were upregulated and mediated rapid increases in DNA methylation, while Dnmt inhibition protected neurons against apoptosis [66]. The increased Dnmt protein expression and DNA hypermethylation were paralleled in motor neurons of ALS patients [66]. Therefore, tight regulation of DNA methylation is also critical for the survival of neurons, as an imbalance to create either a hypo- or hypermethylated genomic state can cause neurodegeneration. The effects on transcription of particular genes is relevant to this finding, as repression of pro-survival genes and inappropriate activation of cell death genes could both lead to neuronal loss. Accordingly, altered methylation of genes that may contribute to ALS pathobiology was observed in human ALS brain-derived DNA, including both hypo- and hypermethylated genes [67]. Thus, disturbance of the steady state levels of DNA covalent modification, as also described above for histone acetylation and methylation, can contribute to altered chromatin remodeling activity that leads to neurodegenerative processes. This further emphasizes the importance of strict control of chromatin dynamics for cellular homeostasis and the physical and functional integrity of neurons.
While examples of dysregulated DNA methylation have not been documented for neurodegeneration affecting the retina, a recent study describes the expression of DNA methyltransferases in mouse retinal progenitors and mature neurons, highlighting distinct patterns of localization in different neuronal cell types that may reflect different developmental requirements for chromatin remodeling and/or differences in chromatin architecture that underlie cell-specific properties [68]. The role of specific Dnmts in mediating the function and maintenance of retinal neurons awaits further studies including cell type-specific ablation.
ATP-dependent nucleosome remodelers mediate survival of specific cell types
ATP-dependent chromatin remodeling proteins function to non-covalently alter the structure of nucleosomes through modulation of DNA-histone interactions, thereby regulating chromatin compaction and DNA accessibility to proteins involved in diverse cellular processes such as DNA replication, recombination, repair, and transcription. The energy of ATP hydrolysis is utilized to disrupt nucleosomes by promoting histone sliding, repositioning, exchange, or eviction [4]. These nucleosome remodeling factors function as part of large multi-protein complexes and are recruited to target genes based on interactions with other subunits of these complexes that can recognize and bind to specific DNA sequences and/or histone and DNA modifications [69].
The importance of ATP-dependent chromatin remodeling proteins in neurodevelopment is demonstrated by the identification of mutations in the genes that encode these proteins, or proteins with which they interact, in human developmental disorders associated with intellectual disability [70–78]. A number of studies suggest that nucleosome remodelers also function to maintain cell viability, including neural tissues. The ATRX protein was one of the first chromatin remodeling factors associated with human genetic disease, with mutations in the gene causing α–thalassaemia mental retardation, X-linked (ATR-X) syndrome [71]. ATR-X syndrome patients exhibit a diverse range of clinical features including microcephaly and cognitive deficits indicating a critical role for ATRX in proper brain structure and function. Consistent with the human phenotype, conditional deletion of Atrx in the developing mouse forebrain resulted in reduced brain size and a significant loss of neuronal cells during corticogenesis [79, 80]. Hypocellularity was also detected at postnatal stages in the hippocampus, with mutant mice completely lacking a dentate gyrus. The neuronal loss occurred due to inappropriate apoptosis of neuroprogenitors and could be recapitulated in isolated neuroprogenitor primary cultures derived from the knockout mice, suggesting the involvement of cell autonomous cell death mechanisms [79]. In contrast, overexpression of Atrx in transgenic mice resulted in the generation of excessive neuroprogenitors [81], further supporting a role for Atrx in neuronal homeostasis.
In the mouse retina, loss of Atrx leads to a selective reduction in amacrine and horizontal cell populations in the postnatal period [82]. Various amacrine cell subtypes are affected, including glycinergic, cholinergic, and dopaminergic neurons [82]. Atrx is therefore important for the survival of inhibitory interneurons in both the retina and the brain, as loss of GABAergic interneurons is also observed in forebrain-specific knockout mice [80]. However, the survival of neuroprogenitors is unaffected in the Atrx-knockout retinas [82], implying differential spatial and temporal neuroprotective functions of Atrx. Interestingly, conditional deletion of Atrx in mouse testis causes prenatal apoptosis of proliferating Sertoli cells [83]. Physical interaction of Atrx with the testis-specific androgen receptor was shown to regulate transcription of tissue-specific target genes [83]. Thus Atrx-mediated maintenance of specific cell types in different tissues including the retina may result from the transcriptional regulation of tissue-specific Atrx targets that impinge on pro-survival pathways.
Epigenetic mechanisms of neurotoxicity
While there is significant documentation of epigenetic factors associated with neuronal survival, there are few well-characterized mechanisms. Several potential triggers of neurotoxicity have been proposed to underlie degeneration of neurons in the retina and brain. These include oxidative stress [84], DNA damage [85, 86], mitochondrial dysfunction [87, 88], lack of neurotrophic support [89], and excitotoxicity [90]. Chromatin remodeling proteins have been implicated in mediating survival responses to some of these neurotoxic stimuli.
Oxidative stress results from the production of cytotoxic reactive oxygen species derived from cellular metabolism of oxygen. High metabolic activity, high proportions of polyunsaturated fatty acids that undergo lipid peroxidation, and exposure to visible light that induces photo-oxidation render the retina particularly susceptible to oxidative damage [91]. The inability of neurons to protect against or repair the damage caused by reactive oxygen species leads to their death. Bmi1 is a member of the Polycomb group family of chromatin modifiers, and its target genes include those involved in neuronal survival as well as antioxidant defenses [92]. Mice with Bmi1 deficiency exhibit retinal defects and increased neuronal death in the brain accompanied by increased apoptotic gene expression, reduced expression of antioxidant genes, and elevated levels of reactive oxygen species [92].
DNA damage is recognized as a significant mediator of neuronal cell death [85, 86]. Increases in both nuclear and mitochondrial DNA lesions have been reported for various neurodegenerative conditions including retinal degeneration [93–95]. Chromatin remodeling is a fundamental process in the protection of genomic integrity by DNA repair mechanisms [96], and thus it follows that chromatin remodeling proteins function in neuroprotective responses to DNA damage. Inhibition of HDAC1 was shown to result in double strand DNA breaks and neuronal death, while HDAC1 overexpression protected against DNA damage and neurotoxicity in cultured neurons [34]. In the pcd mouse model, Purkinje cell degeneration is associated with a large scale reorganization of chromatin and propagation of histone modifications that cause gene silencing (via trimethylated histone H4K20) and that signal DNA damage (phosphorylation of the histone variant H2AX) [42]. Defects in the DNA repair pathway and accumulation of DNA damage in these mice are presumed to trigger neuronal death.
Mitochondrial dysfunction is associated with various forms of retinal and brain degeneration, and mutation of mitochondrial genes can lead to neurodegenerative diseases [88, 97]. Mitochondria in mouse and human tissues were found to contain DNA methyltransferases and methylated mitochondrial DNA [66]. This study further showed that DNMT3a protein levels were upregulated in motor cortex mitochondria from ALS patients, suggesting that epigenetic regulation of mitochondrial genes may contribute to neurodegenerative phenotypes.
Neurotrophins are growth factors important for supporting neuronal function and survival and exert their neurotrophic effects through receptor-mediated intracellular signaling pathways. A number of these factors have demonstrated roles in neuroprotection of various retinal cell types in vitro and when administered to mouse models of retinal degeneration [89, 98]. Brain-derived neurotrophic factor (BDNF) has been reported to prevent retinal ganglion and amacrine cell death [99–101] and can also rescue photoreceptor loss in animal models of retinal degeneration [102, 103]. BDNF expression is controlled by a number of epigenetic factors [104] and can be modulated by DNA methylation and histone modification [105]. Neurotrophins such as BDNF in turn directly induce epigenetic changes to regulate the transcription of their target genes [106, 107]. Given the functional interaction between BDNF and MeCP2 [108] and the physical interaction of MeCP2 with Atrx [75], it is tempting to speculate that the amacrine cell loss observed in Atrx-knockout mouse retinas may result from misregulation of a common pathway involving all three factors that functions to promote retinal neuron survival.
Excitotoxicity is caused by the hyperactivity of glutamate receptors on the cell surface of neurons due to excessive levels of the excitatory neurotransmitter glutamate, which in turn produces multiple adverse effects that lead to neuronal cell death. Excitotoxic neuronal death is a common feature of acute and chronic neurodegenerative conditions affecting various regions of the CNS including the inner retina [90, 109]. Studies are emerging to implicate epigenetic mechanisms in excitotoxic processes. Glutamatergic inputs can induce chromatin remodeling in neurons, and this is inhibited by NMDA receptor antagonists [110]. Glutamate receptor expression is also subject to epigenetic regulation. The expression of glutamate receptor subunits is regulated by the transcriptional repressor REST/NRSF, which requires ATP-dependent chromatin remodeling mediated by Brg1 and histone deacetylation for gene silencing [111]. Increased REST expression was associated with suppression of the AMPA receptor subunit GluR2 and rescue of ischemia-induced neuronal cell death [112, 113]. Brg1-mediated recruitment of HDAC1 to the NMDA receptor subunit NR2B promoter leads to NR2B repression [114], while HDAC inhibition increases NR2B expression and NMDA receptor activity [115]. NR2B expression was also shown to be regulated by the histone methyltransferase Setdb1 [116], indicating multiple epigenetic levels of transcriptional control of glutamate receptors. In addition, the astroglial glutamate transporter EAAT2/GLT1 prevents glutamate-induced excitotoxicity, and its expression is regulated by DNA methylation in response to neuronal stimulation [117].
Conclusions
Complementing the established role for epigenetic regulation in neuronal development and function are studies detailing the importance of chromatin remodeling for long-term survival. We have summarized the current knowledge of epigenetic strategies employed by neurons to withstand a lifetime of genetic and environmental insults in order to evade death. As these mechanisms continue to emerge, it is becoming clear that chromatin regulation of neuroprotection is complex and is impacted by a wide range of neurotoxic stimuli. Failure of these mechanisms leads to cellular dysfunction and neuronal loss, the key hallmarks of human neurodegenerative diseases.
The development of effective treatments for retinal degeneration and other neurodegenerative disorders requires a thorough understanding of the molecular events that control the survival of neurons and their demise in these diseases. The finding that changes in DNA and histone covalent modifications contribute to altered gene expression that leads to neuronal cell compromise can be exploited to generate therapeutic approaches targeted to chromatin modifying activities. For example, the association of reduced histone acetylation levels with neurodegenerative conditions has led to the extensive investigation of HDAC inhibitors as well as the proposal of HAT activators as therapeutics for neuronal degeneration [13, 118, 119]. However, the finely tuned epigenetic landscape of neurons, as evidenced by the interplay of different chromatin remodeling enzymes, poses significant challenges to the development of effective treatment regimes. Therapeutic efforts further need to address the differential temporal and spatial requirements for specific chromatin modifications or processing, as exemplified by the disparity of Atrx-mediated nucleosome remodeling in the embryonic forebrain and in the postnatal retina. It will be necessary to fully characterize the activities of individual chromatin modifying agents on specific genes in distinct neuronal cell populations, and in response to diverse neurotoxic triggers, to ensure specificity of targeted neuroprotective strategies and to avoid confounding off-target effects. With a heightened focus on investigation and discovery into the epigenetic programs controlling neuron survival and homeostasis in the retina and CNS, the promise of such directed treatments grows ever closer.
References
Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell. 2004;116:259–72.
Urnov FD, Wolffe AP. Chromatin remodeling and transcriptional activation: the cast (in order of appearance). Oncogene. 2001;20:2991–3006.
Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21:175–86.
Bell O, Tiwari VK, Thoma NH, et al. Determinants and dynamics of genome accessibility. Nat Rev Genet. 2011;12:554–64.
Agathocleous M, Harris WA. From progenitors to differentiated cells in the vertebrate retina. Annu Rev Cell Dev Biol. 2009;25:45–69.
Reese BE. Development of the retina and optic pathway. Vision Res. 2011;51:613–32.
Cvekl A, Mitton KP. Epigenetic regulatory mechanisms in vertebrate eye development and disease. Heredity. 2010;105:135–51.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705.
Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–19.
Rouaux C, Loeffler JP, Boutillier AL. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 2004;68:1157–64.
Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–50.
Sleiman SF, Basso M, Mahishi L, et al. Putting the ‘HAT’ back on survival signalling: the promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs. 2009;18:573–84.
Furukawa T, Morrow EM, Li T, et al. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999;23:466–70.
Corbo JC, Lawrence KA, Karlstetter M, et al. CRX ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Res. 2010;20:1512–25.
Peng GH, Chen S. Crx activates opsin transcription by recruiting HAT-containing co-activators and promoting histone acetylation. Hum Mol Genet. 2007;16:2433–52.
Palhan VB, Chen S, Peng GH, et al. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:8472–7.
Yvert G, Lindenberg KS, Picaud S, et al. Expanded polyglutamines induce neurodegeneration and trans-neuronal alterations in cerebellum and retina of SCA7 transgenic mice. Hum Mol Genet. 2000;9:2491–506.
La Spada AR, Fu YH, Sopher BL, et al. Polyglutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron. 2001;31:913–27.
Kizilyaprak C, Spehner D, Devys D, et al. The linker histone H1C contributes to the SCA7 nuclear phenotype. Nucleus. 2011;2:444–54.
Abou-Sleymane G, Chalmel F, Helmlinger D, et al. Polyglutamine expansion causes neurodegeneration by altering the neuronal differentiation program. Hum Mol Genet. 2006;15:691–703.
Sancho-Pelluz J, Alavi MV, Sahaboglu A, et al. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010;1:e24.
Gaub P, Joshi Y, Wuttke A, et al. The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain. 2011;134:2134–48.
Chen B, Cepko CL. Requirement of histone deacetylase activity for the expression of critical photoreceptor genes. BMC Dev Biol. 2007;7:78.
Salminen A, Tapiola T, Korhonen P, et al. Neuronal apoptosis induced by histone deacetylase inhibitors. Brain Res Mol Brain Res. 1998;61:203–6.
Boutillier AL, Trinh E, Loeffler JP. Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem. 2003;84:814–28.
Rouaux C, Jokic N, Mbebi C, et al. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–49.
Song C, Kanthasamy A, Anantharam V, et al. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol Pharmacol. 2010;77:621–32.
Parker JA, Arango M, Abderrahmane S, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–50.
Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–79.
de Oliveira RM, Pais TF, Outeiro TF. Sirtuins: common targets in aging and in neurodegeneration. Curr Drug Targets. 2010;11:1270–80.
Shindler KS, Ventura E, Dutt M, et al. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J Neuroophthalmol. 2010;30:328–39.
Jaliffa C, Ameqrane I, Dansault A, et al. Sirt1 involvement in rd10 mouse retinal degeneration. Invest Ophthalmol Vis Sci. 2009;50:3562–72.
Kim D, Frank CL, Dobbin MM, et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron. 2008;60:803–17.
Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–9.
Pandey UB, Nie Z, Batlevi Y, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63.
Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11:62.
Barski A, Cuddapah S, Cui K, et al. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–37.
Steger DJ, Lefterova MI, Ying L, et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol Cell Biol. 2008;28:2825–39.
LaVail MM, Blanks JC, Mullen RJ. Retinal degeneration in the pcd cerebellar mutant mouse. I. Light microscopic and autoradiographic analysis. J Comp Neurol. 1982;212:217–30.
Blanks JC, Mullen RJ, LaVail MM. Retinal degeneration in the pcd cerebellar mutant mouse. II. Electron microscopic analysis. J Comp Neurol. 1982;212:231–46.
Baltanas FC, Casafont I, Lafarga V, et al. Purkinje cell degeneration in pcd mice reveals large scale chromatin reorganization and gene silencing linked to defective DNA repair. J Biol Chem. 2011;286:28287–302.
Rao RC, Tchedre KT, Malik MT, et al. Dynamic patterns of histone lysine methylation in the developing retina. Invest Ophthalmol Vis Sci. 2010;51:6784–92.
Liu DX, Nath N, Chellappan SP, et al. Regulation of neuron survival and death by p130 and associated chromatin modifiers. Genes Dev. 2005;19:719–32.
Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106.
Bitoun E, Davies KE. The robotic mouse: understanding the role of AF4, a cofactor of transcriptional elongation and chromatin remodeling, in Purkinje cell function. Cerebellum. 2009;8:175–83.
Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97.
Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481–514.
Fan G, Beard C, Chen RZ, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–97.
Hutnick LK, Golshani P, Namihira M, et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–88.
Golshani P, Hutnick L, Schweizer F, et al. Conditional Dnmt1 deletion in dorsal forebrain disrupts development of somatosensory barrel cortex and thalamocortical long-term potentiation. Thalamus Relat Syst. 2005;3:227–33.
Nguyen S, Meletis K, Fu D, et al. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236:1663–76.
Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–5.
Gurney ME. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med. 1994;331:1721–2.
Weydt P, Hong SY, Kliot M, et al. Assessing disease onset and progression in the SOD1 mouse model of ALS. Neuroreport. 2003;14:1051–4.
Lalonde R, Le Pecheur M, Strazielle C, et al. Exploratory activity and motor coordination in wild-type SOD1/SOD1 transgenic mice. Brain Res Bull. 2005;66:155–62.
Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–7.
Xu GL, Bestor TH, Bourc’his D, et al. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature. 1999;402:187–91.
Hansen RS, Wijmenga C, Luo P, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–7.
Tohgi H, Utsugisawa K, Nagane Y, et al. Reduction with age in methylcytosine in the promoter region -224 approximately -101 of the amyloid precursor protein gene in autopsy human cortex. Brain Res Mol Brain Res. 1999;70:288–92.
Wu J, Basha MR, Brock B, et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9.
Bihaqi SW, Huang H, Wu J, et al. Infant exposure to lead (Pb) and epigenetic modifications in the aging primate brain: implications for Alzheimer’s disease. J Alzheimers Dis. 2011;27:819–33.
Mastroeni D, McKee A, Grover A, et al. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS One. 2009;4:e6617.
Mastroeni D, Grover A, Delvaux E, et al. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiol Aging. 2010;31:2025–37.
Endres M, Meisel A, Biniszkiewicz D, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–81.
Chestnut BA, Chang Q, Price A, et al. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31:16619–36.
Morahan JM, Yu B, Trent RJ, et al. A genome-wide analysis of brain DNA methylation identifies new candidate genes for sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:418–29.
Nasonkin IO, Lazo K, Hambright D, et al. Distinct nuclear localization patterns of DNA methyltransferases in developing and mature mammalian retina. J Comp Neurol. 2011;519:1914–30.
Hargreaves DC, Crabtree GR. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420.
Troelstra C, van Gool A, de Wit J, et al. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell. 1992;71:939–53.
Gibbons RJ, Picketts DJ, Villard L, et al. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell. 1995;80:837–45.
Meira LB, Graham Jr JM, Greenberg CR, et al. Manitoba aboriginal kindred with original cerebro-oculo- facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene. Am J Hum Genet. 2000;66:1221–8.
Vissers LE, van Ravenswaaij CM, Admiraal R, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36:955–7.
Harikrishnan KN, Chow MZ, Baker EK, et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet. 2005;37:254–64.
Nan X, Hou J, Maclean A, et al. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc Natl Acad Sci U S A. 2007;104:2709–14.
Verstappen G, van Grunsven LA, Michiels C, et al. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum Mol Genet. 2008;17:1175–83.
Yoshimura K, Kitagawa H, Fujiki R, et al. Distinct function of 2 chromatin remodeling complexes that share a common subunit, Williams syndrome transcription factor (WSTF). Proc Natl Acad Sci U S A. 2009;106:9280–5.
Kernohan KD, Jiang Y, Tremblay DC, et al. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev Cell. 2010;18:191–202.
Berube NG, Mangelsdorf M, Jagla M, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–67.
Seah C, Levy MA, Jiang Y, et al. Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. J Neurosci. 2008;28:12570–80.
Berube NG, Jagla M, Smeenk C, et al. Neurodevelopmental defects resulting from ATRX overexpression in transgenic mice. Hum Mol Genet. 2002;11:253–61.
Medina CF, Mazerolle C, Wang Y, et al. Altered visual function and interneuron survival in Atrx knockout mice: inference for the human syndrome. Hum Mol Genet. 2009;18:966–77.
Bagheri-Fam S, Argentaro A, Svingen T, et al. Defective survival of proliferating Sertoli cells and androgen receptor function in a mouse model of the ATR-X syndrome. Hum Mol Genet. 2011;20:2213–24.
Fatokun AA, Stone TW, Smith RA. Oxidative stress in neurodegeneration and available means of protection. Front Biosci. 2008;13:3288–311.
Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res. 2007;614:24–36.
Hetman M, Vashishta A, Rempala G. Neurotoxic mechanisms of DNA damage: focus on transcriptional inhibition. J Neurochem. 2010;114:1537–49.
Fernandez-Checa JC, Fernandez A, Morales A, et al. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: lessons from mouse models. CNS Neurol Disord Drug Targets. 2010;9:439–54.
Barot M, Gokulgandhi MR, Mitra AK. Mitochondrial dysfunction in retinal diseases. Curr Eye Res. 2011;36:1069–77.
Whitmire W, Al-Gayyar MM, Abdelsaid M, et al. Alteration of growth factors and neuronal death in diabetic retinopathy: what we have learned so far. Mol Vis. 2011;17:300–8.
Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–42.
Bramall AN, Wright AF, Jacobson SG, et al. The genomic, biochemical, and cellular responses of the retina in inherited photoreceptor degenerations and prospects for the treatment of these disorders. Annu Rev Neurosci. 2010;33:441–72.
Chatoo W, Abdouh M, David J, et al. The polycomb group gene Bmi1 regulates antioxidant defenses in neurons by repressing p53 pro-oxidant activity. J Neurosci. 2009;29:529–42.
Weissman L, de Souza-Pinto NC, Stevnsner T, et al. DNA repair, mitochondria, and neurodegeneration. Neuroscience. 2007;145:1318–29.
Yang JL, Weissman L, Bohr VA, et al. Mitochondrial DNA damage and repair in neurodegenerative disorders. DNA Repair. 2008;7:1110–20.
Jarrett SG, Lin H, Godley BF, et al. Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res. 2008;27:596–607.
Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–9.
Swerdlow RH. The neurodegenerative mitochondriopathies. J Alzheimers Dis. 2009;17:737–51.
Buch PK, MacLaren RE, Ali RR. Neuroprotective gene therapy for the treatment of inherited retinal degeneration. Curr Gene Ther. 2007;7:434–45.
Chaum E. Retinal neuroprotection by growth factors: a mechanistic perspective. J Cell Biochem. 2003;88:57–75.
Cusato K, Bosco A, Linden R, et al. Cell death in the inner nuclear layer of the retina is modulated by BDNF. Brain Res Dev Brain Res. 2002;139:325–30.
Seki M, Tanaka T, Nawa H, et al. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes. 2004;53:2412–9.
Okoye G, Zimmer J, Sung J, et al. Increased expression of brain-derived neurotrophic factor preserves retinal function and slows cell death from rhodopsin mutation or oxidative damage. J Neurosci. 2003;23:4164–72.
Harada C, Guo X, Namekata K, et al. Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat Commun. 2011;2:189.
Cowansage KK, LeDoux JE, Monfils MH. Brain-derived neurotrophic factor: a dynamic gatekeeper of neural plasticity. Curr Mol Pharmacol. 2010;3:12–29.
Ishimaru N, Fukuchi M, Hirai A, et al. Differential epigenetic regulation of BDNF and NT-3 genes by trichostatin A and 5-aza-2′-deoxycytidine in Neuro-2a cells. Biochem Biophys Res Commun. 2010;394:173–7.
Riccio A. Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat Neurosci. 2010;13:1330–7.
Sen N, Snyder SH. Neurotrophin-mediated degradation of histone methyltransferase by S-nitrosylation cascade regulates neuronal differentiation. Proc Natl Acad Sci U S A. 2011;108:20178–83.
Chang Q, Khare G, Dani V, et al. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–8.
Romano C, Chen Q, Olney JW. The intact isolated (ex vivo) retina as a model system for the study of excitotoxicity. Prog Retin Eye Res. 1998;17:465–83.
Li J, Guo Y, Schroeder FA, et al. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. J Neurochem. 2004;90:1117–31.
Battaglioli E, Andres ME, Rose DW, et al. REST repression of neuronal genes requires components of the hSWI.SNF complex. J Biol Chem. 2002;277:41038–45.
Tanaka H, Calderone A, Jover T, et al. Ischemic preconditioning acts upstream of GluR2 down-regulation to afford neuroprotection in the hippocampal CA1. Proc Natl Acad Sci U S A. 2002;99:2362–7.
Calderone A, Jover T, Noh KM, et al. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–21.
Qiu Z, Ghosh A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron. 2008;60:775–87.
Nicolai J, Burbassi S, Rubin J, et al. CXCL12 inhibits expression of the NMDA receptor’s NR2B subunit through a histone deacetylase-dependent pathway contributing to neuronal survival. Cell Death Dis. 2010;1:e33.
Jiang Y, Jakovcevski M, Bharadwaj R, et al. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J Neurosci. 2010;30:7152–67.
Yang Y, Gozen O, Vidensky S, et al. Epigenetic regulation of neuron-dependent induction of astroglial synaptic protein GLT1. Glia. 2010;58:277–86.
Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–68.
Selvi BR, Cassel JC, Kundu TK, et al. Tuning acetylation levels with HAT activators: therapeutic strategy in neurodegenerative diseases. Biochim Biophys Acta. 2010;1799:840–53.
Acknowledgements
This work was funded by Foundation Fighting Blindness-Canada and Canadian Institutes of Health Research operating grants to D. J. P.
Conflict of interest
The authors declare no competing conflicts, financial or otherwise.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Lagali, P.S., Picketts, D.J. Matters of life and death: the role of chromatin remodeling proteins in retinal neuron survival. j ocul biol dis inform 4, 111–120 (2011). https://doi.org/10.1007/s12177-012-9080-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12177-012-9080-3