
Abstract
An accurate, rapid, and reliable multiresidue QuEChERS method based on gas chromatography coupled with tandem mass spectrometry was developed for the determination of 235 pesticides in challenging, dry, complex herb matrices (Centaurea cyanus L., Matricaria chamomilla L., Thymus vulgaris L.). Sample mass and the type of cleanup sorbent used to estimate the procedure’s effectiveness were optimized. Purification steps with ChloroFiltr, ENVI-Carb, GCB, octadecyl, PSA, and Z-Sep as cleanup sorbents and a step without purification were compared. To minimize the matrix effect and obtain acceptable recoveries for pesticides, 2 g of herb sample and 10 mL acetonitrile, followed by d-SPE cleanup step with a combination of 150 mg PSA/45 mg ENVI-Carb/900 mg MgSO4, and additionally 50 μL of 5% formic acid and some droplets of dodecane, were needed. Matrix effects for the vast majority of pesticides were reduced (> 20%), showing suppression or enhancement. Most recoveries were in the range of 70–120% (RSD < 18%), reaching the quantification limit of 0.001 to 0.002 mg kg−1. There was excellent linearity within the range from 0.001 to 2.00 μg mL−1, and a correlation coefficient higher than 0.999 was obtained. Expanded measurement uncertainty was estimated to be between 4 and 43%. Finally, the developed method was successfully employed to identify and quantify pesticide residues in the analysis of 46 real herb samples.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nowadays, herbs are used in several applications around the world, such as in pharmaceutical products, in food and cosmetic additives, as herbal tea, and in folk medicinal uses. Herbs may contain anthocyanidins, flavonoids, vitamins, and mineral salts (including manganese content) as well as substances which exhibit antioxidant effects. They have many health benefits, such as anti-inflammatory and cholesterol-lowering properties and antispasmodic and anxiolytic activities, and they improve appetite and digestion (Lozano et al. 2012).
Like other plants, herbs are sensitive to fungal diseases (e.g., gray mold), and they are attacked by harmful pests (e.g., aphis) and unwanted weeds. These factors diminish crop yields and its quality; consequently, chemical plant protection is indispensable (Matyjaszczyk 2011). However, pesticide residues may pose potential health risks. Moreover, they are accumulated at different stages of cultivation and during postharvest storage (Hajou et al. 2004).
In order to ensure the safety of herbal products, depending on the intended purpose of the herbs (pharmaceutical products, food), maximum residue limits (MRLs) have been standardized by several international organizations, such as the European Union (EU) (EC Regulation No 396 2005), the European PharmacopoeiaPharmacopeia Commission (EP 8.0 European Directorate for the Quality of Medicines and HealthCare 2014), and the Codex Alimentarius Commission (CA 2016). According to the European Food Safety Authority (EFSA) report (EFSA 2015), 65.1% of the analyzed herbs contained residues of which 9.4% exceeded MRLs established by the EU. This clearly shows that monitoring of pesticide residues in herbs is a very important issue.
Herbs, especially dry herbs, represent a challenge due to the high amount of matrix co-extractives which pose critical analytical problems. Herbs contain a broad spectrum of essential oils, flavonoids, phenolics, lipids, natural pigments, and sugars (Lozano et al. 2012; Ghani 2014). Co-extracted matrix ingredients exhibit both partitioning behavior and chromatographic characteristics similar to some pesticides, which may bring about serious matrix effects (ME) and significant interference in mass spectrometry analysis. According to European guidelines (SANTE 2015), the matrix effect is the influence of one or more undetected components in samples on the measurement of an analyte’s concentration or mass. ME is one of the greatest difficulties in pesticide residue analysis, particularly in the analysis of complex dry samples.
Some methods to eliminate matrix effects are described in the literature, such as sample preparation, dilution of the extract samples, analytical protectants, addition of internal standards, use of different gas chromatography (GC) injection techniques, GC priming, and the matrix-matched standards method (Zrostlíková et al. 2001; Wang et al. 2011; Li et al. 2015; Liu et al. 2016; Nanita et al. 2017). The most common, relatively cheap, and simple method to compensate for matrix effects in gas chromatography is to use matrix-matched calibration standards (Kwon et al. 2012; Lehotay et al. 2010). Also, SANTE (2015) guidelines suggest matrix-matched calibration for pesticide residue analyses as a method of accounting for the matrix effect. Van de Steene et al. (2006) pointed out that due to variable matrix effect from sample to sample, a representative matrix should be used to prepare the calibration solutions. Unfortunately, it has been proved that finding a representative matrix may be very complicated or even impossible.
One of the methods of minimizing the matrix effect, mentioned above, is sample preparation which is required to isolate and quantify pesticide residues in troublesome matrices. Sample preparation is prone to errors and consumes about 80% of the analysis time (Gondo et al. 2016). On the one hand, recoveries for most pesticides need to be in the range of 70–120% (SANTE 2015). On the other hand, the amount of co-extractives needs to be as little as possible to minimize matrix effects and related instrumental problems.
Sample preparation for the analysis of pesticide residues in herbs commonly involves Soxhlet extraction (Rao et al. 2011), microwave-assisted extraction (MAE) (Wan et al. 2010), matrix solid-phase dispersion (MSPD) (Łozowicka et al. 2013), solid-phase microextraction (SPME) (Hwang and Lee 2000), ultrasonic extraction (Tong et al. 2014), and so on (Table 1). Some research works have reported pesticide residues in herbal material but were mainly based on surveying and monitoring market samples (Rao et al. 2011). However, these methods are expensive, laborious, and time-consuming and require multiple steps, resulting in loss of analytes and errors in analysis. The need for a robust, inexpensive, and straightforward method that would provide high-quality results with a minimal number of steps and reduced reagent consumption for an extensive range of analytes and matrices at low detection limits led Anastassiades et al. (2003a, b) to develop the QuEChERS (quick, easy, cheap, effective, rugged, safe) sample preparation technique which provides a method fitting the aforementioned criteria.
A few papers related to pesticide detection in herbs based on QuEChERS methodology have been published (Table 1). Nguyen et al. (2010) developed the QuEChERS method for 234 pesticides in three kinds of Korean herbs using gas chromatography mass spectrometry (GC-MS). The procedure entailed extraction with acetonitrile containing 0.5% acetic acid (HAc) followed by the dispersive solid-phase extraction (d-SPE) cleanup step employing bulk sorbents: MgSO4, primary secondary amine (PSA), and graphitized carbon black (GCB). Similarly, Dai et al. (2011) validated the QuEChERS method for determination of 23 pesticides in three medicinal herbs, entailing acetonitrile extraction and d-SPE cleanup with MgSO4, PSA, and GCB sorbents and finally analysis by GC-MS. Liu et al. (2016) reported a QuEChERS method for the analysis of 74 pesticides in the Chinese materia medica. The pesticides were extracted with hexane and cleaned up with MgSO4, octadecyl (C18), or Florisil, followed by determination by gas chromatography tandem mass spectrometry detection (GC-MS/MS). Despite the continuous appearance of many modifications of QuEChERS methods, there are very limited studies about the influence of these modifications on the matrix effect in scientific literature. Therefore, existing knowledge must be built upon.
This paper aims to improve the cleanup step to effectively minimize the matrix effect while maintaining excellent recovery in the analysis of 235 pesticides in various dried herbs using the QuEChERS method and detection by GC-MS/MS. Purification steps based on different d-SPE sorbent combinations and a step without cleanup were compared. Two grams of the sample and extraction with acetonitrile in combination with sorbents for d-SPE cleanup (PSA/ENVI-Carb/MgSO4) may be used prior to GC-MS/MS. In addition, the resulting procedure was applied for quantification of pesticide residues in 46 herb samples.
Materials and Methods
Materials and Reagents
Acetone, acetonitrile (AcN), and n-hexane were analytical grade and provided for pesticide residue analysis by J.T. Baker (Deventer, The Netherlands). Water was purified by a Milli-Q (Millipore, Billerica, MA, USA) system. Water was cooled to a temperature of about 4 °C. The QuEChERS sorbent kits (1) Z-Sep+, 200 mg; (2) Z-Sep/C18, 12/300 mg; (3) Z-Sep+/PSA/ENVI-Carb/MgSO4, 60/50/10/150 mg; (4) PSA/ENVI-Carb/MgSO4, 150/45/900 mg; (5) PSA/DSC-18/ENVI-Carb/MgSO4, 400/400/400/1200 mg; (6) GCB/PSA/MgSO4, 25/2.5/150 mg; (7) ChloroFiltr/PSA/MgSO4, 50/50/150 mg; and (8) ChloroFiltr/PSA/MgSO4, 50/50/150 mg, were supplied by Agilent Technologies (Santa Clara, CA, USA), UCT (Bristol, UK), and Sigma-Aldrich (Steinheim, Germany).
Analytical reagent-grade magnesium sulfate, sodium chloride, trisodium citrate dihydrate (Na3C6H5O7⋅2H2O), and disodium hydrogen citrate sesquehydrate (Na2HC6H5O7⋅1.5H2O) salt were obtained from Agilent Technologies (Santa Clara, CA, USA). Formic acid (98% purity) was supplied by Merck (Darmstadt, Germany).
The standard pesticides (purity > 95%) were purchased from Dr. Ehrenstorfer Laboratory (Augsburg, Germany). The internal standard (IS), triphenyl phosphate (TPP), was obtained from Sigma-Aldrich (Steinheim, Germany).
Preparation of Standards
The stock standard solution was prepared separately in a concentration around 1000 μg mL−1, using acetone as solvent. Intermediate stock standard mixtures in acetone, containing 10 μg mL−1 of each compound, were prepared by mixing appropriate quantities of the individual stock solutions. A working standard mixture of 0.001/0.002–1.0/2.0 μg mL−1 was prepared in an n-hexane/acetone mixture (9:1, v/v) from the above stock standard by serial dilution. The standard working solution was used to prepare matrix-matched standards and to spike samples in the validation study. The IS was prepared as described above. The stock, working standard solutions, and IS were stored in amber glassware under appropriate conditions, i.e., at − 20 °C until the time of analysis.
Sample Preparation
Two grams of herb was weighed into a 50-mL centrifuge tube. Ten milliliters of cold purified water was added into the tube and shaken by hand for a few seconds to hydrate the samples. After standing for 10 min, 50 μL of 20 μg mL−1 IS and 10 mL acetonitrile were added. The sample was shaken for 1 min. The tube was placed in a − 20 °C freezer for 15 min. Next, 4 g MgSO4, 1 g NaCl, 1 g Na3C6H5O7⋅2H2O, and 0.5 g Na2HC6H5O7⋅1.5H2O were added. The tube was shaken vigorously by hand for 1 min to prevent the formation of crystalline agglomerates during MgSO4 hydration and then centrifuged for 5 min at 4500 rpm. Four-milliliter aliquots of the acetonitrile layer were transferred into a cleanup tube containing PSA/ENVI-Carb/MgSO4 sorbent. The tube was shaken for 1 min and centrifuged at 4500 rpm for 10 min. Two milliliters was downloaded and transferred into a flask and 50 μL of 5% formic acid and some droplets of dodecane were added. Afterwards, the supernatant was evaporated to near dryness using a rotary evaporator at a temperature of about 40 °C. The residue was redissolved in 2 mL of n-hexane/acetone (9:1, v/v) and filtered through a 0.45-μm nylon filter into an autosampler vial for GC analysis.
GC-MS/MS Conditions
Chromatographic analyses were performed in gas chromatograph coupled to triple quadrupole system MS/MS (Agilent Technologies, Palo Alto, CA, USA). The chromatograph was equipped with an Agilent 7693 autosampler. The GC system’s specifications and the triple quadrupole system’s parameters are summarized in Table 2. Data acquisition, control, and data processing were performed using MassHunter Workstation Software (B.06.00). Multiple reaction monitoring (MRM) transitions and other acquisition parameters can be found in the Supplementary Material (Table S1). Retention time locking (RTL) was used to eliminate the need for adjusting the time-segment windows of MRM groups, using chlorpyrifos methyl as the locking compound for a retention time of 16.593 min.
Validation Study
Two hundred and thirty-five pesticides were validated in three kinds of herb matrices, Centaurea cyanus L., Matricaria chamomilla L., and Thymus vulgaris L., using the QuEChERS method and GC-MS/MS analysis. The following parameters were determined according to European guidelines (SANTE 2015) during validation of the analytical method: linearity, recovery, precision, limit of quantification (LOQ), limit of detection (LOD), matrix effects (ME), and uncertainty (U).
Linearity was studied using pure solvent and matrix extracts of Centaurea cyanus L., Matricaria chamomilla L., and Thymus vulgaris L. in six different concentrations: 0.001/0.005, 0.005/0.01, 0.01/0.02, 0.05/0.1, 0.5/1.0, and 1.0/2.0 μg mL−1. The LOQ was set at the minimum concentration that could be quantified with acceptable values of recovery (70–120%) and RSD (≤ 20%). In certain cases with multiresidue methods, recoveries outside this range may be accepted (60–130%) but demonstrate good precision (RSD ≤ 20%) (SANTE 2015). LOD was calculated using signal-to-noise ratio (S/N) criteria, in all cases; LOD = 3 S/N.
For recovery determination, the homogenized samples (2 ± 0.01 g) were spiked with working standard solution at three concentration levels: 0.005/0.01, 0.05/0.1, and 1.0/2.0 mg kg−1. The spiked samples were set aside for 30 min and then processed according to the procedure described above. Precision was expressed in terms of relative standard deviation (RSD %) and calculated for each spiking level (n = 5).
In order to evaluate the influence of matrix components in the detector’s response, the peak area of standard in the matrix and the standard in the solvent at the same concentration level were used. Matrix effects were calculated as follows: %ME = ((peak area (matrix standard)/peak area(solvent standard)) − 1) × 100. The matrix effect could be negative or positive and would be classified in one of three categories: soft matrix effect, medium matrix effect (20–50% positive values and − 20 to − 50% negative values), and strong matrix effect (more than 50% positive and less than −50% negative). Soft matrix effect is considered as ME (%) values within repeatability range values (− 20 to 20%) accepted in pesticide analysis (Ferrer et al. 2011).
The data from the validation study were used to estimate the measurement uncertainty associated with the analytical results. The major uncertainty sources in the uncertainty budget were the repeatability of recoveries from the spiked samples and uncertainty of the average recovery calculated for rectangular distribution. Finally, relative expanded uncertainty was calculated by using coverage factor k = 2 at a confidence level of 95% (Walorczyk et al. 2015).
Results and Discussion
Optimization of Extraction and Cleanup Method
This study presents a modification of one of the most widely described multiresidue methodologies—the QuEChERS approach, which has many advantages including speed, cost, ease of use, good performance characteristics, and wide applicability range (matrices and analytes). Over the years, since its first introduction by Anastassiades et al. (2003a, b), the method has undergone various modifications and enhancements to ensure efficient extraction of pH-dependent compounds (e.g., phenoxyalcanoic acids) (Kaczyński et al. 2016), to minimize degradation of susceptible compounds (e.g., base and acid labile pesticides) (Lehotay et al. 2005b), and to expand the spectrum of food matrices amenable to the method (Łozowicka et al. 2016). Therefore, the QuEChERS method was optimized in order to achieve quick and effective extraction and a clean herbal extract.
In recent years, the significance of assessing matrix effects has increased relatively to recovery, which is a tool commonly used to estimate pesticide extraction efficiency. ME is described as one of the major sources of errors in gas chromatography (Kwon et al. 2012; Li et al. 2015) and may adversely affect the determination of pesticide residues in complex matrices.
In this study, recovery and matrix effect were chosen as criteria to estimate the method’s effectiveness. Thus, the challenge was to assess both parameters for each pesticide (235 pesticides in total) in herb samples prepared by various procedures. Some factors that can influence the performance of QuEChERS, such as sample mass and cleanup step or no cleanup step, were investigated.
Firstly, to ensure adequate partitioning, interactions between pesticides and the matrix were weakened by the addition of distilled water (10 mL) and contact for 10 min before the extraction step. Some papers recommend the addition of water to low-moisture samples before extraction (Anastassiades et al. 2003a, b). Liu et al. (2016), Ghani (2014), and Chen et al. (2012) added water to their herb samples, which made it possible to increase extraction efficiency.
Secondly, the effect of sample mass (1, 2, 5 g) on pesticide recoveries was evaluated. This study indicated that recoveries were in the range of 60–128, 31–135, and 35–149% when the mass of samples was 2, 1, and 5 g, respectively, for herb matrices. Pesticides extracted from 1 g of dry sample showed the same values as extracted from 5 g, but less efficient recovery than for the 2-g sample. However, more pesticides had recoveries below 40% when 1 g sample mass was used compared to 5 g. In the case of the highest sample mass (5 g), more pesticides had recoveries above 130%. Nevertheless, recoveries remained between 60 and 130% for most of the tested compounds (about 89%). Based on this experiment, 2 g sample mass was employed as the optimum amount in this study. Applications of different sample weights have been published (Nantia et al. 2017; Storelli 2014), and there are some reports where 2 g sample mass was chosen (Liu et al. 2016; Lozano et al. 2012) and gave satisfactory results.
Citrate-buffering salts were introduced in order to achieve a constant pH value during the extraction of herb matrices to improve the stability of base-sensitive compounds. These compounds undergo hydrolysis at a pH close to neutral. Under these conditions, the pH value of the matrix could be maintained within the range of 5.0–5.5, which was conducive to the stability of problematic pH-dependent pesticides. Even for troublesome (base-sensitive) pesticides, i.e., captan, chlorothalonil, or tolylfluanid, citrate-buffered QuEChERS enabled acceptable recoveries in all three matrices.
Herb samples contain many complex components, including essential oils, flavonoids, phenolic (Lozano et al. 2012), lipids, natural pigments, and sugars (Ghani 2014), which make analysis more complicated. Thus, the aim of this study was to reduce the co-extracted matrix components as much as possible. To achieve the best efficiency, the no cleanup step was compared to cleanup using eight different d-SPE sorbent combinations, which remove interferences and impact analyte recoveries (Fig. 1).

Visual comparison of extracts: (1) without and with cleanup, (2) Z-Sep+, (3) Z-Sep/C18, (4) Z-Sep+/PSA/ENVI-Carb/MgSO4, (5) PSA/ENVI-Carb/MgSO4, (6) PSA/DSC-18/ENVI-Carb/MgSO4, (7) ChloroFiltr/PSA/MgSO4, (8) ChloroFiltr/PSA/MgSO4/C18, and (9) GCB/PSA/MgSO4
The step without cleanup showed that more than 10% of the analytes had recoveries higher than 130% (i.e., in the range of 130–148%) and over 30% exhibited significant ME (above 50%). It would seem that the procedure without the purification step was inherently associated with co-extractives. So, cleanup of the herbal extract was needed to minimize the matrix effect. Therefore, the next modification with a cleanup step was tested. Only Ghani (2014) applied the QuEChERS method for determination of 10 pesticides in aniseeds without a purification step (Table 1).
During the experiments, it was found that various d-SPE sorbent combinations had a significant influence on the purification and recovery rates of analytes and many studies considered the use of different d-SPE sorbents in herb samples (Nguyen et al. 2010; Chen et al. 2012; Liu et al. 2016). Mostly, PSA, GCB, octadecyl (C18), and Florisil were described (Table 1). PSA can act as both a polar phase and weak anion exchanger with the ability to remove fatty acids, sugars, and other matrix co-extractives (organic acids, anthocyan pigments) which might adversely affect chromatographic performance (Kinsella et al. 2009; Anastassiades et al. 2003a, b). GCB has been reported to be a highly effective sorbent for sample cleanup (Kinsella et al. 2009). This is a nonporous reversed-phase sorbent and removes planar molecules such as natural pigments (e.g., chlorophyll, hemoglobin, and carotenoids), sterols, and nonpolar interferences (Anastassiades et al. 2003a, b). On the other hand, the use of high amounts of GCB (> 10 mg per 1 mL of acetonitrile extracts) may lead to unacceptable losses of some planar pesticides (Walorczyk et al. 2015). In this study, when the combination of GCB/PSA/MgSO4 was used, relatively weak recoveries (for about 20% of pesticides) were obtained, probably because of being subjected to strong GCB adsorption. Some nonpolar pesticides, especially those with a planar structure (chlorothalonil, cyprodinil, fenazaquin, hexachlorobenzene, mepanipirym, pirymetanil, prochloraz, quinoxyfen, thiabendazole) had poor recoveries. About 25% of the tested compounds had ME above 50%. Similarly, Liu et al. (2016), during optimization of the QuEChERS method, established that using PSA/GCB/MgSO4 as a sorbent for simultaneous determination of 135 pesticides in Chinese herbal medicines gave substantially lower recoveries (40.3–60.2%) for some particular pesticides (methamidophos, aldicarb, bifenazate, and bitertanol).
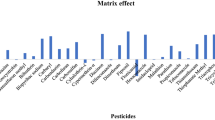
Other sorbents considered for use in this study are zirconium dioxide-based sorbents (Z-Sep and Z-Sep+), ChloroFiltr, and ENVI-Carb, and there is a lack of reports on the subject of pesticide determination in herbs using these kinds of sorbents. The retention mechanism of Z-Sep sorbents involves Lewis acid/base interactions. Due to its properties, it can reduce the amount of lipids and some pigments (Lozano et al. 2014) and provide an optimum balance between planar pesticide recovery and color removal. ChloroFiltr is a polymeric-based sorbent designed for the removal of chlorophyll without sacrificing the recovery of planar analytes (Walorczyk et al. 2015). ENVI-Carb is used to remove trace chemical compounds such as pigments, polyphenols, and other polar compounds. ENVI-Carb is a strong sorbent with a carbon surface comprised of hexagonal ring structures, interconnected and layered into graphitic sheets, and the nonporous nature of the carbon phase allows for rapid processing; adsorption does not require analyte diffusion into pores. The combination of ChloroFiltr/PSA/MgSO4 exhibits a wider spectrum of pigment adsorption. Over 80% of pesticides had acceptable recoveries, and 20% showed ME above 50%. Unfortunately, they were not applicable to this research. Similar results were achieved for combinations with sorbents based on zirconium dioxide. Recoveries in the acceptable range of 60–130% were achieved for about 75% of the analytes. In addition, the matrix effects were not sufficiently reduced (over 20% of pesticides above 50% ME). Excellent results were achieved using the PSA/ENVI-Carb/MgSO4 combination. This combination provided satisfactory recoveries (60–128%), and over 65–72% exhibited soft ME (< ±20%) (Figs. 2 and 3).

Recoveries of 235 pesticides for the modified QuEChERS procedures with and without cleanup

Matrix effects of 235 pesticides for the modified QuEChERS procedures with and without cleanup
In addition, octadecyl like PSA and GCB is the sorbent most commonly used to remove co-extractives from herbs (Table 1). C18 is a hydrophobic reversed-phase sorbent due to its extreme retentive nature toward nonpolar compounds such as lipids and pigments, and it also has stronger adsorption to nonpolar pesticides (Lehotay et al. 2005a). Octadecyl with a combination of ChloroFiltr, ENVI-Carb, and Z-Sep was checked, and the results showed decreased recovery for some pesticides (Fig. 2). Some nonpolar pesticides (alpha-HCH, beta-HCH, lindane, p,p′-DDE, p,p′-DDD, o,p′-DDT, p,p′-DDT) were retained on this sorbent, leading to lower analyte recoveries. About 20% of compounds showed strong matrix effects in all cases. These results indicated a higher presence of co-extractives compared with the extracts after cleanup with C18, which resulted in the occurrence of strong matrix effects. Thus, there is no need to employ C18 in the cleanup step.
As shown in this study, in such a complex matrix like herbs, 2 g of the sample and extraction with acetonitrile in combination with cleanup sorbents (PSA/ENVI-Carb/MgSO4) may be used prior to GC-MS/MS analysis to provide sufficient pigment removal (Fig. 1) while maintaining excellent recovery (60–130%—according to SANTE (2015) guidelines) and reducing matrix effects for the majority of pesticides tested in herb matrices (Figs. 2 and 3).
After d-SPE cleanup, formic acid in acetonitrile (5% v/v, 50 μL) was added to stabilize base-sensitive pesticides, including captan, chlorotalonil, dichlofluanid, dicofol, folpet, and tolyfluanid, which are susceptible to degradation in contact with the basic PSA sorbent. The pH of the final extracts measured after PSA cleanup reaches values in the range from 8 to 9, which endanger the stability of base-sensitive pesticides. The addition of formic acid brings the extract’s pH to a value of about 5 and seems to be the easiest solution to this problem.
Validation Study
The optimized analytical method for determination of 235 pesticides in herb samples using GC-MS/MS was evaluated. The investigated pesticides represent various chemical classes and a wide range of physicochemical properties, including volatility, polarity, chemical and thermal stability, etc. Their polarity ranges from the nonpolar esfenvalerate (logP 6.22), to the polar acephate (logP − 0.89), their solubility from the least soluble deltamethrin (logS − 3.7) to the most soluble acephate (logS 5.9), and their volatility from the least volatile fluquinconazole (logV.P. − 5.19) (V.P., vapor pressure) to the most volatile dichlorvos (logV.P. 3.32). Their molecular weights ranged from 141.13 (methamidophos) to 541.44 (acrinathrin) (PPDB 2016). The pesticides belong to over 50 different classes, i.e., chloronitrile, organochlorine, organophosphate, and pyrethroid (Supplementary Table S1). A series of experiments, covering linearity, precision, recovery, LOQ, LOD, and uncertainty, were performed to validate the modified QuEChERS method under optimized conditions using Centaurea cyanus L., Matricaria chamomilla L., and Thymus vulgaris L.
LOD and LOQ are important validation parameters and were used to evaluate the sensitivity of the analytical methods. LOQs ranged from 0.001 to 0.002 mg kg−1 and were equal to or lower than the European MRLs (EC Regulation No. 396/2005 with amendments). Obtained LOD values ranged from 0.0003 to 0.0007 mg kg−1 (Supplementary Table S2).
Good linearity results, with satisfactory correlation coefficients, were obtained for all pesticides, ranging from 0.99000 to 0.99999 (Supplementary Table S2).
Recoveries obtained for 235 pesticides were satisfactory and ranged from 60 to 127% with RSD 1–15% for Centaurea cyanus L., 60 to 128% with RSD 1–18% for Matricaria chamomilla L., and 60 to 128% with RSD 1–16% for Thymus vulgaris L. (Supplementary Table S3). In all three matrices, a similar group of problematic compounds was observed to have recovery values in the 60–130% range. Since consistent results (≤ 20% RSD) were achieved for these pesticides, the results could be corrected according to the recovery factors known from analyses. According to SANTE (2015) guiding criteria, pesticides resulting in the recovery range 60–130% can be used in routine multiresidue analysis. The fact that the same compounds presented problems in each of the different matrices suggests that the extraction conditions, not matrix components, were responsible for the low recoveries of these compounds. Among these compounds, dichlorvos, dichlobenil, and methamidophos are volatile and have the shortest retention times, and captan, chlorotalonil, dichlofluanid, dicofol, folpet, and tolyfluanid easily degrade during sample preparation and/or GC injection. Consistent recoveries of these problematic pesticides were typically below 70% but above 60%. Only 6% of tested pesticides showed recovery values above 120%, but below 130%.
Almost all pesticides had similar recoveries at three levels for all three matrices. Moreover, the recoveries of some pesticides in Matricaria chamomilla L. and in Thymus vulgaris L. are higher than in Centaurea cyanus L., i.e., bifenthrin, clomazone, dimoxystrobin, etoxazol, imazalil, iprodione, and picoxystrobin but still within the acceptable range. The reason for these differences is associated with the presence of varying proportions of chlorophyll in herbal matrices. On the other hand, some pesticides, i.e., malaoxon, paraoxon ethyl, and methyl, had considerably lower recoveries (below 70%) in Matricaria chamomilla L. and Thymus vulgaris L. compared to recoveries in Centaurea cyanus L. (Supplementary Table S3).
Expanded measurement uncertainties were estimated employing a “top-down” empirical model to be between 5 and 35% for Centaurea cyanus L., 4 and 39% for Matricaria chamomilla L., and 7 and 43% for Thymus vulgaris L. (coverage factor k = 2, confidence level 95%). This is less than the maximum threshold value of 50% recommended by European Union guidelines (SANTE 2015) (Supplementary Table S3).
Matrix Effects
In multiresidue analysis of pesticides in plant products by gas chromatography, co-extracted matrix components often increase the response, resulting in inaccurate quantification. Co-extracted matrix ingredients can compete with target pesticides for access to the active site during the injection process, which may result in enhancement of the detector’s signal. Meanwhile, (semi) polar or thermal-sensitive analytes may decompose at active sites in the liners, column, and detector, giving losses and distorted peak shapes, which may result in suppression of the chromatographic signal (Hajšlová et al. 1998).
The variability of matrix effects was estimated for the validated and optimized QuEChERS method. In the present study, interactions of pesticides with the matrix were observed, and matrix effect values are summarized in Supplementary Table S3, ranging from − 59 to 69% for Centaurea cyanus L., − 69 to 63% for Matricaria chamomilla L., and − 59 to 65% for Thymus vulgaris L. Among the 235 pesticides analyzed, 72% for Centaurea cyanus L., 65% for Matricaria chamomilla L., and 66% for Thymus vulgaris L. exhibited soft matrix effects. About 9% of the tested pesticides had strong matrix effects in all three matrices. ME was comparable in all three selected dried herbs but was more significant when they were in lower concentrations. Analogously, Nantia et al. (2017) showed that ME obtained for five fresh herbal products were quite similar. Therefore, we can confirm that matrix effects are not only associated with pesticides but also the concentration level.
Pesticides with amino (–R-NH–), azole (–N=), benzimidazole, carbamate (–O-CO-NH–), carboxyl (–COOH), hydroxyl (–OH), imidazole, organophosphate (–P=O), and urea (–NH-CO-NH–) groups are the analytes most susceptible to the matrix effect (Anastassiades et al. 2003a). These pesticides have functional groups which interact with silanol groups or metal ions on glass surfaces (liners, glass wool, etc.), so they usually show high matrix effects (Anastassiades et al. 2003a). For example, diniconazole (azole group), imibenconazole (benzimidazole), carbaryl (carbamate), acephate (organophosphate), and other classes like amitraz, chloridazon, and fenhexamid exhibited strong matrix-induced enhancement/suppression effects in all three herb matrices. On the other hand, some pesticides from the aforementioned groups susceptible to ME had a low matrix effect. In addition, compounds with high polarity are among the pesticides having the functional groups listed above as well as acephate, dicrotophos, dimethoate, methamidophos, and mevinphos. Other pesticides with high molecular mass (over 400 g mol−1), such as imibenconazole, propaquizafop, and tau fluvalinate, were also susceptible to ME (Supplementary Table S3). Compounds with high molecular mass used as neat standards generally gave poor peak shapes, which made it more difficult to integrate them and generate a response increase in matrix-matched standards (Sánchez-Brunete et al. 2005).
Signal suppression is less common in GC analysis; in this study, only 7% of pesticides showed signal suppression in all tested matrices. The majority of pesticides presented a similar tendency (enhancement or suppression) and intensity in three matrices. The exceptions are fenamiphos and propham which showed signal suppression in Matricaria chamomilla L. and Thymus vulgaris L. and enhancement in Centaurea cyanus L. Matricaria chamomilla L. and Thymus vulgaris L. gave slightly higher ME results than Centaurea cyanus L. This could be explained by a greater amount of interfering substances extracted from cornflower than from chamomile, causing signal enhancement.
In summary, variation of ME depends on several factors, such as the physical-chemical properties of pesticides, the matrix itself, and the interactions between them.
GC-MS/MS Optimization
The use of MRM mode based on QqQ mass spectrometry allows low analyte detectability and is, at present, one of the most selective approaches for trace analysis. However, for multiclass, multiresidue methods, a precise optimization of MS/MS parameters is needed in order to maximize the signal for each pesticide. The first step was to select the parent ions (precursor ions), which were chosen after examination of the full scan spectra of each pesticide. The base peak was not always preferred (highest intensity), and a more selective one was chosen for several pesticides. For some pesticides, three parent ions were selected, one for each transition.
Secondly, four transitions were taken for each pesticide after the fragmentation of each parent ion was checked in product scan mode. Finally, MRM collision energy was optimized for each selected transition using matrix-matched solutions, and the three most selective transitions were chosen. Supplementary Table S1 provides the selected conditions implemented in the MRM program used for GC-MS/MS analysis.
Application to Real Samples
Real samples were applied in the analysis in order to prove the suitability of the optimized method. Forty-six samples of herbs were randomly analyzed, namely Centaurea cyanus L. (4 samples), Chelidonium majus (1), Cirsium Mill. (1), Echinacea purpurea L. (1), Epilobium L. (2), Equisetum L. (2), Filipendula ulmaria (1), Glechoma hederacea L. (2), Hypericum perforatum (1), Malva L. (1), Marrubium vulgare L. (1), Matricaria chamomilla L. (1), Origanum vulgare (2), Pulmonaria L. (5), Sambucus L. (1), Taraxacum officinale (2), Thymus vulgaris (1), Tilia (6), Urtica dioica L. (1), Valeriana officinalis L. (1), and Veronica L. (9). Ten samples were positive and nine of 235 pesticides were found (Table 3) in the range 0.005–1.103 mg kg−1. In five samples, the detected levels were above the MRLs specified by the European Union (EC Regulation No. 396/2005 with amendments). Recovery factors were used to calculate pesticide concentration only in the case of pesticides indicating recoveries outside the range of 70–120% (within the range of 60–69 and 121–130%). Figure 4 shows chromatograms of matrix-matched standards and real samples containing diazinon 0.005 mg kg−1 and chlorpyrifos 0.04 mg kg−1 detected in Hypericum perforatum.

Chromatograms of a matrix Hypericum perforatum-matched standards of 235 pesticides and b real sample of Hypericum perforatum containing diazinon (0.005 mg kg−1) and chlorpyrifos (0.04 mg kg−1)
Conclusion
A modified QuEChERS method coupled with GC-MS/MS was developed for the simultaneous determination of 235 pesticides in various kinds of herbs: Centaurea cyanus L., Matricaria chamomilla L., and Thymus vulgaris L. This study discusses the impact of certain parameters, such as sample mass (1, 2, 5 g) and type of cleanup sorbent on recoveries and matrix effects, with the aim of choosing suitable methods for multiresidue analyses. Finally, the modified QuEChERS method based on acetonitrile extraction followed by PSA/ENVI-Carb/MgSO4 cleanup was validated in terms of linearity, recovery, precision, LOQ, ME, and U. The application of PSA/ENVI-Carb/MgSO4 did not have a negative influence on recoveries and was efficient in the removal of pigments and other co-extracted compounds. Recoveries of the tested pesticides were between 60 and 128% and RSDs were consistently < 18%. It should be noted that this combination ensured the best matrix effect values. Low ME was observed for the majority of pesticides showing suppression or enhancement (> 20%). The proposed method meets EU criteria and MRLs and is thus useful for routine analyses of pesticide residues in herbs. This method was applied for monitoring of pesticides in 46 samples of herbs.
References
Attallah ER, Barakat DA, Maatook GR, Badawy HA (2012) Validation of a quick and easy (QuEChERS) method for the determination of pesticides residue in dried herbs. J Food Agric Environ 10(1):755–762
Anastassiades M, Lehotay SJ, Stajnbaher D, Schenck FJ (2003a) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86(2):412–431
Anastassiades M, Mastovska K, Lehotay SJ (2003b) Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J Chromatogr A 1015:163–184
CA Codex Alimentarius (2016) Food and Agriculture Organization of the United Nations. https://www.fao.org/fao-who-codexalimentarius/codex-home/en/. Accessed 22 August 2016
Chen L, Song F, Liu Z, Zheng Z, Xing J, Liu S (2012) Multi-residue method for fast determination of pesticide residues in plants used in traditional Chinese medicine by ultra-high-performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr A 1225:132–140
Dai R, Ren X, He X, Huo Y (2011) Convenient analytical method for quantitative determination of 23 pesticide residues in herbs by gas chromatography–mass spectrometry. B Environ Contam Tox 86:559–564
Du B, Li X, Liu H (2007) Determination of organochlorine pesticide residues in herbs by capillary gas chromatography. Life Sci J 4(1):40–42
Du G, Xiao Y, Yang HR, Wang L, Song YL, Wang YT (2012) Rapid determination of pesticide residues in herbs using selective pressurized liquid extraction and fast gas chromatography coupled with mass spectrometry. J Sep Sci 35:1922–1932
EC Regulation No 396/2005 Of the European Parliament and of the Council of 23 February 2005 on maximum residue levels of pesticides in or on food and feed of plant and animal origin and amending Council Directive 91/414/EEC. Off J Eur Union L 70/1, 16.3.2005, with later amendments. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32005R0396&from=en. Accessed 04 May 2017
EFSA European Food Safety Authority (2015) Scientific report of EFSA. The 2013 European Union report on pesticide residues in food. EFSA J 13(3):4038. https://www.efsa.europa.eu/sites/default/files/scientific_output/files/main_documents/4038.pdf. Accessed 22 August 2016
EP 8.0 European Directorate for the Quality of Medicines and HealthCare (2014) European Pharmacopoeia, 8th edition, European Directorate for the Quality of Medicines and HealthCare, Strasbourg, France
Ferrer C, Lozano A, Agüera A, Girón AJ, Fernández-Alba AR (2011) Overcoming matrix effects using the dilution approach in multiresidue methods for fruits and vegetables. J Chromatogr A 1218(42):7634–7639
Ghani SBA (2014) Determination of multiclass pesticides in dry herbs using GC-ECD. J Appl Sci Agr 9(3):955–959
Gondo TT, Obuseng VC, Mmualefe LC, Okatch H (2016) Employing solid phase microextraction as extraction tool for pesticide residues in traditional medicinal plants. J Anal Methods Chem. https://doi.org/10.1155/2016/2890219
Hajou RM, Afifi FU, Battah AH (2004) Comparative determination of multi-pesticide residues in Pimpinella anisum using two different AOAC methods. Food Chem 88:469–478
Hajslova J, Holadova K, Kocourek V, Poustka J, Godula M, Cuhra P, Kempny M (1998) Matrix-induced effects: a critical point in gas chromatographic analysis of pesticide residues. J Chromatogr A 80:283–295
Ho YM, Tsoi YK, Leung KSY (2013) Highly sensitive and selective organophosphate screening in twelve commodities of fruits, vegetables and herbal medicines by dispersive liquid–liquid microextraction. Anal Chim Acta 775:58–66
Hu Y, Wan L, Zhang J, Yang F, Cao J (2012) Rapid determination of pesticide residues in Chinese materia medica using QuEChERS sample preparation followed by gas chromatography–mass spectrometry. Acta Pharm Sin B 2(3):286–293
Hwang BH, Lee MR (2000) Solid-phase microextraction for organochlorine pesticide residues analysis in Chinese herbal formulations. J Chromatogr A 898:245–256
Kaczyński P, Łozowicka B, Jankowska M, Hrynko I (2016) Rapid determination of acid herbicides in soil by liquid chromatography with tandem mass spectrometric detection based on dispersive solid phase extraction. Talanta 152:127–136
Kinsella B, O’Mahony J, Malone E, Moloney M, Cantwell H, Furey A, Danaher M (2009) Current trends in sample preparation for growth promoter and veterinary drug residue analysis. J Chromatogr A 1216:7977–8015
Kwon H, Lehotay SJ, Geis-Asteggiante L (2012) Variability of matrix effects in liquid and gas chromatography–mass spectrometry analysis of pesticide residues after QuEChERS sample preparation of different food crops. J Chromatogr A 1270:235–245
Lehotay SJ, Mastovská K, Lightfield AR (2005a) Use of buffering and other means to improve results of problematic pesticides in a fast and easy method for residue analysis of fruit and vegetables. J AOAC Int 88(2):615–629
Lehotay SJ, Mastovská K, Yun SJ (2005b) Evaluation of two fast and easy methods for pesticide residue analysis in fatty food matrixes. J AOAC Int 88(2):630–638
Lehotay SJ, Son K, Kwon H, Koesukwiwat U, Fu W, Mastovska K et al (2010) Comparison of QuEChERS sample preparation methods for the analysis of pesticide residues in fruits and vegetables. J Chromatogr A 1217(16):2548–2560
Li W, Liu Y, Duan J, Saint CP, Mulcahy D (2015) The role of methanol addition to water samples in reducing analyte adsorption and matrix effects in liquid chromatography-tandem mass spectrometry. J Chromatogr A 1389:76–84
Liu XQ, Li YF, Meng WT, Li DX, Sun H, Tong L, Sun GX (2016) A multi-residue method for simultaneous determination of 74 pesticides in Chinese material medica using modified QuEChERS sample preparation procedure and gas chromatography tandem mass spectrometry. J Chromatogr B 1015–1016:1–12
Lozano A, Rajski Ł, Belmonte-Valles N, Uclés A, Uclés S, Mezcua M, Fernández-Alba AR (2012) Pesticide analysis in teas and chamomile by liquid chromatography and gas chromatography tandem mass spectrometry using a modified QuEChERS method: validation and pilot survey in real samples. J Chromatogr A 1268:109–122
Lozano A, Rajski Ł, Uclés A, Belmonte-Valles N, Mezcua M, Fernández-Alba AR (2014) Evaluation of zirconium dioxide-based sorbents to decrease the matrix effect in avocado and almond multiresidue pesticide analysis followed by gas chromatography tandem mass spectrometry. Talanta 118:68–83
Łozowicka B, Jankowska M, Rutkowska E, Hrynko I, Kaczynski P, Micinski J (2013) The evaluation of a fast and simple pesticide multiresidue method in various herbs by gas chromatography. J Nat Med 68:95–111
Łozowicka B, Ilyasova G, Kaczynski P, Jankowska M, Rutkowska E, Hrynko I, Mojsak P, Szabunko J (2016) Multi-residue methods for the determination of over four hundred pesticides in solid and liquid high sucrose content matrices by tandem mass spectrometry coupled with gas and liquid chromatograph. Talanta 151:51–61
Matyjaszczyk E (2011) Selected aspects of plant protection in Poland, five years on from EU accession. Outlook Agr 40(2):119–123
Nantia EA, Moreno-González D, Manfo FPT, Gámiz-Gracia L, García-Campaña AM (2017) QuEChERS-based method for the determination of carbamate residues in aromatic herbs by UHPLC-MS/MS. Food Chem 216:334–341
Nguyen TD, Lee KJ, Lee MH, Lee GH (2010) A multiresidue method for the determination 234 pesticides in Korean herbs using gas chromatography mass spectrometry. Microchem J 95:43–49
PPDB Pesticide Properties Database (2016) University of Hertfordshire. https://sitem.herts.ac.uk/aeru/ppdb/en/index.htm. Accessed 22 August 2016
Rao MM, KumarMeena A, Galib E (2011) Detection of toxic heavy metals and pesticide residue in herbal plants which are commonly used in the herbal formulations. Environ Monit Assess 181:267–271
Qian G, Rimao H, Feng T, Wu X, Xuede L, Haiqun C, Yanhong S, Jun T (2010) A multiresidue method for 20 pesticides in Radix paeoniae Alba of Chinese herb by gas chromatography with electron-capture detection. B Environ Contam Tox 84:779–783
Sadowska-Rociek A, Surma M, Cieślik E (2013) Application of QuEChERS method for simultaneous determination of pesticide residues and PAHs in fresh herbs. B Environ Contam Tox 90(4):508–513
Sanchez-Brunette C, Albero B, Martin G, Tadeo JL (2005) Determination of pesticide residues by GC–MS using analyte protectants to counteract the matrix effect. Anal Sci 12:1219–1296
SANTE (2015) Document No. SANTE/11945/2015. Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed. https://ec.europa.eu/food/plant/docs/plant_pesticides_mrl_guidelines_wrkdoc_11945_en.pdf. Accessed 22 August 2016
Sarkhail P, Yunesian M, Ahmadkhaniha R, Sarkheil P, Rastkari N (2012) Levels of organophosphorus pesticides in medicinal plants commonly consumed in Iran. Daru 20:9
Słowik-Borowiec M (2015) Validation of a QuEChERS-based gas chromatographic method for multiresidue pesticide analysis in fresh peppermint including studies of matrix effects. Food Anal Method 8(6):1413–1424
Storelli MM (2014) Evaluation of toxic metal (Hg, Cd, Pb), polychlorinated biphenyl (PCBs), and pesticide (DDTs) levels in aromatic herbs collected in selected areas of southern Italy. Environ Sci Pollut R 21(2):946–953
Tong H, Tong Y, Xue J, Liu D, Wu X (2014) Multi-residual pesticide monitoring in commercial Chinese herbal medicines by gas chromatography–triple quadrupole tandem mass spectrometry. Food Anal Method 7(1):135–145
Tuzimski T (2011) Determination of analytes in medical herbs extracts by SPE coupled with two-dimensional planar chromatography in combination with diode array scanning densitometry and HPLC-diode array detector. J Sep Sci 34(1):27–36
Van De Steene JC, Mortier KA, Lambert WE (2006) Tackling matrix effects during development of a liquid chromatographic–electrospray ionisation tandem mass spectrometric analysis of nine basic pharmaceuticals in aqueous environmental samples. J Chromatogr A 1123:71–81
Walorczyk S, Drożdżyński D, Kierzek R (2015) Two-step dispersive-solid phase extraction strategy for pesticide multiresidue analysis in a chlorophyll-containing matrix by gas chromatography-tandem mass spectrometry. J Chromatogr A 1412:22–32
Wan YQ, Mao XJ, Yan AP, Shen MY, Wu YM (2010) Simultaneous determination of organophosphorus pesticides in Chinese herbal medicines by microwave-assisted extraction coupled with dispersive-solid phase extraction and gas chromatography. Biomed Chromatogr 24:961–968
Wang Y, Jin HY, Ma SC, Lu J, Lin RC (2011) Determination of 195 pesticide residues in Chinese herbs by gas chromatography–mass spectrometry using analyte protectants. J Chromatogr A 1218:334–342
Zrostlíková J, Hajšlová J, Godula M, Maštovská K (2001) Performance of programmed temperature vaporizer, pulsed splitless and on-column injection techniques in analysis of pesticide residues in plant matrices. J Chromatogr A 937:73–86
Zhuang WE, Gong ZB (2012) Gel permeation chromatography purification and gas chromatography-mass spectrometry detection of multi-pesticide residues in traditional Chinese medicine. Am J Analyt Chem 3(1):24–32
Funding
This study has been performed from statutory topics (Grant number SBI-05 development and implementation of methods for the determination of pesticide residues in plant material using GC-MS/MS and LC-MS/MS) funded by the Ministry of Science and Higher Education, Poland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ewa Rutkowska declares that she has no conflict of interest. Bożena Łozowicka declares that she has no conflict of interest. Piotr Kaczyński declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rutkowska, E., Łozowicka, B. & Kaczyński, P. Modification of Multiresidue QuEChERS Protocol to Minimize Matrix Effect and Improve Recoveries for Determination of Pesticide Residues in Dried Herbs Followed by GC-MS/MS. Food Anal. Methods 11, 709–724 (2018). https://doi.org/10.1007/s12161-017-1047-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-017-1047-3