
Abstract
Hydroxycinnamate coenzyme A (CoA) thioesters are substrates for biosynthesis of lignin and hydroxycinnamate esters of polysaccharides and other polymers. Hence, a supply of these substrates is essential for investigation of cell wall biosynthesis. In this study, three recombinant enzymes, caffeic acid 3-O-methyltransferase, 4-coumarate-CoA ligase 1, and 4-coumarate-CoA ligase 5, were cloned from wheat, tobacco, and Arabidopsis, respectively, and were used to synthesize 14C-feruloyl-CoA, caffeoyl-CoA, p-coumaroyl-CoA, feruloyl-CoA, and sinapoyl-CoA. The corresponding hydroxycinnamoyl-CoA thioesters were high-performance liquid chromatography purified, the only extraction/purification step necessary, with total yields between 88–95%. Radiolabeled 14C-feruloyl-CoA was generated from caffeic acid and S-adenosyl-14C-methionine under the combined action of caffeic acid 3-O-methyltransferase and 4-coumarate-CoA ligase 1. About 70% of 14C-methyl groups from S-adenosyl methionine were incorporated into the final product. The methods presented are simple, fast, and efficient for the preparation of the hydroxycinnamate thioesters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant cell walls contain large amounts of phenolic compounds, which are key factors in biomass recalcitrance. Lignin, which is found in all vascular plants, is a polymer of monolignols derived from hydroxycinnamic acids (p-coumaric, ferulic, and sinapic acid) and their coenzyme A (CoA) thioesters. Grasses and other commelinid monocots are unique in having ferulate esters covalently bound to xylans. These ferulate esters are bound to O5 of arabinofuranose residues attached to the backbone of arabinoxylanan and glucuronoarabinoxylan. Esters of p-coumaric acid are also found in grass cell walls, but it is not clear if they are directly attached to polysaccharides in the same way. Ferulate esters have not been reported on hemicelluloses from other plant groups. However, ferulic acid esters are known to be present in pectin of plants belonging to Chenopodiaceae, e.g., spinach and sugar beet, and there is some evidence to suggest that ferulate esters of pectin are present in stomata in a wide range of species [6]. The ferulate esters in grasses are important for recalcitrance as demonstrated by experiments where the ferulic acid ester content has been lowered in transgenic plants.
The feruloylation reactions are not understood and have been difficult to study. Incorporation of ferulate esters onto xylan takes place in the Golgi vesicles [4, 14], but an active xylan feruloyl transferase activity has not been detected. One report has shown transfer of ferulic acid from feruloyl-CoA to a trisaccharide acceptor by a rice enzyme preparation [17], but a similar activity has not been reported from other studies, and the feruloyl transferase has not been identified. The combined evidence indicates that the feruloylation reaction in vivo is rather complex. Wheat Golgi vesicles incorporate significant amounts of ferulic acid into xylan in vivo, but incubation of isolated wheat Golgi with feruloyl-CoA led only to feruloyl transfer onto protein, but not to any detectable feruloylated polysaccharide [14]. Incorporation of ferulic acid into protein has also been observed in parsley membranes [7]. Furthermore, bioinformatics has been used to identify several feruloyl transferase candidate genes, which are co-expressed with xylan biosynthetic genes in rice and maize [12]. However, these proteins are all predicted to be cytoplasmic. We hypothesize that the feruloylated protein observed in the wheat Golgi vesicles is an intermediate in the biosynthesis of arabinoxylan. Interestingly, a similar apparent intermediate was found in wheat Golgi when incubated with UDP-arabinose in the absence of UDP-xylose. Arabinose was transferred onto arabinoxylan only when both nucleotide sugars were provided [15].
One of the reasons for the slow progress in the study of feruloylation is that it is difficult to obtain substrates. Naturally occurring hydroxycinnamates are commercially available, but the thioesters are not. They can be synthesized chemically using published methods [1, 16], but for most biochemistry laboratories, an enzymatic synthesis is more convenient. In addition, since the yields of enzymatic synthesis are higher than for chemical synthesis, the enzymatic method is more cost effective [1]. Most importantly, for biosynthetic studies, it is highly advantageous to have the radiolabeled substrates available, and chemical synthesis of the radiolabeled substrates is not realistic. We have previously developed a method to prepare radiolabeled ferulic acid and feruloyl-CoA, but the method was very cumbersome and resulted in poor yield [13]. In this paper, we present methods to prepare hydroxycinnamate-CoA thioesters, 14C-ferulic acid, and 14C-feruloyl-CoA in high yield. The methods do not require any specialized equipment or expertise except for standard reverse-phase high-performance liquid chromatography (HPLC).
Methods
Cloning, Expression, and Purification of Caffeic Acid O-Methyltransferase and 4-Coumarate-CoA Ligase Enzymes
TaCOMT sequence (Genbank: EF413031) without native stop codon was amplified from complementary deoxyribonucleic acid (cDNA) derived from wheat seedlings with TaCOMT-fwd (5′-CACCATGGGGTCGATCGCCGCCGGC-3′) and TaCOMT-rev (5′-CTTAGTGAACTCGATGGCCCATGCGT-3′). Nb4CL1 sequence (Genbank: U50845) was obtained from cDNA derived from Nicotiana benthamiana stem tissue using Nb4CL-fwd (5′-CACCATGCCAATGGAGACTACTACAGAAACA-3′) and Nb4CL-rev (5′-ATTTGGAACACCAGCAGCCAGTC-3′). At4CL5 (Genbank: NM_113018) was amplified from cDNA derived from Arabidopsis thaliana stem tissue, second internode, using At4CL5-fwd (5′-CACCATGGTGCTCCAACAACAAACG-3′) and At4CL5-rev (5′-TTTAGAGCACATGGTTTCCAATTTAGC-3′). The resulting PCR products were introduced into pENTR SD/D TOPO cloning vector (Invitrogen, Carlsbad, CA), according to the manufacturer's protocol and verified by sequencing. After recombination into pET-DEST42 expression vector (Invitrogen), which introduces C-terminal V5 epitope and 6× His tags, the constructs were introduced into BL-21 Star™ (DE3) chemically competent Escherichia coli (Invitrogen), according to the manufacturer's instructions. Single bacterial colonies grown on LB agar containing 100 μg ml−1 carbenicilline were isolated to inoculate 5 ml liquid cultures supplemented with 100 μg ml−1 carbenicilline and grown overnight at 37°C. The overnight cultures were used to inoculate 0.5–l LB cultures containing 100 μg ml−1 carbenicilline, which were grown at 22°C until OD600 reached ∼0.3. Expression was induced by addition of 1 mM isopropyl-β-d-thiogalactopyranoside, and the cultures were further grown at 22°C overnight. Recombinant proteins were affinity-purified twice using Ni-NTA agarose (Quiagen, Hilden, Germany) according to the manufacturer's instructions, desalted with Zeba Spin desalting columns (Thermo Scientific, Rockford, IL), and concentrated using Vivaspin sample concentrators (GE Healthcare, Piscataway, NJ). Recombinant proteins were stored at −20°C in 10 mM phosphate buffer pH 7.2, containing 20% glycerol. Protein concentrations were determined according to Bradford [2]. Typical yields were 0.8 mg recombinant protein for Nb4CL-1, 0.3 mg for At4CL-5, and 0.6 mg for TaCOMT, respectively. The purified proteins (1 μg) were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 7–15% gradient polyacrylamide gels stained with SyproRuby (Invitrogen) or blotted onto nitrocellulose membranes (GE Healthcare). Blots were probed with a 1:8,000 dilution of anti-polyhistidine antibody (Sigma-Aldrich, St. Louis, MO), followed by a 1:20,000 dilution of goat anti-mouse IgG conjugated to HRP (Sigma-Aldrich), before applying ECL Plus detection reagent (GE Healthcare). Blots were imaged using ChemiDoc-It 600 Imaging System (UVP, Upland, CA).
Caffeic acid O-methyltransferase Activity Assays
Caffeic acid O-methyltransferase (COMT) reaction was carried out in the dark in 1 ml 50 mM sodium phosphate buffer (pH 7.1) containing approximately 200 μM caffeic acid (the exact amount were determined by HPLC and calculated according to a standard curve), 500 μM S-adenosyl-l-methionine (SAM), and 10 μg purified TaCOMT. After incubation for 5 h at 25°C, the reaction was terminated by boiling at 95°C for 10 min. After centrifugation, aliquots of the supernatant were subjected to HPLC analysis. No differences, which may result from possible decomposition upon heat treatment, were observed compared to non-heat-inactivated reactions. However, heat-inactivation and centrifugation prior to HPLC helps to prolong the life of the HPLC column.
HPLC
HPLC analysis was carried out using a Dionex Ultimate 3000 and UV detection (Dionex, Sunnyvale, CA). For analytical purposes, samples were separated on a reverse-phase C18 column (Synergy 4u Fusion-RP 80A 250 × 2.0 mm, Phenomenex, Torrance, CA). Prior to injection (10 μl), the samples were spin filtered (0.45 μm). A flow of 0.3 ml min−1 and a gradient of solvent A (0.5% TFA) and solvent B (acetonitrile) was applied (0–5 min, 10% B isocratic; 5–25 min, 10–30% B linear; 25–40 min, 30% B isocratic; 40–45 min, 30–35% B linear; 45–46 min, 35–100% B linear; 46–51 min, 100% B isocratic; 51–53 min 100–10% B linear; 53–60 min 10% B isocratic). Purification of synthesized CoA thioesters was carried out on a semi-preparative C18 column (Synergy 4u Fusion-RP 80A 250 × 10.0 mm, Phenomenex) running the same sequence but with a flow of 5 ml min−1 and using a manual injection valve. Corresponding fractions were collected using a Foxy® Jr. fraction collector.
4-Coumarate-CoA Ligase Reactions
4-Coumarate-CoA ligase (4CL) reactions were carried out in the dark in 1 ml 50 mM sodium phosphate buffer (pH 7.1) containing 400 μM caffeic, trans-p-coumaric, trans-ferulic, or sinapic acid, respectively, 5 mM MgCl2, 2.5 mM ATP, 800 μM CoA, and 10 μg purified Nb4CL1 or At4CL5. The reactions were incubated at 25°C and the change in absorbance was continuously monitored according to the reported absorption maxima of the products (346 nm for feruloyl-CoA and caffeoyl-CoA, 333 nm for p-coumaroyl-CoA and 352 nm for sinapoyl-CoA [9, 16]). After termination by boiling at 95°C for 10 min, samples were spin filtered and subjected to HPLC purification as described above. Fractions were lyophilized and dissolved in an appropriate volume of 50 mM sodium phosphate buffer (pH 7.1), to give a final concentration of 2 mM (about 200 μl). The extinction coefficients used for quantification were ε 346 = 18 mM−1 cm−1 for caffeoyl-CoA, ε 333 = 21 mM−1 cm−1 for p-coumaroyl-CoA, ε 346 = 19 mM−1 cm−1 for feruloyl-CoA, and ε 352 = 20 mM−1 cm−1 for sinapoyl-CoA [9, 16].
Characterization of Thioesters
The collected thioesters were analyzed by HPLC at different wavelengths (260, 320, 333, 346, and 352 nm), and the characteristic spectra of cinnamoyl thioesters were monitored. Products were hydrolyzed in 1 N KOH at room temperature for 24 h and analyzed by HPLC as described above. Electrospray ionization-mass spectrometer (ESI-MS) analyses were performed as follows: Purified feruloyl-CoA, caffeoyl-CoA, p-coumaroyl-CoA, and sinapoyl-CoA were made up to concentrations of 50, 67, 67, and 65 μM, respectively, in 50% methanol. The hydroxycinnamoyl-CoAs were separated on a ZIC®-HILIC (hydrophilic interaction liquid chromatography) column (150 × 2.1 mm, 3.5 μm particle size, Merck SeQuant) using an Agilent Technologies 1200 Series HPLC system (Agilent Technologies, CA, USA). A sample injection volume of 2 μL was used throughout. The column compartment was set to 40°C. The column was eluted isocratically with a mobile phase composition of 35% 50 mM ammonium acetate in water and 65% acetonitrile. The flow rate was 0.15 ml min−1 for the separation of caffeoyl-CoA, coumaroyl-CoA, and sinapoyl-CoA, and 0.1 ml min−1 for the separation of feruloyl-CoA. The HPLC system was coupled to an Agilent Technologies 6210 time-of-flight mass spectrometer (LC-TOF MS), by a one sixth post-column split. Compound identification was based on accurate mass measurements and empirical formula generation. ESI was conducted in the negative ion mode, and a capillary voltage of −3,500 V was utilized. MS experiments were carried out in full scan mode at 0.85 spectra per second and a cycle time of 1.176 s, for the detection of [M–H]− ions. The instrument was tuned for a range of 50–1,700 m/z. Prior to LC-TOF MS analysis, the TOF MS was calibrated via an ESI-L-low concentration tuning mix (Agilent Technologies, CA, USA). Internal reference mass calibration was utilized throughout the chromatographic run via an API TOF reference mass solution kit (Agilent Technologies, CA, USA). Data acquisition and analysis were performed by the MassHunter software package (Agilent Technologies, CA, USA).
Synthesis of 14C-Labeled Feruloyl-CoA
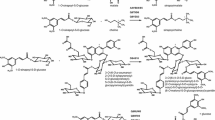
In order to prepare 14C-labeled feruloyl-CoA, TaCOMT and Nb4CL were used sequentially (Fig. 1). Purified TaCOMT (10 μg) was incubated in 1 ml containing 50 mM sodium phosphate buffer (pH 7.1), 100 μM caffeic acid, and 2.5 μCi [methyl-14C]-S-adenosyl-L-methionine (50 mCi/mmol, MP Biomedicals, Solon, OH, USA). After incubation for 20 h at 25°C in the dark, the reaction was terminated by boiling at 95°C for 10 min. After allowing the sample to cool down, 5 mM MgCl2, 2.5 mM ATP, 200 μM CoA, as well as 10 μg purified Nb4CL1, were added, and the reaction was further incubated for ≥12 h at 25°C. The sample was spin filtered and the radiolabeled feruloyl-CoA was HPLC purified as described above. Fractions were collected and aliquots were analyzed using a scintillation counter. Fractions containing feruloyl-CoA were lyophilized and resuspended in an appropriate volume of 50 mM sodium phosphate buffer (pH 7.1). Purified 14C-feruloyl-CoA was aliquoted and stored at −80°C.

Reaction scheme for the enzymatic synthesis of 14C-feruloyl-CoA
Results and Discussion
Synthesis and Purification of Hydroxycinnamoyl Thioesters
Heterologous Expression and Enzymatic Properties of Nb4CL1 and At4CL5
Tobacco 4-coumarate-CoA ligase 1 (4CL1) has been shown to efficiently convert caffeic acid, p-coumaric acid, and ferulic acid into their corresponding thioesters [8]. In contrast to 4CL1, Arabidopsis 4-coumarate-CoA ligase 5 (4CL5) has been reported to be able to utilize sinapic acid as substrate [3, 5]. In order to synthesize caffeoyl-CoA, p-coumaroyl-CoA, feruloyl-CoA, and sinapoyl-CoA, we heterologously expressed 4CL1 and 4CL5 in E. coli. SDS-PAGE analysis and SyproRuby stain as well as Western blotting of affinity-purified recombinant Nb4CL1 protein revealed a single band of about 65 kDa, which is in agreement with the predicted mass of 63.8 kDa (59.8 kDa plus C-terminal peptide containing the V5 epitope and the polyhistidine tag of approximately 4 kDa; Fig. 2a). SDS-PAGE analysis of affinity-purified recombinant At4CL5 protein revealed multiple bands in the range between about 68 and 80 kDa, the lowest one corresponding to its expected molecular mass of about 67 kDa (62.6 kDa plus C-terminal peptide containing the V5 epitope and the polyhistidine tag of approximately 4 kDa; Fig. 2a). In contrast to 4CL1, only low amounts of 4CL5 were produced in E. coli, and we were not able to improve the yield by modification of expression or purification conditions. In all samples, a non-specific 75-kDa protein was observed, which was not detected by Western blot analyses. Recombinant Nb4CL1 and At4CL5 were tested for enzymatic activity with caffeic, p-coumaric, ferulic, and sinapic acid as substrates. The biochemical function as 4-coumarate-CoA ligase could be verified for both proteins (Fig. 2b and c). The preferred substrate for Nb4CL1 was ferulic acid > p-coumaric acid > caffeic acid, and there was no detectable activity with sinapic acid (Fig. 2b), which is consistent with previous results [1, 8]. In contrast, At4CL5 showed slightly different relative activities as previously reported [3, 5]. Ferulic and caffeic acid were preferred, followed by p-coumaric acid and sinapic acid (Fig. 2c). However, in practice, these differences could be ignored, since all four naturally occurring 4CL substrates were completely converted to their corresponding CoA ester when incubated overnight (≥12 h).

4-Coumarate-CoA ligase reaction and purification of hydroxycinnamoyl-CoA thioesters. SDS-PAGE analysis of recombinant Nb4CL1 and At4CL5. Purified protein (1 μg) was resolved by 7–15% PAGE and stained with SyproRuby (left) or analyzed by Western blotting (right) (a). Time course of the formation of hydroxycinnamoyl-CoA thioesters using 10 μg Nb4CL1 (b) or At4CL5 (c), and 400 μM of the appropriate hydroxycinnamic acid. Concentrations of hydroxycinnamoyl-CoA thioesters were determined spectrophotometrically. HPLC analyses and ESI-MS spectra of At4CL5 reactions with caffeic acid (d–f), p-coumaric acid (g–i), ferulic acid (j–l), and sinapic acid (m–o) as substrate. The shown ESI-MS spectra are for the minor B peaks in the chromatograms. The spectra for the major A peaks were essentially identical but with even less noise (see also Table 2)
Synthesis and Purification of CoA Thioesters
For preparative scale synthesis of CoA thioesters, Nb4CL1 or At4CL5 was incubated with 400 nmol of the appropriate hydroxycinnamic acid as substrate and 800 nmol CoA in a total volume of 1 ml. After termination of the reaction, the thioesters caffeoyl-CoA, p-coumaroyl-CoA, feruloyl-CoA, and sinapoyl-CoA were separated by reverse-phase HPLC. They had the retention times of 15.3, 18.3, 19.0, and 20.2 min, respectively. The corresponding peaks were collected and dried under reduced pressure. As shown in Table 1, the final amount of CoA thioesters was 0.3–0.4 mg, which corresponds to 88–95% yield, calculated based on the total amount of hydroxycinnamic acid used as substrate (Table 1). The reaction could be scaled up to 1 μmol with similar yields (data not shown). However, if large amounts of CoA thioesters are desired (multi-milligram scale), HPLC purification would become the limiting step and therefore the purification method developed by Beuerle and Pichersky [1] may be preferred.
Characterization of Thioesters
Aliquots of the purified thioesters were rechromatographed using the analytical system and monitored at different wavelengths. In each sample, two separated peaks, both with the characteristic spectrum of caffeoyl-CoA p-coumaroyl-CoA, feruloyl-CoA, and sinapoyl-CoA, respectively, were detected. LC-TOF MS analysis of both peaks showed that in all cases, the compounds exhibited exact mass measurements with high mass accuracies, and as a result, each CoA-thioester's identity was confirmed with a high degree of confidence (Fig. 2, Table 2). It was therefore concluded that the two peaks correspond to the cis and trans isomers of the thioesters. The substrates were predominantly the trans isomer (Fig. 2e, h, k, n) but nevertheless, some cis–trans isomerization could not be avoided even though samples and reactions were always shielded from light. Neither HPLC nor LC-TOF MS analysis showed detectable levels of any products besides the expected hydroxycinnamoyl-CoAs.
Except for caffeoyl-CoA, all thioesters could be hydrolyzed into their corresponding hydroxycinnamic acids. Two peaks were detected in case of p-coumaroyl-CoA and feruloyl-CoA, both with the characteristic spectrum of the corresponding hydroxycinnamic acid most likely corresponding to their cis/trans isomers (data not shown).
Heterologous Expression and Enzymatic Properties of TaCOMT
Radiolabeled ferulic acid is not commercially available. Therefore, in order to produce 14C-feruloyl-CoA, two enzymatic steps were employed, with caffeic acid utilized as the initial substrate. In a previous attempt to make 14C-feruloyl-CoA an enzyme preparation from wheat seedling, protein extracts containing 4CL as well as COMT activities was used [13]. Due to high thioesterase activity present in the enzyme preparation, the final yield did not exceed 10% (based on the amount of 14C-methyl from 14C-SAM incorporated into the corresponding thioester) [13]. Furthermore, it was necessary to carry out the enzymatic reactions separately and purify the 14C-ferulic acid by HPLC prior to the 4CL reaction. Caffeic acid 3-O-methyltransferase (TaCOMT) has been cloned previously from wheat [10] and was shown to exhibit strong activity towards a variety of substrates including caffeic acid and caffeoyl-CoA while utilizing SAM as a methyl donor [11]. To overcome the problems associated with using enzymes extracted from plants, TaCOMT was cloned from wheat seedlings and was heterologously expressed in E. coli. SDS-PAGE and SyproRuby stain as well as Western blotting confirmed the presence of the recombinant protein with a molecular mass of approximately 40 kDa, which is consistent with its predicted mass of 42.5 kDa (including C-terminal peptide containing the V5 epitope and the polyhistidine tag of approximately 4 kDa). In addition, C-terminal fragments between 25 and 28 kDa, which might represent side products due to internal initiation of translation or possible degradation during protein expression or purification, were detected in stained gels as well as by Western blotting (Fig. 3a). COMT was tested for enzymatic activity using caffeic acid as substrate and SAM as methyl donor. Subsequently, an almost complete conversion of caffeic acid into ferulic acid was observed in reactions starting with approximately 200 μM caffeic acid and saturating concentrations of SAM (1 mM) after 5 h incubation (Fig. 3b).

Synthesis and purification of 14C-labeled feruloyl-CoA. SDS-PAGE analysis of recombinant TaCOMT. Purified protein (1 μg) was resolved by 7–15% PAGE and stained with SyproRuby (left) or analyzed by Western blotting (right) (a). Time course of the formation of ferulic acid from approximately 200 nmol caffeic acid and 1.0 μmol SAM. The exact concentrations were determined by HPLC (b). Ferulic acid formation from approximately 200 nmol caffeic acid and different concentration of SAM after 5 h incubation. The exact concentrations were determined by HPLC (c). Preparation of 14C-labeled feruloyl-CoA from 100 nmol caffeic acid and 50 nmol 14C-SAM. HPLC analysis of COMT reaction after 8 h incubation with (e) and without 14C-SAM (d). Purification of 14C-labeled feruloyl-CoA after 4CL reaction (g). Fractions were collected every minute and analyzed using a scintillation counter. HPLC analysis of purified 14C-feruloyl-CoA (f)
SAM Concentration
Different SAM concentrations were tested in non-radioactive COMT reactions. Not surprisingly, when used in excess, a maximal yield of ferulic acid was obtained. The absolute yield of ferulic acid was almost 100% when using 0.5–1 mM concentrations of SAM and 0.2 mM caffeic acid as substrate (Fig. 3b and c). When using equal amounts of caffeic acid and SAM, about 50% of SAM methyl groups were incorporated into ferulic acid after 5 h incubation (Fig. 3c). The final yield could be improved by incubation for ≥8 h to up to 70%.
Preparation and Purification of 14C-labeled Feruloyl-CoA
For the final production of radioactive feruloyl-CoA, the yield of ferulic acid relative to the amount of 14C-labeled SAM used must be considered in order to minimize the use of the radiolabeled substrate for both safety regulation and economic reasons. For 14C-labeling experiments, standard concentrations of 100 μM caffeic acid and 50 μM 14C-SAM, equivalent to 2.5 μCi and 50 nmol, were determined to give a maximum yield of ferulic acid. After termination of the COMT reaction, an aliquot was analyzed by HPLC (Fig. 3e). According to ferulic acid standards, 35 nmol ferulic acid were obtained. In control reactions without SAM, caffeic acid was not converted into ferulic acid (Fig. 3d). Thus, 70% of SAM methyl groups were incorporated into ferulic acid. After inactivation of COMT, and in order to obtain 14C-labeled feruloyl-CoA, Nb4CL1 and cofactors were added, and the sample was further incubated for ≥8 h before being subjected to HPLC purification. The purified 14C-feruloyl-CoA corresponded to 68% of total counts used in the reaction (Fig. 3g), consistent with the calculated amount of 35 nmol and yield of 70% based on ferulic acid production.
Conclusion
In a previous attempt, radiolabeled feruloyl-CoA was synthesized using a crude protein extract derived from wheat seedlings. Due to high thioesterase activity, the final yield of labeled feruloyl-CoA esters did not exceed 10% [13]. In this study, we combined two enzymatic reactions in a single tube starting with caffeic acid as the substrate, 14C-SAM as the methyl donor, and recombinant caffeic acid 3-O-methyltransferase, which led to the incorporation of 70% of 14C-methyl groups from SAM into ferulic acid. In a second step, 4CL was used to prepare the corresponding thioesters without any significant losses. Moreover, recombinant Nb4CL1 and At4CL5 were used to prepare non-labeled hydroxycinnamoyl-CoA thioesters with excellent yield and purities. For preparation of small amounts of hydroxycinnamate thioesters, the methods presented are simpler, faster, and more efficient than previously published methods. 4CL and COMT clones are available upon request.
Abbreviations
- 4CL:
-
4-Coumarate-CoA ligase
- CoA:
-
Coenzyme A
- COMT:
-
Caffeic acid O-methyltransferase
- SAM:
-
S-adenosyl-l-methionine
- IgG:
-
Immunoglobulin G
- HRP:
-
Horseradish peroxidase
- Ni-NTA:
-
Nickel nitrilotriacetic acid
- TFA:
-
Trifluoroacetic acid
References
Beuerle T, Pichersky E (2002) Enzymatic synthesis and purification of aromatic coenzyme A esters. Anal Biochem 302:305–312
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Costa MA, Bedgar DL, Moinuddin SG, Kim KW, Cardenas CL et al (2005) Characterization in vitro and in vivo of the putative multigene 4-coumarate:CoA ligase network in Arabidopsis: syringyl lignin and sinapate/sinapyl alcohol derivative formation. Phytochemistry 66:2072–2091
Fry SC, Willis SC, Paterson AEJ (2000) Intraprotoplasmic and wall-localised formation of arabinoxylan-bound diferulates and larger ferulate coupling-products in maize cell-suspension cultures. Planta 211:679–692
Hamberger B, Hahlbrock K (2004) The 4-coumarate:CoA ligase gene family in Arabidopsis thaliana comprises one rare, sinapate-activating and three commonly occurring isoenzymes. Proc Natl Sci USA 101:2209–2214
Jones L, Milne JL, Ashford D, McCann MC, McQueen-Mason SJ (2005) A conserved functional role of pectic polymers in stomatal guard cells from a range of plant species. Planta 221:255–264
Kohler A, Kauss H (1997) Transfer of hydroxycinnamoyl residues to microsomal proteins from parsley. Phytochemistry 44:225–228
Lee D, Douglas CJ (1996) Two divergent members of a tobacco 4-coumarate:Coenzyme A ligase (4CL) gene family. Plant Physiol 112:193–205
Lüderitz T, Schatz G, Grisebach H (1982) Enzymic synthesis of lignin precursors. Purification and properties of 2-coumarate:CoA ligase from cambial sap of spruce (Picea abies L.). Eur J Biochem 123:583–586
Ma QH, Xu Y, Lin ZB, He P (2002) Cloning of cDNA encoding COMT from wheat which is differentially expressed in lodging-sensitive and -resistant cultivars. J Exp Bot 53:2281–2282
Ma QH, Xu Y (2008) Characterization of a caffeic acid 3-O-methyltransferase from wheat and its function in lignin biosynthesis. Biochimie 90:515–524
Mitchell RAC, Dupree P, Shewry P (2007) A novel bioinformatics approach identifies candidate genes for the synthesis and feruloylation of arabinoxylan. Plant Physiol 144:43–53
Obel N, Scheller HV (2000) Enzymatic synthesis and purification of caffeoyl-CoA, p-coumaroyl-CoA, and feruloyl-CoA. Anal Biochem 286:38–44
Obel N, Porchia A, Scheller HV (2003) Intracellular feruloylation of arabinoxylan in wheat. Evidence for feruloyl-glucose as precursor. Planta 216:620–629
Porchia AC, Sørensen SO, Scheller HV (2002) Arabinoxylan biosynthesis in wheat. Characterization of arabinosyltransferase activity in Golgi membranes. Plant Physiol 130:432–441
Stöckigt J, Zenk MH (1975) Chemical syntheses and properties of hydroxycinnamoyl-coenzyme A derivatives. Z Naturforsch 30:352–358
Yoshida-Shimokawa T, Yoshida S, Kakegawa K, Ishii T (2001) Enzymic feruloylation of arabinoxylan-trisaccharide by feruloyl-CoA: arabinoxylan-trisaccharide O-hydroxycinnamoyl transferase from Oryza sativa. Planta 212:47–74
Acknowledgements
This work was supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rautengarten, C., Baidoo, E., Keasling, J.D. et al. A Simple Method for Enzymatic Synthesis of Unlabeled and Radiolabeled Hydroxycinnamate-CoA. Bioenerg. Res. 3, 115–122 (2010). https://doi.org/10.1007/s12155-010-9085-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12155-010-9085-3