
Abstract
In the present study, a simultaneous sample preparation method for the determination of 3-monochloropropane-1,2-diol (3-MCPD) and polycyclic aromatic hydrocarbons (PAHs) in different foodstuffs has been evaluated. Sample preparation procedure involved acetonitrile extraction/partitioning and cleanup by dispersive solid phase extraction with the final detection by gas chromatography coupled with mass spectrometry. The experiment demonstrated that freezing out of the sample extracts prior to dispersive solid phase extraction (d-SPE) cleanup step gave better results in removal of undesirable matrix constituents although with slightly lower recovery values. The addition of higher amount of MgSO4 has contributed to increased values of polycyclic aromatic hydrocarbon recovery, in contrast to 3-MCPD for which the recoveries were insignificantly lower when MgSO4 was added. Finally, a simultaneous chromatographic measurement has revealed that quantification of PAHs was not effective due to signal enhancement resulted from the presence of derivatisation products. However, the presented concept can be streamlined and an interesting analytical approach providing good validation parameter values: recoveries within the range 50–110 % for PAHs and 76–107 % for 3-MCPD, precision not exceeding 16.5 % (PAHs) and 7.7 % (3-MCPD) and quantification limits lower than 0.9 μg kg−1 for PAHs and 9.3 μg kg−1 for 3-MCPD.
Similar content being viewed by others


Introduction
Nowadays, researchers and specialists involved in analysis of food contaminants seek new universal methods of sample preparation that could be used for simultaneous determination of many undesirable components in food samples. These simultaneous analytical procedures shorten time needed to sample preparation and reduce the amount of reagents. Nevertheless, the main challenge is the assurance of reliable analytical results of target contaminant determination considering their different physicochemical properties such as polarity, volatility and solubility in a wide range of solvents. The procedures of simultaneous preparation of food samples and determination of various contaminants therein have been developed initially for halogenated contaminants and polycyclic aromatic hydrocarbons in fish and shrimps (Kalachova et al. 2011), polycyclic aromatic hydrocarbons (PAHs) and pesticides in fresh herbs (Sadowska-Rociek et al. 2013), 3-monochloropropane-1,2-diol (3-MCPD) and acrylamide in different foods (Xu et al. 2013), chloropropanols, acrylamide and ethyl carbamate in fermented products, flavouring and related foods (Mo et al. 2014). However, foregoing studies are related mainly to compounds having similar polarity and lipophilicity. So far, no attempts of simultaneous sample preparation step concerning the compounds with totally different features such as 3-MCPD and PAHs have been undertaken.
As mentioned before, these methods should comply with all the criteria set for the analysis of individual compound but, at the same time, should be fast, simple and safe for operators and the environment, with a small amount of samples and hazardous reagents needed. One of such methods is the Quick, Easy, Cheap, Effective, Rugged and Safe (QuEChERS) approach that has been used extensively with slight modifications of solvents and sorbents for more than 12 years. Besides pesticides, the method is commonly applied for the determination of other organic contaminants such as mycotoxins (Rejczak and Tuzimski 2015), pharmaceuticals (Núñez et al. 2015), polycyclic aromatic hydrocarbons (Rejczak and Tuzimski 2015) and, finally, 3-monochloropropane-1,2-diol (Sadowska-Rociek and Cieślik 2015a) in different food samples.
These two last groups of compounds are often found together in the same food products that have been previously heat-treated in the processes of baking, grilling, smoking or roasting. Hence, the presence of PAHs and 3-MCPD can be observed in bakery products such as sweet and savoury biscuits, grilled or smoked meat, fish and cheese but also in cereal and instant coffee. Because of potential carcinogenic and toxic properties of PAHs (IARC 2014) and 3-MCPD (European Commission 2001), they were considered harmful to human health and therefore maximum levels in specific foods for them were established by European legislation (European Commission 2006, European Commission 2011a). This entails an obligation to monitor their presence in food using appropriate analytical methods.
However, previously mentioned products, especially food of animal origin, contain high content of fat that may influence on an analytical signal but also contaminate the components of gas chromatograph and mass spectrometer. Aside from the use of expensive sorbents such as Z-Sep (Rajski et al. 2013), storing the extract in a freezer (below −20 °C) is a cheap alternative to reduce the amount of fatty matrix interferences. After freezing, an aliquot can be easily taken and then the extract can be quickly filtered (Nácher-Mestre et al. 2014). Nonetheless, the process has to be done quickly after removing the extracts from the freezer to prevent the precipitated matter from re-dissolving. In the original QuEChERS method, designated for pesticide residue determination in food of plant origin (European Committee for Standardization 2008), the freeze out method is described by removal of the precipitate by centrifugation. However, this demands for a centrifuge with a cooling option to maintain low temperatures (Norli et al. 2011). Another disadvantage is that freezing as a cleanup step may deteriorate the content of the analytes in the extract because some of the compounds, especially lipophilic ones such as PAHs, may be removed together with fat. Thus, in case of these compounds, thorough and detailed investigation of freezing usefulness should be carried out. To our best knowledge, freezing out and fat removal have not been applied yet under determination of PAHs and 3-MCPD in food.
In this work, we evaluated the application of QuEChERS method for the simultaneous sample preparation step in the determination of PAHs and 3-MCPD in selected foodstuffs. The experiment involved the assessment of usefulness of freezing out step, the necessity of MgSO4 addition and the possibility of simultaneous gas chromatography coupled with mass spectrometry (GC-MS) analysis of investigated compounds. In the last stage of the study, the developed procedure has been subjected to validation protocol.
Materials and Methods
Chemicals and Reagents
Hexane, chloroform, acetone and acetonitrile, HPLC grade for liquid chromatography LiChrosolv®, were purchased from Merck KGaA, Germany. Magnesium sulphate anhydrous p.a. and sodium chloride p.a. were purchased from Chempur S.A., Poland. Primary secondary amine (PSA), strong anion exchange (SAX), and C18 (octadecyl) SPE Bulk Sorbent were derived from Agilent Technologies, USA. EPA 525 PAH Mix-B (containing acenaphthylene, anthracene, benzo[a]anthracene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, benzo[g,h,i]perylene, chrysene, dibenzo[a,h]anthracene, fluorene, indeno[1,2,3-c,d]pyrene, phenanthrene and pyrene), chrysene-d12 (internal standard), anthracene-d10 (syringe standard), 3-MCPD, 3-monochloropropane-1,2-diol-d5 (3-MCPD-d5; internal standard), 3-monobromochloropropane-1,2-diol (3-MBPD; syringe standard) and phenylboronic acid (PBA) (derivatisation agent) were obtained from Sigma-Aldrich, USA.
Deionised water (18 MΩ) was produced by a Milli-Q system (Millipore, USA). A sodium chloride solution of 200 mg mL−1 (20 %) was prepared in deionised water. Stock (1 mg mL−1), intermediate (100 μg mL−1) and working (2 μg mL−1) standard solutions of each chloropropanol were prepared in 20 % NaCl. PBA solution was prepared by dissolving 5 g PBA in 20 mL mixture of acetone and water (19:1, v/v). Stock (1 mg mL−1), intermediate (100 μg mL−1) and working (1 μg mL−1) PAH standard solutions, chrysene-d12 and anthracene-d10 were prepared in hexane.
Instrumentation
PAH and 3-MCPD analyses were performed using Varian IonTrap 4000 GC/MS (Varian, Inc., USA) with a CP-8410 auto-injector (Bruker, USA) with DB-5MS column (30 m × 0.25 mm × 0.25 μm; Agilent Technologies, USA). The injector temperature was set at 270 °C, and injection volume was 1.0 μL. Each injection was performed in triplicate. The GC oven was operated with the following temperature program: 50 °C (1.0 min), 15 °C min−1, 300 °C (6.0 min) for PAH analysis and 50 °C (1.0 min), 10 °C min−1, 210 °C (1.0 min), 30 °C min−1, 250 °C (5.0 min) for the determination of chloropropanols. The analyses were carried out with a solvent delay of 8.0 min. Helium 5.0 (Linde Gas, Poland) was used as the GC carrier gas at a flow rate of 1.0 mL min−1. The emission current of the ionisation filament was set at 15 μA. The ion trap mass spectrometer was operated in the internal ionisation mode. The trap and the transfer line temperatures were set at 180 and 230 °C for 3-MCPD analysis, 220 and 270 °C for PAH analysis and 200 and 250 °C respectively for simultaneous analysis of PAHs and 3-MCPD. Analyses were conducted in the selected ion monitoring (SIM) mode based on the use of one quantitative ion of PAHs and PBA derivatives of 3-MCPD. Confirmation ions and retention times were also used to ensure identification of the analytes (Table 1). Acquisition and processing data were performed using Varian Start Workstation software and NIST 2.0 library.
MS1 Minishaker (IKA, Germany) and MPW 350 R Centrifuge (MPW Med. Instruments, Poland) were used during sample preparation. AccublockTM (Labnet, USA) with nitrogen 5.0 (Linde Gas, Poland) was accomplished to evaporate solvents and concentrate the extracts.
Sample Preparation
In the experiment, the samples of traditional Polish smoked cheese, smoked ham, smoked fish, sweet and savoury biscuits (crackers), cereal coffee and natural instant coffee delivered from a local market were used for the preparation of blank and spiked samples. Recovery studies involved three samples being spiked at the level of 20 μg kg−1 with the PAH standard solution, chrysene-d12 solution, 3-MCPD solution and 3-MCPD-d5 solution. Blank samples and reagent blanks were prepared similarly to the fortified samples.
In brief, the final extraction process was performed using water and acetonitrile followed by addition of NaCl and MgSO4 (Fig. 1). The slurry obtained was thoroughly shaken by hand and centrifuged. Then, in case of samples with high fat content (fish, cheese, ham, biscuits and crackers), an additional low-temperature clean step was performed before dispersive solid phase extraction (d-SPE). An aliquot of the supernatant was transferred into a glass vial and stored for overnight in a freezer (−18 °C). Thereafter, the extract was immediately filtrated in a freezer by filter paper to remove precipitated co-extractives. Next, the supernatants of coffee extracts or previously obtained filtrates were cleaned up by appropriate sorbents (according to the scheme at Fig. 1) and MgSO4. The tubes were subsequently hand-shaken and centrifuged. Afterwards, the extract was divided into two parts. One part of the extract was evaporated under a stream of N2 to dryness; the residues were dissolved in NaCl solution and derivatised with PBA. The second part of the extract was subjected to the dispersive liquid–liquid microextraction (DLLME) procedure. Finally, the received solutions were analysed by GC-MS.

Schema of the sample preparation procedure
Standards Preparation
3-MCPD Analysis
A series of standard solutions in NaCl solution were prepared by dilution of the standard mixture solution at the following concentrations: 0, 0.5, 2, 5, 10, 20, 40, 70 and 100 ng mL−1. The calibration solutions were treated in a similar way to the samples. The GC-MS chromatogram of standards at the concentration of 40 ng mL−1 in hexane is displayed in Fig. 2a.

GC-MS chromatograms of target compounds at the concentrations of 100 ng mL−1; a mixture of chloropropanols (temperature of ion trap: 180 °C, temperature of transfer line: 230 °C), b mixture of PAHs (temperature of ion trap: 220 °C, temperature of transfer line: 270 °C), c mixture of chloropropanols and PAHs together (temperature of ion trap: 200 °C, temperature of transfer line: 250 °C); (1) 3-monochloropropane-1,2-diol-d5; (2) 3-monochloropropane-1,2-diol; (3) 3-monobromopropane-1,2-diol; (4) acenaphthylene; (5) fluorene; (6) phenanthrene; (7) anthracene-d10; (8) anthracene; (9) pyrene; (10) benzo[a]anthracene; (11) chrysene-d12; (12) chrysene; (13) benzo[b]fluoranthene; (14) benzo[k]fluoranthene; (15) benzo[a]pyrene; (16) indeno[1,2,3-c,d]pyrene; (17) dibenzo[a,h]anthracene; (18) benzo[g,h,i]perylene
PAH Analysis
A series of standard solutions in hexane and in examined matrices were prepared by dilution of the standard mixture solution at the following ranges: 0.1, 1, 5, 10, 40, 70 and 100 ng mL−1. Each standard solution contained 100 μL of the anthracene-d10 solution. GC-MS chromatogram of standard solution at the concentration of 40 ng mL−1 in hexane is presented in Fig. 2b.
Calibration curves for all compounds were constructed by plotting integrated peak areas, divided by the area of syringe standard, against concentrations of compounds. Peak areas have been reduced by the area of the peaks of compounds derived from blank matrix (or hexane).
Results and Discussion
Optimisation of Sample Preparation
The main goal of our research was to evaluate a simultaneous sample preparation step for determination of PAHs and 3-MCPD in the selected foods. The proposed analytical procedure was partially based on our previously developed methods for PAHs and 3-MCPD analyses (Sadowska-Rociek and Cieślik 2015a, b), including the selection of sorbents and volumes of solvents used in DLLME technique, but some other necessary optimisations of our proposed concept were performed.
In the first stage of the experiment, an effectiveness of freezing out on different steps of the sample preparation was tested. The experiment was conducted for the samples with a higher fat content: smoked fish, smoked cheese, smoked ham, crackers and biscuits. As mentioned before, no data concerning effectiveness of freezing out have been available for PAHs and 3-MCPD analyses. Thus, two variants were investigated: (1) freezing the extract sample after the extraction and prior to cleanup using d-SPE, and (2) freezing after d-SPE. The yield of both variants was assessed on the basis of the recovery values, and the presence of interferents in the sample chromatograms was also taken into consideration. The percent recoveries were calculated by subtracting values in a blank sample from values in a spiked sample and in respect to existing limits: 50–120 % for PAHs and 75–110 % for 3-MCPD (European Commission 2011b).
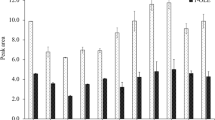
Recoveries of PAHs and 3-MCPD calculated for two tested variants are displayed in Fig. 3. To facilitate the interpretation, the results for PAH markers (European Commission 2011a) were only presented. The recovery values were comparable in both variants, being slightly higher for the samples that had been frozen out after the process of d-SPE, in particular for benzo[b]fluoranthene and 3-MCPD. This would have suggested that this variant was more recommended. However, considering the GC-MS chromatograms (Fig. 4, example of smoked fish) of the samples wherein the freezing was applied in both versions, it has been concluded that freezing out the extract prior to d-SPE had led to removal of more undesired components of the matrix. Therefore, in view of above results, it was decided to apply the process of freezing out the sample extracts before d-SPE cleanup step, although with insignificantly lower recovery values.

Recoveries of compounds obtained in two variants of freezing out; (1) before d-SPE, (2) after d-SPE; B[a]a, benzo[a]anthracene; Chr, chrysene; B[b]f, benzo[b]fluoranthene; B[a]p, benzo[a]pyrene; 3-MCPD, 3-monochloropropane-1,2-diol

GC-MS chromatograms of smoked fish sample being frozen out; a before d-SPE, b after d-SPE
The next step of the method optimisation was to verify whether the addition of MgSO4 is necessary in the sample preparation process. In original QuEChERS method, anhydrous MgSO4 is added twice to remove residual water from acetonitrilic layer through its binding. In almost all studies relating to the usage of QuEChERS method for the determination of contaminants in foods, MgSO4 has been always added. In our recent research, concerning the determination of 3-MCPD in coffee samples, MgSO4 was not used, and the recovery rates of 3-MCPD were still within acceptable limits. Similar studies for PAHs, however, have not been conducted yet. The experiment was planned using the simplest central composite design (CCD) 2k + 1 (Li et al. 2015) where k was equal to 2 because two factors were investigated: (1) amount of MgSO4 utilised in extraction step (0–4 g) and (2) amount of MgSO4 added in d-SPE clean up (0–0.9 g). Hence, five variants (A–E) were tested: four combinations of minimal and maximal values of MgSO4 amount and one point with the mean values of the scopes (Table 2).
Figure 5 depicts the recoveries in tested combinations. Because the results obtained were similar for all food products, the graph presents entirely the average recoveries of all matrices to make the plot more concise. The PAH recoveries within established limits for all compounds were obtained only in the samples where MgSO4 was used both during extraction and cleanup (variant B). In the samples without the addition of MgSO4 (variant D), recovery values were below an acceptable limit (50 %) with the exception of acenapththylene and fluorene. In the variant A, the recoveries were also acceptable but not for all compounds. Variant C resulted in proper recoveries only for light compounds. Finally, for variant E, the recoveries were comparable with the variant B being within established range of 50–120 %, except for indeno[c,d]pyrene and dibenzo[a,h]anthracene. Hence, it has been stated that the addition of MgSO4 under the extraction step affects more the recovery values than the addition in cleanup step. In contrast, the 3-MCPD recoveries were within acceptable limits for all variants being slightly lower in the samples where MgSO4 was applied.

Recoveries of compounds with different amount MgSO4 added to the samples; Acp, acenaphthylene; Flu, fluorene; Phen, phenanthrene; Ant, anthracene; Pyr, pyrene; B[a]a, benzo[a]anthracene; Chr, chrysene; B[b]f, benzo[b]fluoranthene; B[k]f, benzo[k]fluoranthene; B[a]p, benzo[a]pyrene; I[cd]p, indeno[1,2,3-c,d]pyrene; D[ah]a, dibenzo[a,h]anthracene; B[ghi]P, benzo[g,h,i]perylene; 3-MCPD, 3-monochloropropane-1,2-diol
Surprisingly, comparing the appearance of PAH chromatograms, especially these obtained from natural instant coffee (Fig. 6), it has been discovered that the extracts of the samples with the addition of magnesium sulphate (B, C and E) contained more undesirable compounds than the sample extracts where MgSO4 was not used. This phenomenon could have been explained by an insufficient purity of magnesium sulphate, but this hypothesis has not been confirmed in the analyses of reagent samples. Another supposition was that elevated temperature arising from the addition of MgSO4 could have released certain compounds such as caffeine (the biggest peak in Fig. 6) from coffee samples. However, previously performed studies in this area (Forsberg et al. 2011) reported that increased extraction temperatures resulted from the addition of magnesium sulphate can rather disrupt analyte–matrix interactions and therefore improve the extraction yield, which is in opposite with our findings. However, in case of certain matrix components, it seems that the presence of MgSO4 contributes to transfer these substances together with the analytes into acetonitrile extract. Finally, the increased amount of co-extractives in the samples with MgSO4 added could have been an effect of improved performance of the extraction process. Anyhow, to summarise, it was found that due to the appropriate value of PAH recovery, the use of MgSO4 is a crucial factor during simultaneous sample preparation step despite of the presence of some interferents in the extracts. However, the outcomes give rise to further investigation of the use of MgSO4 in QuEChERS method.

GC-MS chromatograms of natural coffee samples with different amount MgSO4 added; a–e tested variants (see Table 2)
Finally, the possibility of simultaneous chromatographic analysis of PAHs and 3-MCPD was investigated. For this purpose, the sample preparation process was modified. Two milliliters of solution after d-SPE was derivatised as in case of 3-MCPD analysis, and the extract was subjected to GC-MS assay with slight changes of mass spectrometer temperatures and initial column temperature program leading to revised retention times for chloropropanols. Theoretically, this extract should contain PBA derivatives of chloropropanols and PAHs that are inert and chemically stable so it should not undergo the derivatisation process.
A series of standard solution of PAHs and chloropropanols in hexane were analysed simultaneously and also in two other analyses, dedicated for PAHs and chloropropanols separately. The differences between the slope coefficients of calibration curves (f factor) are presented in Fig. 7. The coefficients of calibration slope obtained in separate analysis of PAHs and chloropropanols were established as 100 % to facilitate the comparison. Comparable values (up to 10 %) were obtained only for 3-MCPD and for light PAHs (acenapththylene, fluorene, phenantherene), pyrene and benzo[b]fluoranthene. For other PAHs, studied coefficients were significantly higher as the effect of the signal enhancement resulted from the presence of some compounds having identical ions as polycyclic aromatic hydrocarbons. For example, the signal for benzo[a]anthracene, chrysene and chrysene-d12 was increased by the presence of ions in the scopes 226–240 (Fig. 2c). These interfering compounds were formed probably from phenylboronic acid under derivatisation process.

Comparison of slope coefficients (f factor) for separate analyses and simultaneous analysis of 3-MCPD and PAHs; Acp, acenaphthylene; Flu, fluorene; Phen, phenanthrene; Ant, anthracene; Pyr, pyrene; B[a]a, benzo[a]anthracene; Chr, chrysene; B[b]f, benzo[b]fluoranthene; B[k]f, benzo[k]fluoranthene; B[a]p, benzo[a]pyrene; I[cd]p, indeno[1,2,3-c,d]pyrene; D[ah]a, dibenzo[a,h]anthracene; B[ghi]P, benzo[g,h,i]perylene; 3-MCPD, 3-monochloropropane-1,2-diol
Another pitfall of simultaneous GC-MS measurement was the optimisation of ion trap and transfer line temperature. According to the principles of mass spectrometer operating conditions, the temperature of ion trap should be 50–75 °C below the highest temperature for the GC column temperature ramp without compromising chromatographic performance and the transfer line temperature should be no lower than 30 °C below the highest temperature of the GC column oven program in the active method (Varian Inc. 2009). Hence, relevant but different values of temperature were optimised and set for both analyses of chloropropanols and PAHs separately. Because of some differences between these temperatures for chloropropanols and PAHs, for simultaneous analysis, a compromise had to be approved involving the use of average temperature values. However, this compromise and the necessity of temperature column changes at the beginning of the analysis resulted in probably worse chromatographic performance (see Fig. 2b, c).
A simultaneous GC-MS measurement was also performed for the spiked samples of investigated matrices. These results were compared with the results obtained in separate analyses of PAHs and 3-MCPD (Table 3). The recovery values calculated for analyses conducted separately for chloropropanols and PAHs ranged from 76 to 107 % and from 50 to 110.3 %, respectively fulfilling the requirements described in European Commission 2011b. In case of chromatographic analyses performed simultaneously for chloropropanols and PAHs, comparable values of recovery in all investigated matrices were observed only for 3-MCPD. For polycyclic aromatic hydrocarbons, the results were lower and did not reach the limits 50–120 %. Among PAHs, acenapththylene was the only compound with similar recoveries received in simultaneous and separate analyses. However, this remark has not been reported for this cereal coffee and smoked cheese. For other compounds, the recovery rates achieved in simultaneous GC-MS analysis were very low and some compounds were not detected at all (dibenzo[a,h]antracene and benzo[g,h,i]perylene). The acceptable recovery values for most compounds were noticed merely for sweet biscuits, while for cereal coffee, only the recovery of indene[c,d]pyrene was reported within established limits. These findings led to the conclusion that although a simultaneous sample preparation step for the determination of 3-MCPD and PAHs was possible, the chromatographic assay did not give appropriate results and it was necessary to perform two separate GC-MS analyses.
Validation Protocol
The developed analytical procedure was subjected to validation process that involved method specificity (matrix effects), linearity, limit of detection, limit of quantification, precision, accuracy and uncertainty according to the criteria established by the European Commission (2011b) and also interpreting the general requirements for the competence of testing and calibration laboratories laid down in ISO standard 17025 (ISO/IEC 2005).
The matrix effects were estimated on the percentage (%ME) of the difference between the slope values of the matrix match calibration curve and the solvent one (Table 4). %ME was in the range 93–107 % for PAHs and 87–103 % for 3-MCPD (data not shown). Assuming that matrix effects occur when the %ME is higher than 20 % (Farajzadeh et al. 2014), the matrix effects were not observed.
Linearity of the method (the correlation coefficient of calibration curve) was calculated for solvent and matrix-matched calibration. The obtained values were higher than 0.99 for all compounds and matrices (Table 4). Limit of detection (LOD) and limit of quantification (LOQ) were estimated on the basis of the signal of the background noise measured from the standard chromatograms at the lowest calibration level. The limit of detection was calculated as three times higher than the level of noise, and the limit of quantification was equal to three times of the detection limit. LOQs were lower than 0.9 μg kg−1 for PAHs and 9.3 μg kg−1 for 3-MCPD.
The repeatability expressed as a relative standard deviation (RSDr) was calculated from six spiking samples analysed on the same day with the same instrument and the same operator while reproducibility (RSDR) included preparation and analysis from three different days of six spiking samples, by different laboratory technicians, using different series of reagents and calibration curves. The values of HORRATr and HORRATR were calculated based on RSDr and RSDR, according to Horwitz equation (European Commission 2011b). In case of PAHs, the achieved results of repeatability and reproducibility for all matrices were below 12.4 and 16.5 %, respectively, with HORRAT values lower than 2 for each of the compounds, which was in accordance with EU criteria. For 3-MCPD, RSDr and RSDR were lower than 5.6 and 7.7 %, respectively in all examined products not exceeding thereby the values established by European Commission (2011b).
The method accuracy was determined with the recovery using spiked samples, because no certified PAH and 3-MCPD reference materials of all matrices were available. All results were found within acceptable limits and ranged from 50 to 110 % for PAHs and 76 to 107 % for 3-MCPD.
Estimation of measurement uncertainty was accomplished according to the EURACHEM/CITAC guideline (EURACHEM/CITAC 2012) by identifying and quantifying the uncertainty components of whole analytical process. The relative expanded uncertainties (rU c) expressed as a percent value (p = 0.05; k = 2) for all PAHs were in the range 9.5–17.8 % and for 3-MCPD in the range 11.8–14.9 (Table 4) which are below the limits established by EU.
Conclusions
The outcomes resulted from the experiment reported in this study indicate that the simultaneous sample preparation for the determination of PAHs and 3-MCPD combined with freezing out of the fat sample extracts can be an effective and simple analytical approach providing appropriate validation parameter values. However, the results showed that separate chromatographic measurement and use of MgSO4 during extraction and cleanup were imperative to ensure appropriate levels of PAH recoveries.
The simultaneous sample preparation in the determination of PAHs and 3-MCPD is evidently beneficial from an economical point of view. First of all, it reduces the cost of reagents and other materials, mainly sorbents, which are the most expensive. Simultaneous preparation of samples generates also less waste and shortens the time required, which is especially important in case of a larger batch of samples, for example in routine surveys of food within the monitoring studies.
It should be emphasised as well that simultaneous sample preparation procedures are especially relevant when it comes to a very small amount of sample that might not be enough for a number of different assays or dividing the sample into smaller ones is for certain reasons difficult or impossible.
The study also demonstrated the flexibility of QuEChERS method as a technique for sample preparation in the analysis of various organic contaminants in food. Besides individual analyses such as pesticide residue analysis, PAHs, and mycotoxins, this sample treatment can be successfully applied for the simultaneous extraction of analytes with different physicochemical properties (polarity, water solubility, etc.), and cleanup of the undesirable matrix components. However, some other factors (e.g. chromatographic conditions during GC-MS analysis) cause the necessity to use separate routes to be followed during the analysis. Thus, our findings may be some preliminary foundation in this area of interest and encourage other researchers to develop simultaneous sample preparation methods with overcoming the difficulties mentioned above.
References
EURACHEM/CITAC Guide CG 4 (2012) Quantifying Uncertainty in Analytical Measurement, 3rd edn
European Commission (2001) Opinion of the Scientific Committee on Food on 3-monochloro-propane-1,2-diol (3-MCPD) updating the SCF opinion of 1994. European Commission, Brussels. ec.europa.eu/food/fs/sc/scf/out91_en.pdf Accessed 28 December 2015
European Commission (2006) Commission Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs
European Commission (2011a) Commission Regulation (EU) No 835/2011 of 19 August 2011 amending Regulation (EC) No 1881/2006 as regards maximum levels for polycyclic aromatic hydrocarbons in foodstuffs
European Commission (2011b) Commission Regulation (EU) No 836/2011 of 19 August 2011 amending Regulation (EC) No 333/2007 laying down the methods of sampling and analysis for the official control of the levels of lead, cadmium, mercury, inorganic tin, 3-MCPD and benzo(a)pyrene in foodstuffs
European Committee for Standardization (2008) European Standard EN 15662. Foods of Plant Origin – Determination of Pesticide Residues Using GC-MS and/or LC-MS/MS Following Acetonitrile Extraction/Partitioning and Clean-up by Dispersive SPE (QuEChERS method)
Farajzadeh MA, Khoshmaram L, Nabil AAA (2014) Determination of pyrethroid pesticides residues in vegetable oils using liquid–liquid extraction and dispersive liquid–liquid microextraction followed by gas chromatography–flame ionization detection. J Food Comp Anal 34:128–135
Forsberg ND, Wilson GR, Anderson KA (2011) Determination of parent and substituted polycyclic aromatic hydrocarbons in high-fat salmon using a modified QuEChERS extraction, dispersive SPE and GC-MS. J Agric Food Chem 59:8108–8116
IARC, International Agency for Research on Cancer (2014) IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. VOLUME 92 Some Non-heterocyclic Polycyclic Aromatic Hydrocarbons and Some Related Exposures. IARC, Lyon. http://www.iarc.fr Accessed 28 December 2015
International Organization for Standardization (2005) ISO/IEC 17025:2005 General requirements for the competence of testing and calibration laboratories
Kalachova K, Pulkrabova J, Drabova L, Cajka T, Kocourek V, Hajslova J (2011) Simplified and rapid determination of polychlorinated biphenyls, polybrominated diphenyl ethers, and polycyclic aromatic hydrocarbons in fish and shrimps integrated into a single method. Anal Chim Acta 707:84–91
Li M, Liu Y, Fan B, Lu J, He Y, Kong Z, Zhu Y, Jian Q, Wang F (2015) A chemometric processing-factor-based approach to the determination of the fates of five pesticides during apple processing. LWT- Food Sci Technol 63:1102–1109
Mo W, He H, Xu X, Huang B, Ren Y (2014) Simultaneous determination of ethyl carbamate, chloropropanols and acrylamide in fermented products, flavoring and related foods by gas chromatography triple quadrupole mass spectrometry. Food Control 43:251–257
Nácher-Mestre J, Serrano R, Portolés T, Berntssen MHG, Pérez-Sánchez J, Hernández F (2014) Screening of pesticides and polycyclic aromatic hydrocarbons in feeds and fish tissues by gas chromatography coupled to high-resolution mass spectrometry using atmospheric pressure chemical ionization. J Agric Food Chem 62:2165–2174
Norli HR, Christiansen A, Deribe E (2011) Application of QuEChERS method for extraction of selected persistent organic pollutants in fish tissue and analysis by gas chromatography mass spectrometry. J Chromatogr A 1218:7234–7241
Núñez M, Borrull F, Fontanals N, Pocurull E (2015) Determination of pharmaceuticals in bivalves using QuEChERS extraction and liquid chromatography-tandem mass spectrometry. Anal Bioanal Chem 407:3841–3849
Rajski T, Lozano A, Uclés A, Ferrer C, Fernández-Alba AR (2013) Determination of pesticide residues in high oil vegetal commodities by using various multi-residue methods and clean-ups followed by liquid chromatography tandem mass spectrometry. J Chromatogr A 1304:109–120
Rejczak T, Tuzimski T (2015) A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem 13:980–1010
Sadowska-Rociek A, Cieślik E (2015a) Assessment of 3-MCPD levels in coffee and coffee substitutes by simplified QuEChERS method. J Verbrauch Lebensm 10:117-122
Sadowska-Rociek A, Surma M, Cieślik E (2013) Application of QuEChERS method for simultaneous determination of pesticide residues and PAHs in fresh herbs. Bull Environ Contam Toxicol 4:508–513
Sadowska-Rociek A, Cieślik E, Sieja K (2015b) Determination of polycyclic aromatic hydrocarbons in chocolate using the combination of Quick Easy Cheap Effective Rugged Safe method and dispersive liquid-liquid microextraction. J Food Nutr Res 54:289-297
Varian Inc (2009) Software Operation Manual 4000 GC/MS Version 6. Varian, Inc, USA
Xu X, He H, Zhu Y, Feng L, Ying Y, Huang B, Shen H, Han J, Ren Y (2013) Simultaneous determination of 3-monochloropropane-1,2-diol and acrylamide in food by gas chromatography-triple quadrupole mass spectrometry with coupled column separation. Anal Chim Acta 760:93–99
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of Interest
Anna Sadowska-Rociek, Ewa Cieślik and Krzysztof Sieja declare that they have no conflict of interest.
Funding
This study was funded by Ministry of Science and Higher Education of Republic of Poland within the statutory R & D activities (DS-3707/15/KTGiK)
Informed Consent
Not applicable.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sadowska-Rociek, A., Cieślik, E. & Sieja, K. Simultaneous Sample Preparation Method for Determination of 3-Monochloropropane-1,2-Diol and Polycyclic Aromatic Hydrocarbons in Different Foodstuffs. Food Anal. Methods 9, 2906–2916 (2016). https://doi.org/10.1007/s12161-016-0486-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-016-0486-6